Article Text

Abstract
Background Novel therapeutic strategies in ovarian cancer (OC) are needed as the survival rate remains dismally low. Although dendritic cell-based cancer vaccines are effective in eliciting therapeutic responses, their complex and costly manufacturing process hampers their full clinical utility outside specialized clinics. Here, we describe a novel approach of generating a rapid and effective cancer vaccine using ascites-derived monocytes for treating OC.
Methods Using the ID8 mouse ovarian tumor model and OC patient samples, we isolated ascites monocytes and evaluated them with flow cytometry, Luminex cytokine and chemokine array analysis, ex vivo cocultures with T cells, in vivo tumor challenge and T cell transfer experiments, RNA-sequencing and mass spectrometry.
Results We demonstrated the feasibility of isolating ascites monocytes and restoring their ability to function as bona fide antigen-presenting cells (APCs) with Toll-like receptor (TLR) 4 lipopolysaccharide and TLR9 CpG-oligonucleotides, and a blocking antibody to interleukin-10 receptor (IL-10R Ab) in the ID8 model. The ascites monocytes were laden with tumor antigens at a steady state in vivo. After a short 48 hours activation, they upregulated maturation markers (CD80, CD86 and MHC class I) and demonstrated strong ex vivo T cell stimulatory potential and effectively suppressed tumor and malignant ascites in vivo. They also induced protective long-term T cell memory responses. To evaluate the translational potential of this approach, we isolated ascites monocytes from stage III/IV chemotherapy-naïve OC patients. Similarly, the human ascites monocytes presented tumor-associated antigens (TAAs), including MUC1, ERBB2, mesothelin, MAGE, PRAME, GPC3, PMEL and TP53 at a steady state. After a 48-hour treatment with TLR4 and IL-10R Ab, they efficiently stimulated oligoclonal tumor-associated lymphocytes (TALs) with strong reactivity against TAAs. Importantly, the activated ascites monocytes retained their ability to activate TALs in the presence of ascitic fluid.
Conclusions Ascites monocytes are naturally loaded with tumor antigen and can perform as potent APCs following short ex vivo activation. This novel ascites APC vaccine can be rapidly prepared in 48 hours with a straightforward and affordable manufacturing process, and would be an attractive therapeutic vaccine for OC.
- antigen presentation
- immunologic memory
- genital neoplasms, female
- immunotherapy, active
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
There is an urgent need for novel therapeutic strategies in ovarian cancer (OC) as the survival rate remains dismally low.1 Many studies have shown that prolonged OC survival is correlated with an increased presence of CD3+/CD8+ T cells in tumors.2–5 This suggested that using cancer immunotherapy to boost antitumor T cell responses could benefit patients with OC. We have previously developed a personalized dendritic cell (DC)-based vaccine from peripheral blood monocytes, pulsed with whole tumor lysate and matured ex vivo with cytokines to elicit tumor-specific CD3+ T cells in OC. We have successfully tested this approach in mouse studies and in patients with high-grade serous OC (HGSOC) in clinical trials.6–10 It was efficacious in prolonging survival, however, ex vivo preparations of DCs are laborious, costly and require several days of cell culture.6 11 12Such complex production requirements restrict the availability of these vaccines outside of specialized centers.11–14
As an alternative, we investigated the potential of creating an affordable personalized cancer vaccine by using ascites as a source of tumor antigen-laden myeloid cells which do not require additional antigen pulsing in culture. Indeed, OC is a suitable model for this approach as most patients presented with advanced stage III/IV cancer will have malignant ascites at the time of diagnosis. Hence, this offers a unique opportunity to obtain a large amount of ascites material at the time of initial surgery. Although dominant immunosuppressive characteristics of the ascites environment are well documented, the immunogenic function of ovarian ascites-derived myeloid cells can be restored ex vivo.15–18 Here, we demonstrate that a brief (48 hours) ex vivo treatment with agonists to Toll-like receptor (TLR) 4 lipopolysaccharide (LPS) and TLR9 (CpG-oligonucleotides) plus a blocking antibody to the interleukin-10 receptor (IL-10R Ab) can restore the ability of ascites-derived monocytes to activate autologous tumor-reactive T cells and induce protective anti-tumor immunity when used as a cellular vaccine in a mouse model of OC. The translational relevance of this approach is supported by results obtained with human ascites samples, in which treatment with TLR agonists and a human IL-10R Ab restored the ability of ascites monocytes to elicit a potent cytokine response from autologous peritoneal T cells as well as enhanced ex vivo proliferation of tumor-reactive T cells. The clinical translation of this approach can lead to the development of a rapid, simple and more widely accessible therapeutic vaccine for women with advanced OC.
