Article Text

Abstract
Cellular immunotherapies have emerged as a successful therapeutic approach to fight a wide range of human diseases, including cancer. However, responses are limited to few patients and tumor types. An in-depth understanding of the complexity and dynamics of cellular immunotherapeutics, including what is behind their success and failure in a patient, the role of other immune cell types and molecular biomarkers in determining a response, is now paramount. As the cellular immunotherapy arsenal expands, whole-body non-invasive molecular imaging can shed a light on their in vivo fate and contribute to the reliable assessment of treatment outcome and prediction of therapeutic response. In this review, we outline the non-invasive strategies that can be tailored toward the molecular imaging of cellular immunotherapies and immune-related components, with a focus on those that have been extensively tested preclinically and are currently under clinical development or have already entered the clinical trial phase. We also provide a critical appraisal on the current role and consolidation of molecular imaging into clinical practice.
- Adaptive Immunity
- Immunotherapy, Adoptive
- Receptors, Chimeric Antigen
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Molecular imaging of T cell-based immunotherapy
Adoptive T cell therapy seeks to harness a patient’s immune system to hunt down and kill cancer cells. This new class of ‘living drugs’ includes tumor-infiltrating lymphocytes (TILs), transgenic endogenous T cell receptor (TCR)-T cells, chimeric antigen receptor (CAR)-T cells and regulatory T cells (Tregs) among others.
Distribution, successful targeting, expansion, infiltration, and antitumor activity are only some of the many aspects influencing the way patients respond to immunotherapy. Answering the following questions might help us improve the safety and efficacy of cell-based immunotherapy:
What are the immune cell types contributing to enhance immunotherapy response?
Can we identify and monitor a specific subset of T cells?
What are the kinetics of distribution of cell-based immunotherapies in the human body and are they capable to successfully target the tumor?
What is their activation status at the tumor site?
How long do they persist in the tumor and how does persistence correlate with antitumor response?
Do therapeutic cells continue to expand over time and retain their function?
What are the factors responsible for the heterogeneous response in patients, and can we predict them?
Can we reliably predict and prevent side effects associated with immunotherapy (including on-target off-tumor toxicity, neurotoxicity and cytokine release syndrome (CRS)) in patients?
Non-invasive molecular imaging is a powerful tool that can be successfully employed by scientists and clinicians to answer all above questions and better understand the reasons behind immunotherapy success and, more importantly, its failure. The variety of labeling methods and imaging modalities available can be tailored toward the different types of cellular immunotherapies (figure 1A), immune-related components (metabolism, checkpoints, etc.) and immune cell biomarkers. In this review, we provide an extensive description of all available modalities and labeling strategies suitable for imaging the immunotherapy paradigm and describe their preclinical and clinical application. Priority has been given to clinically relevant seminal studies.

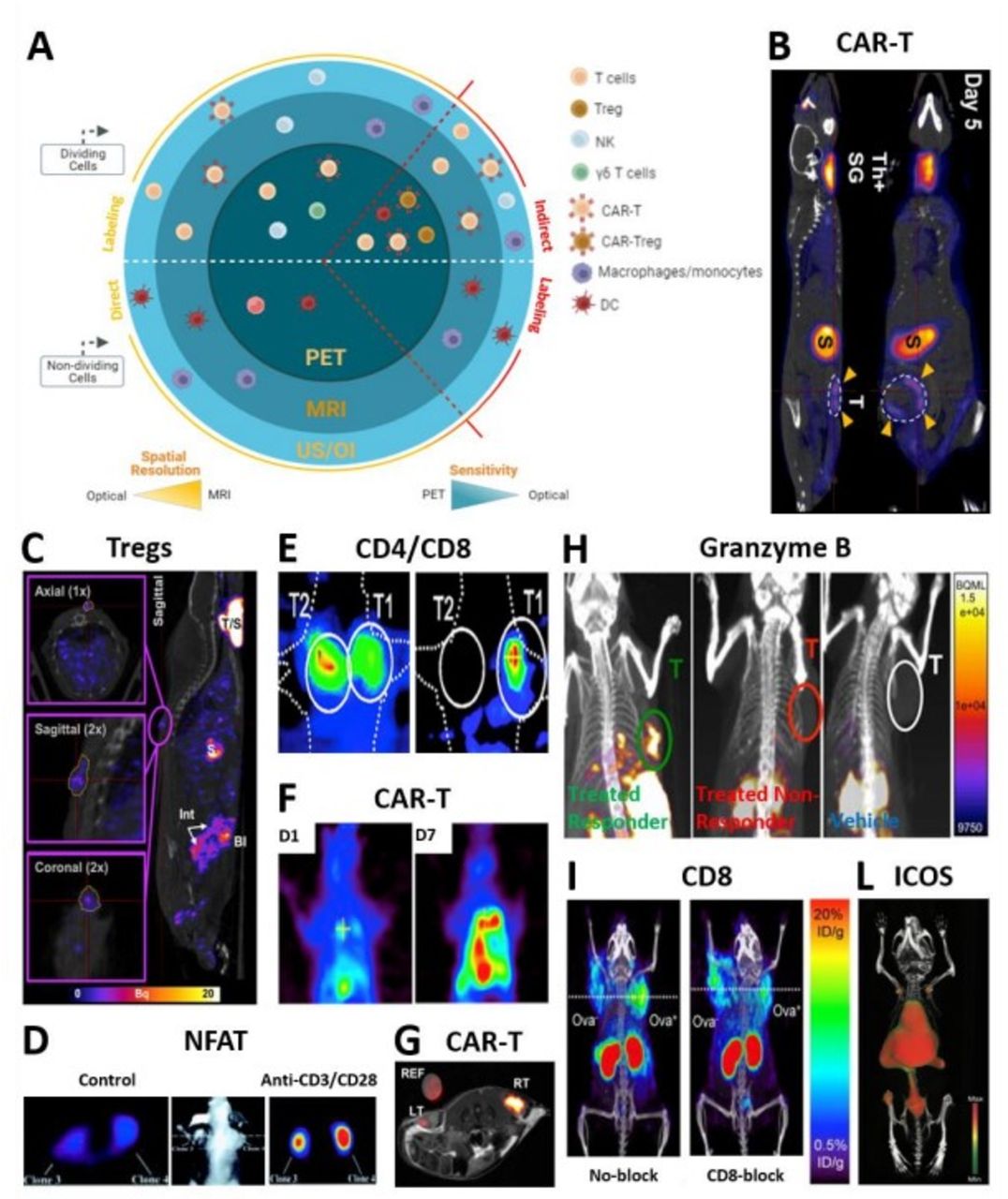
Molecular imaging of cellular immunotherapies and immune-related biomarkers.| T cell trafficking and activation status and biomarkers visualized by different reporter-based and direct imaging modalities. (A) Sketch summarizing all immune cells amenable to molecular imaging through indirect and direct approaches. (B) Coronal and sagittal slices showing intra-tumorally injected panErbB-directed CAR-T cells tumor targeting and retention in a TNBC model by NIS/[18F]BF4- PET-CT 5 days post treatment.27 Indicated with yellow arrows are CAR-T cells residing at the tumor. Other signal is due to the endogenous NIS expression: ThSG is thyroid+salivary glands; S is stomach. (C) In vivo trafficking and persistence at the skin graft of intravenously administered polyclonal Tregs by NIS/99mTcO4- SPECT-CT.33 The sagittal image depicts a representative animal receiving 5×106 hNIS-GFP+Tregs 30 days prior to imaging and residing at the skin graft. (D) Transaxial sections show a representative nude mouse bearing different subcutaneously injected Jurkat cells. The NFAT-mediated TKGFP+reporter responded to TCR activation by intravenous administration of anti-CD3/CD28 antibodies (right) as compared with anti-mouse control antibody (left).43 To allow for tracer clearance from the bloodstream and improve signal-to-noise ratio, HSV1-tk/ [124I]FIAU PET imaging was performed 24 hours post tracer administration. (E) Visualized is selective accumulation of systemically administered EBV-directed CD8+cells in HLA-A0201+EBV BLCL tumor (T1) by HSV1-tk/ [124I]FIAU PET (right). Although hNET/ [123I]MIBG SPECT images show a high accumulation of CD4+cells in corresponding HLA-DRB10701+EBV BLCL (T2) tumors (left), signal is detected also in T1 tumor (right), likely caused by the [124I]FIAU decay.22 Images were taken 4 hours post administration of a mixture of tracers. (F) [18F]-FEAU PET showing increased intrapleural accumulation of HSV-tk+mesothelin-targeted CAR-T cells by day seven post regional infusion in a orthotopic model of ffLuc+mesothelioma.74 The latter correlated to decreased tumor burden as depicted by tumor BLI (not shown). (G) TATP-F68-PFC labeled 1×107 human CAR-T cells visualized by in vivo 19F MRI (RT: right tumor) as compared with F68-PFC control (LT: left tumor) in a mouse glioma model.75 19F image of injected cells is in pseudo-color; T2-weighted 1H image of tumors is in greyscale. (H) anti PD-1 and anti-CTLA-4 immunotherapy efficacy assessed 12 days post treatment by in vivo imaging of granzyme B as a marker of T cell activation using 68Ga-NOTA-GZP probe for PET-CT.41 Sagittal images showing differentiation between responders treated (green), non-responders treated (red) and vehicle treated (white) mice. (I) Representative PET-CT images acquired 22 hours post 89Zr-malDFO-169 cys-diabody injection and showing unblocked and CD8-blocked mice bearing EL4-Ova- (left) and EL4 Ova+ (right) tumors 5 days postadoptive OT-I T cell transfer.38 (L) Coronal-ventral view of CD-19-targeted CAR-T cell activation during anti-tumor response by 89Zr-DFO-ICOS mAb PET-CT. Signal due to successful targeting is detected in lumbar vertebrae, iliac bone, femur and tibia. Signal due to tracer clearance is in heart, spleen and liver. imaging performed 5 days post-CAR-T cell systemic delivery and 48 hours post-tracer administration.76 All figures adapted with permission from publishers. DC, dendritic cell; hNIS, human sodium iodide symporter; NFAT, nuclear factor of activated T cells; PET, positron emission tomography; SPECT, single photon emission computed tomography ; TCR, T cell receptor; TNBC, triple-negative breast cancer.
Immune cell imaging labeling strategies and modalities
Non-invasive molecular imaging of cell-based immunotherapy can be achieved using direct or indirect strategies (refer to Kircher et al for graphical representation).1 2
Direct cell labeling is based on the ex vivo labeling of cells prior to administration into the desired recipient (animal model and/or human) and is particularly used in the context of nuclear-based imaging (positron emission tomography (PET) and single photon emission computed tomography (SPECT)). This approach enables a straight-forward labeling of any immune cell or immune-related component but also comes with multiple drawbacks, including label efflux and dilution during cell expansion. With efflux, the probe leaks out of the cell and redistributes throughout the body, rendering it impossible to discriminate between labeled cells and free probe. As the directly labeled cells continue to actively divide, the label is distributed to the progeny and progressively diluted, making it unsuitable to image highly proliferative cells (e.g., T cells). After their systemic administration, prelabeled T cells generate a detectable signal regardless of their viability status: in fact, damaged and dead cells still lead to signal, resulting in the potential misinterpretation of the patient’s response to immunotherapy.
Indirect cell labeling is a very powerful approach for the non-invasive and repetitive observation of T cell-based immunotherapies and is achieved by ectopic expression of a reporter gene in the cell of interest. For details on historical development and reporter expression cassettes refer to Serganova et al.3 Foreign or synthetic (non-human derived) reporters are more suited for the in vivo highly sensitive monitoring of systemically infused adoptive T cells as they are exclusively expressed in the cells of interest and do not generate background. However, host reporters have the advantage of not eliciting a host immune response. Unlike direct labeling, expression and function of the reporter are strictly linked and are a prerequisite of viable cells, meaning that only intact/healthy cells will result in signal. The permanent integration of the reporter into the host cell genome results in absence of label dilution during cell division and allows the long-term imaging of the rapidly expanding immune cell therapeutics in preclinical models as well as the human body. For preclinical studies, the unlimited imaging window allows to quantify the object of interest without having to sacrifice the animal, thereby significantly reducing the number of animals needed in the study and minimizing the inter-animal variability.
Available imaging strategies that can be tailored toward immunotherapy include: (1) optical, (2) ultrasound (US), (3) MR and (4) nuclear-based imaging. Magnetic particle imaging is an emerging modality in the field of cellular immunotherapies4 and will not be discussed in this review.