Methods
Cell lines and mouse strains
ID8 is an established cell line derived from spontaneous in vitro malignant transformation of C57BL/6 mouse ovarian surface epithelial cells, generously provided by Paul Terranova (University of Kansas). The ID8-VEGF (vascular endothelial growth factor) OC was established from ID8 OC cells transduced with the mouse VEGF 164 isoform, as previously described.19 The ID8 tumor cells were tested for mycoplasma annually and the tumor cells were used at less than 10 passages each time. For T cell proliferation assays, ID8 line transfected to express ovalbumin (OVA) was used.6 In all experiments, tumor cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, California, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin at 37°C and 5% CO2. Tumor and ascites were induced by inoculating female C57BL/6 mic aged 6–8 weeks (Charles Rivers, Massachusetts, USA) with 5×106 tumor cells intraperitoneally (i.p.) in 100 µL of sterile saline. Animals were weighed at least twice weekly and euthanized when they reached 30 g due to ascites accumulation as a surrogate endpoint for survival in compliance with institutional animal care and use committee requirements. Animal experimentation procedures were performed according to the protocols approved by the University of Pennsylvania.
Enrichment of different leucocyte populations for mouse studies
Ascites specimens were first enriched for CD45+ leucocytes using magnetic beads (Miltenyi Biotec, California, USA). To obtain myeloid-derived ascites cells, CD45+ ascites cells were plated for 2 hours and adherent CD45+ cells were considered candidate antigen-presenting cells (APCs) for further analysis. Non-adherent T cells were removed and resuspended in media containing IL−2 (50 IU/mL, BD Biosciences, California, USA) for T cell-related experiments. Peripheral T cells were harvested from blood collected from retro-orbital samples or from spleens using CD3 negative selection (Miltenyi Biotec, California, USA). In experiments that evaluated CD4+ or CD8+ populations, cells were first purified by CD3 negative-selection and followed by CD4 positive-selection magnetic beads (Miltenyi Biotec, California USA).
Treatment with TLR agonists and IL-10R ab
TLR4 agonist LPS (Sigma-Aldrich, Missouri, USA) was used at 50 ng/mL, TLR9 agonist CpG-oligonucleotides (mouse ODN1826 and human ODN2216, both from Invivogen, California, USA) were used at 3 µg/mL, and IL-10R Ab (mouse clone 1B1.3a, BD Pharmingen and human clone 3F9, Biolegend, California, USA) were used at 1 µg/mL.
Interferon production by mouse T cells
Following exposure to TLR agonists±IL-10R Ab for 48 hours in culture, ascites APCs were washed and plated at 1×106 cells/well. T cells were added in a 1:1 or 2:1 ratio. After overnight culture, cell-free supernatant was evaluated for interferon-γ (IFN-γ) (ELISA, Biolegend, California, USA). T cells or ascites APCs alone were used as negative controls, and T cells exposed to phorbol 12‐myristate 13‐acetate and ionomycin or conconavalin A (all from Sigma-Aldrich, Missouri, USA) served as positive controls. To confirm that IFN-γ production resulted from signaling through the T-cell receptor (TCR), 5 µg/mL of a blocking Ab to MHC Class I (clone 28-8-6, Biolegend, California, USA) and/or II (clone M5/114.15.2, eBioscience, Thermo Fisher Scientific, Massachusetts, USA) were added to the ascites APCs for 30 min prior to T cell exposure. Fc receptor blocking Ab (eBioscience, Thermo Fisher Scientific, Massachusetts, USA) was added to reduce non-specific binding. Extended cytokine profiles of cell-free supernatant were obtained with multiparameter analyte profiling (Myriad Rules-Based Medicine Luminex multianalyte profile).
In vivo survival studies
To determine if TLR agonist-treated ascites APCs could induce anti-tumor immune responses in vivo, ascites APCs were administered i.p. in tumor-bearing or tumor-naïve recipient animals. Briefly, ascites APCs were treated with LPS, CpG-oligonucleotides and IL-10R Ab for 48 hours, harvested using 0.5% Trypsin (TrypLE, Gibco, Thermo Fisher Scientific, Massachusetts, USA), washed and resuspended at 1×107 cells/mL sterile saline. Recipient animals were inoculated i.p. with 1×106 cells/100 µL saline, followed by two booster injections of 5×105 cells the following weeks. In tumor-naïve recipients, the first vaccine was administered 30 days before tumor challenge. In tumor-bearing recipients, first vaccine was administered beginning 3–7 days after tumor inoculation. In each case, animals were challenged i.p. with 5×106 ID8-VEGF tumor cells on day 0. Survival was measured from the time of tumor inoculation until the mice reached 30 g due to ascites accumulation.
Adoptive transfer of T cells
The role of T cells in tumor-rejection was tested by adoptive transfer of splenocytes from long-term survivors of prophylactic vaccination. Briefly, spleens were collected on day 216 from mice that received an i.p. inoculation of treated ascites APCs followed by a tumor challenge. Whole CD3+ cells, CD4+ or CD8+ cells were collected as described above. Tumor-naïve recipient animals were exposed to 400 Rads of whole-body irradiation 24 hours prior to receiving 1×106 adoptively transferred T cells i.p. Next day, recipient animals were challenged with 5×106 tumor cells injected i.p. and survival was measured from the day of tumor challenge until animals reached 30 g due to ascites accumulation.
Functional assessment of T cells from vaccinated animals
The functional status of T cells in long-term survivors of prophylactic and therapeutic vaccination studies were evaluated. Briefly, day 4 bone marrow-derived DCs (BMDCs) were pulsed with ID8 tumor lysate and matured with 100 ng/mL IFN-γ and 10 ng/mL LPS on day 5 as previously described.11 Then, BMDCs were washed and cocultured with T cells derived from peritoneal cavity, regional lymph nodes (mesenteric and inguinal) or spleen of vaccinated animals. In all cases, T cells were freshly isolated and enriched by CD3 negative-selection (Miltenyi Biotec, California USA). Following coculturing T cells with tumor antigen-pulsed BMDCs, IFN-γ was measured (ELISA, Biolegend, California, USA). Antibodies to MHC Class I (Biolegend, California, USA) and II (eBioscience, Thermo Fisher Scientific, Massachusetts, USA) were added at 5 µg/mL each to test for TCR engagement in the T cell response. T cells from unvaccinated tumor-bearing animals and tumor-naïve animals were used as negative controls, and T cells cultured with CD3/CD28 beads served as a positive control (Dynabeads, Thermo Fisher Scientific, Massachusetts, USA).
Human specimen isolation, in vitro stimulation and coculture assay
Human ascites specimens were collected from patients undergoing primary surgery for OC. Briefly, ascites samples were retrieved through the biobank of the Center of Experimental Therapeutics at the Department of Oncology, CHUV, Switzerland. Informed consent was obtained from all patients. After removing red blood cells with ammonium-chloride-potassium lysis buffer (Sigma-Aldrich, Missouri, USA), ascites cells were plated for 2 hours. Adherent cells were considered candidate APCs and were further enriched using magnetic CD14 beads (Miltenyi Biotec, California USA). CD14+ ascites APC were plated at 2×106 cells/mL in Roswell Park Memorial Institute (RPMI) 1640 media containing 10% FBS, and treated for 48 hours with TLR agonists and IL-10R Ab prior to evaluation for activation by flow cytometry. In some experiments, treated CD14+ ascites APCs were cocultured with T cells at 4:1 ratio, respectively, for 72 hours and the supernatant evaluated for cytokines by BD Cytometric Bead Array TCR-MHC-dependent T cell activation was assessed by adding 10 ug/mL anti-human HLA-ABC (clone W6/32, Biolegend, California, USA) and HLA-DR (clone Tü36, Biolegend, California, USA) antibodies in presence of human Fc block (BD Biosciences). To simulate the effect of reinfusing these cells i.p., coculture experiments were performed in cell-free supernatant from autologous ascites specimens in lieu of culture media. The effect of the ascites cytokine environment on the ability of treated ascites APC in eliciting an IFN-γ response was determined by comparing these results with the results obtained using culture media.