Optical imaging (OI) is a non-invasive imaging modality using visible, ultraviolet and infrared light and the special properties of photons to acquire biological information. Within this class, fluorescence (FLI) and bioluminescence imaging (BLI) have emerged as powerful modalities. FLI relies on the use of light-activated fluorescent molecules and encompasses a wide range of resolution and imaging depths, from subcellular (<400 µm with intravital microscopy) to small-animal imaging (1–3 mm spatial resolution at <10 cm with FLI mediated tomography).5 FLI reporters are categorized as artificial cell surface molecules, near-infrared, photoactivatable and photoconvertible and (monomeric and non) fluorescent proteins (previously reviewed by Volpe et al).6 In BLI, visible light is emitted when an enzyme (eg, a luciferase) oxidizes its own substrate. The enzyme is introduced in the desired cells by genetic engineering and serves as a reporter. Available luciferase enzymes (either present in nature or chimeras) and substrates can be found in a review by Mezzanotte et al.7
Consideration on optical imaging. BLI and FLI are widely used to track cell-based immunotherapies in preclinical research due to the simplicity of the technique and the affordable costs. Since they do not involve exposure to ionizing radiation, they can be safely used for repeated imaging of immune cells. However, their application to humans is restricted by several key limitations: (1) reporters are typically of non-human origin, thus posing the risk for immunogenicity; (2) limited light tissue penetration; (3) only a qualitative output signal (relative quantification) can be obtained, while other imaging modalities (e.g., positron emission tomography (PET) allow for absolute quantification.
Ultrasound imaging (US) is based on the propagation of mechanical waves from a transducer into the tissue, resulting in echoes that can be resolved to depict objects of interest. US signal is effectively and safely enhanced by contrast agents (encapsulated gas microbubbles or non-microbubbles).
Considerations on ultrasound imaging. US is widely available in preclinical and clinical/diagnostic settings, non-invasive, quantitative and inexpensive compared with other imaging modalities. Depending on the trafficking kinetics of the therapeutic cells, the high temporal resolution of US and fast distribution of contrast agents allows to in vivo track them within minutes after probe administration. The rapid clearance also allows the repetitive injection of the agent without having to wait hours or days, although there may not be clinical benefit of doing so in the context of immunotherapy as no significant changes are expected in such a short time. Importantly, with the expansion of the so-called ‘acoustogenetics’, acoustic reporters may not only used to non-invasively monitor immune therapeutics, but also to remotely control them for therapeutic purposes.8 However, despite the recent development of mammalian-compatible reporters,9 the current unavailability of non-immunogenic reporters is limiting their clinical translation.
Magnetic resonance imaging (MRI) exels in soft tissue contrast and not involving ionizing radiation. In fact, it is based on the perturbation of the nuclear magnetic moment of endogenous nuclei (1H in H2O) in a magnetic field. Negative contrast agents (iron-oxide nanoparticles)10 and reporter genes11 result in the reduction or absence of signal. Paramagnetic agents (eg, gadolinium) are positive contrast agents and increase the MRI signal.
Considerations on magnetic resonance imaging. Magnetic nanoparticles and reporter genes can be both used to label cell-based immunotherapies. Positive contrast agents are not safe and sensitive enough to be clinically translated.12 Different chemical exchange saturation transfer MRI approaches have been developed to image T cell metabolites changes as biomarkers of their activation state and response to immunotherapy.13
Nuclear-based imaging offers whole-body non-invasive imaging capabilities for preclinical and clinical application.14 Direct labeling can be performed using SPECT-compatible and PET-compatible radioactive probes, while the indirect labeling is based on cell engineering with foreign, host and synthetic radionuclide-based reporter genes. The reporter transgene can encode for: (1) transporters (internalizing the probe), (2) receptors (expressed on the plasma membrane of cells and irreversibly binding to the probe) or (3) enzymes (modifying the probe structure and resulting in its entrapment and intracellular accumulation). For a complete list of available nuclear-based reporters and paired probes refer to Volpe et al.14
Consideration on nuclear-based imaging. In addition to the already discussed limitations of direct labeling, a few more considerations are needed. As the overall signal strength of directly labeled cells is only retained until radiotracer decay, the choice of tracers with longer half-life is crucial to extend the already limited time window for imaging. However, this approach would have consequences for the management of patients and their body excreta, thus posing a limitation for its clinical translation. Moreover, the longer decay prolongs the time of radioactivity emission in the patient’s body and may, therefore, damage the healthy tissues and impact on the viability of the labeled immune cells. In fact, clinically used radiotracers (e.g., 111In-oxine,89Zr-oxine), when used at high concentrations, can interfere with cell survival and proliferative ability.15 On the other hand, reporter-based imaging of cellular immunotherapies is not dictated or limited by the radiotracer half-life. In fact, short-lived isotopes are not only used but also recommended to avoid potential toxicity and radio-damage of target cells. However, challenges posed to the strategy include (1) potential immunogenicity of foreign and/or synthetic reporters, (2) elaborate and costly protocols, (3) access to highly skilled research personnel to conduct cell engineering, in vitro assays, development of mouse models and often long and elaborate in vivo imaging experiments and (4) the need of a solid imaging infrastructure and annexed radiochemistry facility.
Application of imaging toward cell-based immunotherapy
Conventional imaging methodologies routinely used in clinical practice focus on assessment of disease response by providing anatomic (e.g., computed tomography (CT), MRI) or metabolic (e.g., [18F]-fluorodeoxyglucose (FDG)-PET, functional MRI) information of the tumor but fail to give a complete representation of the complex spectrum of responses to immunotherapy. As the latter is often accompanied by an initial ‘pseudoprogression’, understanding the distinctive features of tumors treated with immunotherapy and the fate of these ‘living drugs’ is paramount to reliably predict responses.
Here we report seminal studies using molecular imaging for the non-invasive preclinical and clinical imaging (tables 1 and 2) of cellular immunotherapies and immune biomarkers, searched using “immunotherapy”, “cancer”, and “imaging” key words and by literature search on PubMed.
Seminal preclinical nuclear-based, MRI and ultrasound studies employing direct and indirect molecular imaging for the in vivo monitoring of T cell-based immunotherapies and their immune-related components
Past and ongoing clinical trials with their respective molecular targets and imaging agents used for the nuclear-based and MRI-based imaging of T cell-based immunotherapy and related immune components
Imaging T cell trafficking and persistence
Current clinical trials involving infusion of cell-based immunotherapies are performed without knowledge of their whole-body distribution during treatment. Cell-based therapeutics can potentially redistribute and lead to so-called ‘on-target off-site’ toxicity, causing severe side effects or even death. Non-invasive long-term imaging can be used for the monitoring of their spatiotemporal in vivo distribution, persistence at the tumor site, survival and in situ expansion.
Cytolytic T cells (CTLs). Nuclear imaging with FDA-approved 111In-oxine clinical probe16 and 89Zr-oxine15 was employed to track the fate of directly labeled CTLs. A good manufacturing practise (GMP)-compatible production of 89Zr-oxine and white blood cell radiolabeling has been described and could accelerate its application in in-human cell imaging studies.17 Indirect strategies for in vivo tracking of T cells involve the use of BLI (overviews from Costa et al18 and Rabinovich et al)19 or nuclear-based (overview from Volpe et al) reporters.14 Koehne et al demonstrated for the first time the feasibility of long-term imaging of CTLs by HSV1-tk/[131I]FIAU- and [124I]FIAU-PET.20 As the interplay between CD4+and CD8+ T cells has a significant impact on the kinetics, persistence and antitumor activity of adoptive cell therapies,21 a reporter-based method was developed preclinically to independently track the CD4+ and CD8+ T cells by human norepinephrine transporter (hNET)-afforded -SPECT and HSV1-tk-afforded-PET, respectively (figure 1E).22 Notably, a human carcinoembryonic antigen (hCEA) reporter could also be used for the molecular imaging of adoptive T cell therapies, provided that the target tissue is not expressing any hCEA.23
Chimeric antigen receptor (CAR)-T cells. Direct labeling protocols using 89Zr-oxine24 and 89Zr-DFO25 have been developed for the in vivo tracking of CAR-T cells. Many host, foreign and synthetic reporter genes were also employed and provided a greater advantage for the repeated monitoring of CAR-T cells by either optical, ultrasound, nuclear imaging or hybrid. Moroz et al preclinically demonstrated the superiority of hNET/[18F]MFBG reporter/probe combination for the repeated imaging of T cells (sensitivity of<1×105 T cells/cm3).26 CAR-T detection sensitivity was as high as 3000 cells with sodium iodide symporter (NIS)-based [18F]BF4- PET (figure 1B) and revealed the inverse correlation between CAR-T tumor retention and PD-L1 expression in triple-negative breast cancer models.27 tPSMAN9Del-based [18F]DCFPyL PET enabled the reliable detection of 2000 CAR-T cells and the visualization of CAR-T infiltration into local and metastatic lesions in a model of acute lymphoblastic leukemia.28 A novel reporter, eDHFR, derived from E. Coli allowed for successful visualization of GD2-specific CAR-T cells targeting human colorectal xenografts using [18F]TMP-afforded PET.29 A novel DOTA antibody reporter 1 (DAbR1) irreversibly binding to the lanthanoid (S)−2-(4-acrylamidobenzyl)-DOTA (AABD) was tested in xenografts and provided information on the in vivo distribution and homing of systemically administered CD19-directed CAR-T cells at the tumor using 86Y- and 177Lu-AABD.30 The first documented in-human study by Keu et al31 showed the trafficking of CD8+GRm13Z40 (interleukin-13 zetakine receptor alpha 2)-targeted CAR-T cells previously infused into the medial left frontal lobe of patients with high grade glioma, providing insights on their successful homing at the tumor by HSV1-tk /[18F]FHBG PET (figure 2A; NCT01082926).31
{kind=link}
{kind=link}
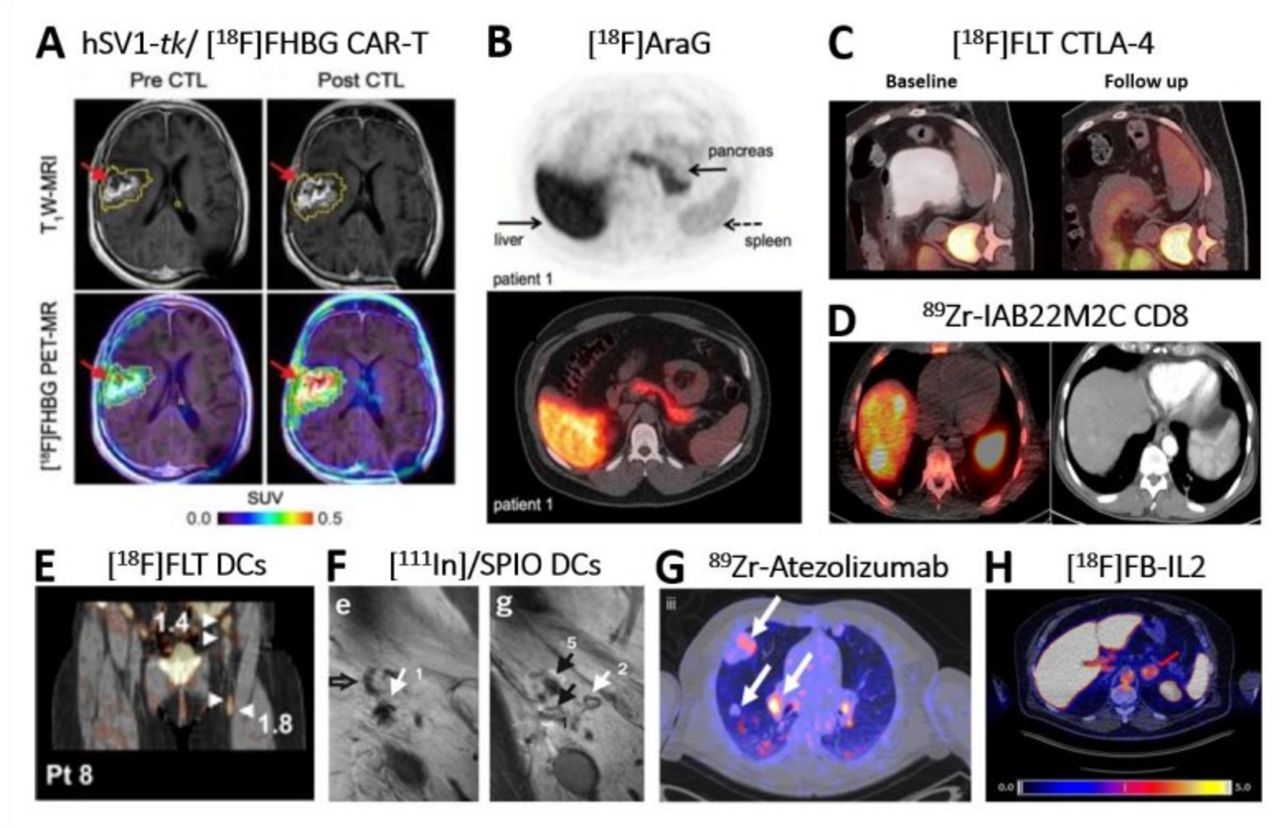
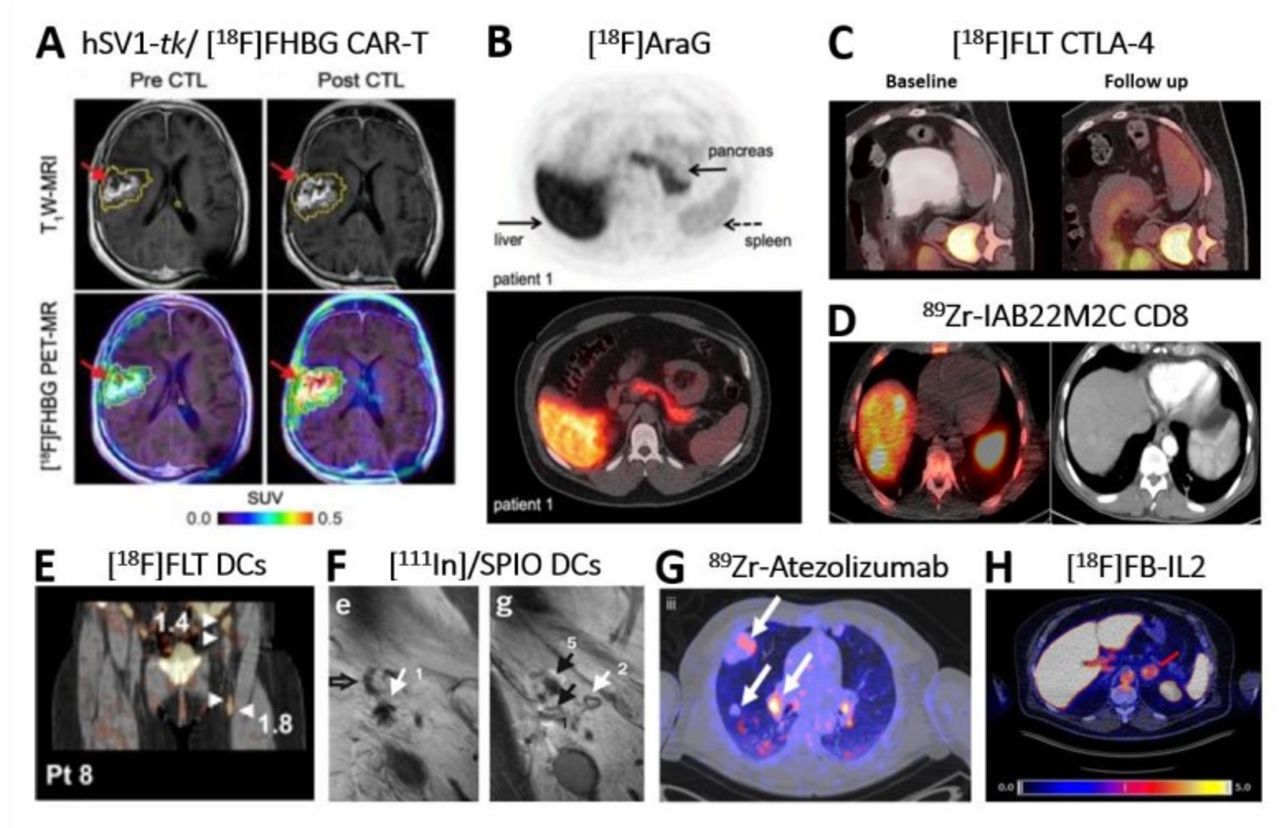
Clinical application of direct and indirect molecular imaging in cell-based Immunotherapy. (A) Non-invasive reporter gene imaging of IL-13 zetakine-directed and HSV1-tk-expressing CAR-T cells in a 66-year-old patient with recurrent frontoparietal glioblastoma. [18F]FHBG PET-CT revealed increased uptake after CAR-T cells infusion 1-week post infusion. Tumor extent was assessed by T1W MRI (top row) before and after CAR-T intratumoral infusion. Images were superimposed with [18F]FHBG PET (bottom row).31 (B) Image of an healthy volunteer showing 18F]F-AraG tracer in vivo distribution 60 min post intravenous administration of 189.07 MBq.99 The tracer exhibits hepatobiliary and renal clearance. shown are transversal PET (top) and fused PET-CT (bottom). (C) [18F]-FLT PET-CT imaging revealed T cell splenic proliferation in a patient with metastatic melanoma undergoing a durable regression post anti-CTLA-4 (tremelimumab) treatment.54 Baseline scan (left) was compared with a 3-month follow-up scan (right). (D) 89Zr-IAB22M2C PET-CT of CD8+T cells performed in a patient with hepatocellular carcinoma on immunotherapy for 12 weeks prior to imaging.39 40 Zr-IAB22M2C-positive lesions in the PET-CT merge (left image) correspond to two liver metastases (left side), spleen (right side) and bone marrow (middle). Additional uptake was seen also in three abdominal lymph node metastases. CT alone is in right image. Images acquired 24 hours post tracer injection. (E) [18F]FLT PET-CT visualizes immune responses in four LNs (see arrows) 3 days after intranodal delivery of [111In]/SPIO-labeled and antigen-loaded DCs and 1 hour after tracer injection.62 In fact, tracer retention was observed in the injected ln and the three draining LNs. (F) In vivo migration of 111In]/SPIO-labeled dendritic cells from LN1 (site of injection; arrow 1/e) to other LNs (arrows 5/g) using MRI.63 Open arrow in left image (E) indicates empty ln (NO SPIO); closed black arrow in right image (G) indicates ln positive for SPIO. Images taken 2 days post intranodal injection of DCs. (G) 89Zr-Atezolizumab PET revealed uptake in different tumor lesions (white arrows) in one representative patient 7 days postinjection.100 Pronounced uptake heterogeneity was seen within the same patient. (H) [18F]FB-IL2 uptake corresponding to activated tumor-infiltrating T cells at an adrenal gland metastasis (red arrow) in a representative melanoma patient undergoing immune checkpoint inhibitor therapy (SUVmax of 5.2).37 All patients enrolled in this study received intravenous bolus injection of~200 MBq [18F]FB-IL2 in 5 min. Transversal PET-CT image shown. All figures adapted with permission from publishers. CAR, chimeric antigen receptor; DC, dendritic cell; LNs, lymph nodes; SPIO, superparamagnetic iron oxide; PET, positron emission tomography.
Imaging regulatory T cells and gamma delta T cells
Regulatory T cells (Tregs). The efficacy of adoptive regulatory T cells (Treg) therapy (based on either polyclonal or the more potent antigen-specific Tregs), was demonstrated in numerous preclinical disease models and clinical trials.32 Nuclear-based reporter gene imaging offers a clinically compatible platform for future Tregs imaging in humans by providing the missing information on their in vivo distribution and persistence and addressing potential safety concerns. Notably, NIS-afforded SPECT-CT allowed tracking of human polyclonal Tregs to the human skin transplant in humanized mice (figure 1C). Authors further demonstrated that Tregs successful trafficking to the skin graft is regulated by Gr-1+ neutrophils and monocytes.33
CAR-Tregs. To improve specificity for the target antigen and therapy success, the field is quickly evolving from polyclonal to CAR-Tregs. As a result, more CARs are being developed and require in vivo testing by molecular imaging. Notably, HLA-A2 CAR-Tregs were recently engineered to constitutively express IL-10 and NIS reporter. In vitro, cells maintained their phenotype and gained an additional suppressive advantage, while NIS could be used for their non-invasive in vivo monitoring.34
Gamma delta T cells. They exert their cytotoxic activity through antigens recognition and indirectly enhance the anti-tumor activity of other immune cells by secreting multiple cytokines. Tracking with 89Zr-oxine in a xenograft model of human triple-negative breast cancer revealed a significantly increased tumor infiltration upon pretreatment with PEGylated liposomal alendronate.35
Functional Imaging of T cells
Upon antigen recognition and engagement, T cells upregulate multiple activation biomarkers (surface, nuclear), leading to the release of anti-tumor cytokines. Those are early predictive biomarkers of immunotherapy response, providing valuable information on T cell activation and expansion (e.g., signal increase over time), and can be targeted by molecular imaging.36 Like cancer cells, activated T cells undergo metabolic reprogramming to survive the hostile tumor microenvironment (TME) and retain their antitumor activity. Those metabolic changes can also be in vivo monitored in real-time using molecular imaging.
T cell activation biomarkers. In vivo imaging of T cell-surface markers (such as the costimulatory molecules OX40 and ICOS (figure 1L), IL-2 (NCT01789827; NCT02922283 in figure 2H),37 TCR and lineage defining molecules (including CD3, CD4, and CD8) was successfully performed using direct labeling with PET radiotracers.36 Preclinical testing of 89Zr-DFO anti-CD8 cys-diabody (figure 1I)38 and 89Zr-Df-IAB22M2C mini-body (figure 2D; trialed in NCT04029181, NCT03107663, NCT03802123)39 revealed CD8+T cells response against cancer. A new 89Zr-DFO-REGN5054 antibody40 for the non-invasive detection of CD8 expression is now in clinical trial (NCT05259709). Other T cell-surface markers associated with T cell activation, such as PD-1, CTLA-4, TIM-3 and LAG-3, are also immune checkpoint molecules and will be discussed in their dedicated section.
Targets related to T cell activation. Granzyme B is a predictive biomarker of successful checkpoint immunotherapy and can be imaged by 68Ga-NOTA-GZP PET (figure 1H; NCT04169321).41 Similarly, a preclinically tested 89Zr-labeled monoclonal antibody targeting IFN-gamma may provide insights on immune T cell activation and function.42
Nuclear Factor of Activated T cells (NFAT)-mediated T cell activation. Ponomarev et al described a TCR-dependent NFAT-HSV1-tk-GFP inducible PET-FLI dual-reporter system under the control of an artificial cis-acting NFAT-specific enhancer (figure 1D).43 44 On TCR-MHC complex engagement, the NFAT-HSV1-tk-GFP starts reporting on NFAT-mediated transcription of IL-2 and other cytokines, thereby providing information on the in vivo activation status of T cells. The combination of NFAT-inducible and constitutive reporters allowed to simultaneously visualize the trafficking, proliferation and activation of donor T cells and T cell precursors during T cell development after allogeneic hematopoietic stem cell transplantation in a mouse model of graft-versus-host disease.45
Metabolic targets. [18F]FDG imaging is routinely used to assess the changes in glucose metabolism and monitor the response in patients undergoing immunotherapy, including checkpoint inhibition.36 Other 18F-based examples are in overview from van der Veen et al.46
Imaging the Tumor Microenvironment (TME)
As the TME plays a critical role in immune surveillance and immune evasion, its major regulators can be considered promising immunotherapeutic targets. Immunosuppressive conditions include the upregulation of several immune checkpoints negatively regulating T cell activity (e.g., PD-1/PD-L1, CTLA-4, TIM-3 and LAG-3). Targeting of checkpoints by so-called ‘immune checkpoint inhibitors’ led to the Food and Drug Administration (FDA) approval for clinical use of (1) PD-1 inhibitors (e.g., pembrolizumab, nivolumab), (2) PD-L1 inhibitors (e.g., atezolizumab, avelumab, durvalumab) and (3) CTLA-4 inhibitor (ipilimumab). All immune checkpoints are amenable to molecular imaging and a wide range of agents has already been developed to non-invasively interrogate their heterogeneous expression and dynamics in preclinical models and clinical trials.36 46
Programmed cell death protein 1 (PD-1/CD279). A preclinical evaluation of 89Zr-labeled FDA-approved Pembrolizumab (Keytruda; humanized IgG4) suggested its use for the selection of patients benefitting from anti-PD-1 therapy.47 Suitability of 89Zr-labeled FDA-approved anti-PD-1 antibody Nivolumab as a diagnostic and disease monitoring tool is being investigated in a clinical trial (2015-004760-11).48 Accurate identification of early responses and resistance to PD-1 blockade was assessed by 89Zr-labeled Pembrolizumab PET (NCT02760225).49
Programmed cell death protein ligand 1 (PD-L1/CD274). Antibody-based therapies targeting PD-L1 aim to monitor its in vivo expression as a predictive biomarker for checkpoint inhibitor therapy success. Examples are the newly developed nanobodies against PD-L150 and BMS-986192 tracer51 for SPECT and PET imaging, respectively. The latter tracer is now in two clinical trials (NCT03843515, NCT3520634).
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4/CD152). Imaging CTLA-4 as a predictive biomarker of anti-CTLA-4 therapy is a useful tool. To reliably monitor CTLA-4 in vivo expression, the FDA-approved CTLA-4 antibody Ipilimumab was radiolabeled with multiple PET tracers and validated in xenograft52 and humanized mouse models.53 In a clinical trial, investigators are studying if high uptake of 89Zr-Ipilinumab correlates to treatment response (NCT033313323). As CTLA-4 blockade prevents engagement to B7 costimulatory molecules and promotes T cell activation and proliferation, [18F]FLT PET can detect the associated changes in DNA synthesis during anti-CTLA-4 therapy in secondary lymphoid organs (e.g., spleen) of patients with metastatic melanoma (figure 2C; NCT00471887).54
Lymphocyte activation gene 3 protein (LAG-3). The in vivo expression of LAG-3 can be assessed using 99mTc labeled nanobodies55 and 89Zr labeled REGN3767 fully human anti-LAG-3 antibody.56 Clinical studies with 89Zr-DFO-REGN3767 are ongoing in patients with diffuse large B-cell lymphoma (NCT04566978) and advanced solid tumors (NCT04706715).
T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3). TIM-3 in vivo expression was monitored in a metastatic melanoma mouse model using the 64Cu-NOTA radiolabeled rat anti-mouse TIM-3-specific monoclonal antibody (RMT3-23) by PET.57 A clinical trial investigated the in vivo distribution of 18F-CFA for the reliable monitoring of immune activation post anti-TIM-3 and anti-PD-1 treatment (NCT03409419). However, no results are available to date.
T cell immunoreceptor with Ig and ITIM domains (TIGIT). Two antibody-based TIGIT-specific 64Cu- and 89Zr-labeled tracers were developed for the preclinical quantification of TIGIT expression on TILs.58 These probes could be used to select which patients are candidates for anti-TIGIT immunotherapy.
Imaging other immune cell types
Natural killer (NK) cells. The lack of monitoring and diagnostic platforms to assess of NK-based immunotherapies efficacy has constituted a major drawback to the development of novel therapeutic strategies. NK cells can be imaged in real-time by MRI.59 As some iron-oxide nanoparticles have already been approved by the FDA (e.g., ferumoxide, ferumoxytol and ferucarbotran), this approach can be directly translated into the clinic for the short-term monitoring of engineered NK cells. However, nuclear-based imaging remains the most promising methodology for their clinical translation due to the increasing number of FDA-approved probes.59 Notably, Sato et al demonstrated that 89Zr-oxine labeling of adoptively transferred NK cells from rhesus macaque, here used as a clinically relevant preclinical model, does not impact neither cell phenotype nor viability or function of therapeutic cells, thereby providing a clinically translatable platform for their in vivo tracking in humans.60
CAR-NK cells. They provide the unique opportunity to engineer off-the-shelf allogeneic products readily available for clinical use. Direct labeling is a readily applicable method to monitor their in vivo fate in patients,59 whereas the challenges posed by the intrinsic properties of NK cells (e.g., poor transduction with retroviral and lentiviral methods) have delayed the development of an indirect and clinically compatible long-term monitoring tool.
Dendritic cells (DCs). In vivo tracking of DCs is typically achieved by direct labeling. Aarntzen et al proved that pretreatment of melanoma patients with an extra dose of 19F-labeled DCs does not improve their relocation to the lymph nodes, a prerequisite for them to trigger an effective immune response.61 The same author used [18F]FLT to visualize T and B cell proliferation as a readout of early immune responses after administration of DC vaccine therapy in melanoma patients with lymph nodal metastases (figure 2E; NCT00243594, NCT00243529).62 Accuracy of intranodal and internodal delivery and in vivo distribution of DCs can be assessed by superparamagnetic iron oxide nanoparticles (SPIONs) MRI (figure 2F).63 A comprehensive list of MRI-based DCs imaging is in overview from Bulte and Shakeri-Zadeh.64 Alternatively, DCs can be imaged using reporter genes, including NIS65 and ferritin.66
Monocytes and macrophages. As highly phagocytic cells, they can efficiently take up SPIONs67 and 19F-PFC68 and be monitored by MRI. Tumor-associated macrophages preferentially incorporate the FDA-approved iron oxide nanoparticle compound ferumoxitol and can be preclinically imaged by MRI.67 CAR-macrophages (CAR-M)69 were pioneered in Gill’s laboratory at UPenn and entered the very first in-human phase 1 multicenter clinical trial for the treatment of metastatic HER2-positive solid tumors (NCT04660929). With more of CAR-M being developed, we expect to see an increase of preclinical and clinical testing employing molecular imaging.
Imaging Cellular Immunotherapies: a Future Perspective
With the constant expansion of the cellular immunotherapy arsenal, imaging the in vivo fate of cellular immunotherapies is paramount for the reliable assessment and prediction of therapeutic responses. As reporter genes have multiple advantages over the direct labeling approach, they could be fully integrated into clinical practice. The clinical studies involving HSV1-tk (figure 2A and table 2) have set the stage for expanding the portfolio of clinically used reporters for cellular immunotherapies imaging. However, some impediments have slowed down the process, including the regulatory concerns of using viral delivery systems to introduce the reporter into the desired therapeutic cells. Since commercially available CAR-T products are now generated through viral delivery (e.g., the lentiviral-based Kymriah and the retroviral-based Yescarta), the skepticism of using them for the delivery of reporter is no longer justified. In addition, vectors could be rendered even safer by using self-inactivating ‘third generation’ versions currently tested in gene therapy.70 To avoid the viral-mediated random positioning of reporter genes into the host genome, safer ways based on gene editing (e.g., ZFPs, TALENs and CRISPR-Cas) are available.71 The delivery at the same time of both the CAR and the reporter (or reporters) is desired, therefore the construction of GMP-compatible cassettes for the delivery of large payloads is needed. Pairing two different reporters (including an inducible one) would allow the simultaneous in vivo assessment of distribution and targeting of cellular immunotherapies with the monitoring of their functional status. However, it would also require the development of clinical imaging protocols specifically tailored to this set up. While they provide good image contrast, xenogenic (e.g., HSV1-tk) and synthetic reporters can elicit a host immune response. Although this may not represent an issue for preclinical studies, particularly when heavily immunocompromised mouse models are used, it is indeed a barrier for their full integration into clinical practice. Host reporters are a valid alternative as they are recognized as ‘self’ by the immune system. To this class belong two promising candidates for clinical translation, namely the human sodium iodide symporter (hNIS) and the human norepinephrine transporter (hNET) with their respective clinically compatible radioactive PET probes, [18F]BF4- and [18F]MFBG.14 The limited endogenous expression, the favorable target-to-background ratio, exquisite detection sensitivity and faster tracer clearance, make NIS a superior reporter for the imaging of cellular immunotherapies. Since NIS tracers do not rely on cyclotron accessibility (e.g., 99mTcO4-, xyxI-) and can be produced by automated synthesis at high molar activities (e.g., 18F-BF4-), their production is greatly simplified and cost effective, making them broadly available for imaging. Moreover, the short half-life of [18F]BF4- greatly facilitates the management of patients in a clinical setting.
As host reporters are endogenously expressed in the human body and are responsible for unfavorable contrast, reporters within this class (e.g., hNIS, hNET, hPSMA) need to be strategically selected depending on the expected distribution of the cell-based immunotherapy and location of the tumor. The human somatostatin receptor subtype 2 (hSSTr2) can be endogenously expressed in several immune cell types, including T cells, and may, therefore, not be suited for the imaging of cellular immunotherapies. With the FDA approval of more compatible probes, we do expect a significant increase in usage of reporters in immunotherapy-related clinical trials, but their full integration into clinical practice is still premature. Direct imaging has, therefore, become the fastest approach for clinical translation. Despite the evident limitations intrinsic to the methodology, the technological advancement of the total-body PET (with its >40 times higher sensitivity than conventional PET) can be used to extend the tracking time of directly labeled cellular immunotherapies and immune-related biomarkers. Many antibodies targeting immune cell therapeutics or immune biomarkers have been successfully conjugated to imaging probes. Some of them are currently being tested in clinical trials, others will soon reach the same stage. However, the applicability of these probes for the in vivo monitoring of cellular immunotherapies and biomarkers will be affected by the pharmacodynamics and pharmacokinetics of the therapeutic compound.
Imaging cellular immunotherapies is already playing a central role in guiding the FDA approval of the next generation of off-the-shelf cell-based immunotherapies and in reliably evaluate response in treated patients. Tumors can respond in a heterogeneous manner to cellular immunotherapy.72 Biomarkers and checkpoints expression can be heterogeneous too.73 While biopsies alone may not be able to catch with the same level of precision the complexity of the immune response, the latter can be reliably captured by molecular imaging between patients or within the same patient and, sometimes, within the same tumor lesion. Therefore, the clinical application of molecular imaging is not exclusively limited to the in vivo monitoring of therapeutic immune cells and their activation status, but also of immune cell biomarkers as a readout of heterogeneous response to immunotherapy, contributing to the identification of patients most likely to benefit from these therapies and thus avoiding unnecessary side effects and reducing the costs associated with ineffective treatment. The increased number of clinical trials of cell-based immunotherapies employing molecular imaging and the constantly expanding clinically approved portfolio of imaging probes will provide justification toward the consolidation of this tool into clinical practice in the years to come. Moreover, radiomics in becoming increasingly important by offering a large array of prospective biomarkers for the interpretation and even prediction of immunotherapy treatment and unfavorable effects. This predictive value increases when combined to other ‘omics’ and/or artificial intelligence.
Summary
Requisites for the successful molecular imaging of cellular immunotherapies:
Absent or limited label dilution on cell division (preserved when using indirect reporter gene strategies).
Label retention as a prerequisite for repetitive non-invasive in vivo imaging.
Labeling strategy and imaging probe should not compromise viability, metabolism and/or function of immune cell therapeutics.
Essential requisites for clinical translation:
Exquisite sensitivity of the imaging modality (depth penetration) and quantification.
Availability of clinically compatible imaging probes.
Lack of reporter gene immunogenicity.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors Conceptualization: AV and VP. Writing original draft: AV. Review and editing: AV, VP, PSA and HS.
Funding AV is supported by the EIO Fellowship Award from the Center for Experimental Immuno-Oncology (FP00001443) and the Tow Foundation Fellowship Award (FP00004141) at Memorial Sloan Kettering Cancer Center. VP’s laboratory work is supported by NIH Grants (R01 CA220524-01A1, R01 CA204924 and R21 CA250478). PSA’s laboratory work is supported by NIH Grants (P30 CA008748, R01 CA236615-01, and R01 CA235667), the U.S. Department of Defense (CA180889 and CA200437), the Mr William H. Goodwin and Alice Goodwin, the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. The authors received further support from the NIH/NCI Cancer Center Support Grant to MSKCC (P30 CA008748).
Competing interests Dr Adusumilli declares research funding from ATARA Biotherapeutics, has served on the Scientific Advisory Board or as a consultant to ATARA Biotherapeutics, Bayer, BioArdis, Carisma Therapeutics, Imugene, ImmPACT Bio, and Johnson & Johnson, and has patents, royalties, and intellectual property on mesothelin-targeted CARs and T-cell therapies. Memorial Sloan Kettering Cancer Center has licensed intellectual property related to mesothelin-targeted CARs and T-cell therapies to ATARA Biotherapeutics and has associated financial interests.
Provenance and peer review Commissioned; externally peer reviewed.