Proliferation of human tumor-associated lymphocytes, tumor reactivity and ELISPOT
After 72 hours coculture of tumor-associated lymphocytes (TALs) with non-stimulated or stimulated CD14+ ascites APCs, media was changed and the TALs expanded for 21 days with IL-2 (3000 U/mL, Proleukin, Novartis Pharma, Switzerland). As a reference for T cell expansion, conventional expansion was performed on non-sorted ascites cells plated at day 0 directly in media and IL-2 (3000 U/mL). Then, the number of cells and their phenotype were evaluated by flow cytometry. 2×105 of expanded TALs were used for TCR sequencing. Tumor-associated antigen (TAA) peptides discovered by immunopeptidomic as being presented on tumor or CD14+ cells from the same ascites were synthesized (PEPotec SRM Peptide, Thermofisher scientific, Massachusetts, USA) and used to evaluate tumor reactivity of expanded TALs by ELISPOT (Mabtech, Ohio, USA). After expansion, TALs were washed in serum-free RPMI and rested overnight in complete RPMI containing IL-2 (150 U/mL) prior to ELISPOT assay. TALS of ×105were exposed to TAA peptides (MHC Class I and II-restricted) at 2 uM for 24 hours and T cell activation evaluated by counting the number of reactive spots.
CD14+ cell isolation from human ascites, RNA extraction and sequencing
Human ascites CD14+ cells were isolated by fluorescence-activated cell sorting, gating first on the live single CD45+ cells and further gating on CD14+CD3negCD19negCD56neg population. 98.3%–99.6% pure CD14+ cells were obtained from three OC ascites samples (online supplementary figure S4). RNA was extracted for sequencing according to the manufacturer’s protocol (Qiagen, Hilden, Germany). Cryopreserved fragments or snap frozen (<20 mg) tumor samples were submerged in 350 µL of RNeasy Lysis Buffer buffer containing 40 µM dithiothreitol (Sigma-Aldrich, Missouri, USA). Tissues were completely homogenized on ice using a pestle (70 mm, Schuett-Biotec, Göttingen, Germany) and passed through a 26G needle syringe five times (BD Microlance, Allschwil, Switzerland). Supernatant was collected by centrifugation at 4°C for 3 min at 18 213 x g for RNA extraction.
Supplemental material
RNA quality was assessed on a Fragment Analyzer (Agilent Technologies, California, USA) and had a RQN between 2.3 and 8.9. RNA-seq libraries were prepared using 400 ng of total RNA and the Illumina TruSeq Stranded Total RNA Library Prep Gold (Illumina; San Diego, USA). Cluster generation was performed with the resulting libraries using the Illumina HiSeq3000/4000 SR Cluster Kit and sequenced on the Illumina HiSeq4000 using HiSeq3000/4000 SBS Kit for 150 cycles. Sequencing data were demultiplexed using the bcl2fastq Conversion Software (V.2.20, Illumina; California, USA). Sequencing reads were aligned to the reference genome with the STAR aligner using the two pass mapping strategy. A number of reads mapping to each protein-coding gene (as defined in the annotation files) were counted using HTSeq-count in union mode. In-house scripts were used for quantifying gene expression in transcripts per million.
Immunoaffinity purification of HLA peptides and liquid chromatography with tandem mass spectrometry (LC-MS/MS) analyses
HLA immunoaffinity purification was performed as previously described.20 21 See online supplementary methods.
Supplemental material
Statistical analysis
A two-tailed Student’s t-test was used to compare means of continuous measurements between two groups. The analysis of variance (ANOVA) was used to compare more than two groups. When the ANOVA was significant, Tukey’s procedure was used to identify which groups were significantly different. Kaplan-Meier survival curves were computed. The log-rank test and the exact two-sided p values were used to test the hypothesis that the survival curves were the same. Differences were considered statistically significant when p<0.05. SAS software (V.9.3), StatXact Procs V.9 for SAS and Graphpad Prism were used.
Results
OC ascites monocytes are laden with tumor antigen but display poor immunogenicity at the steady state
We hypothesized that ascites-associated monocytes could take up tumor antigen in vivo. Following i.p. inoculation of ID8 tumor cells expressing green fluorescent protein (GFP), mice rapidly developed peritoneal carcinomatosis and large volume ascites (mean 41 days). In mice with advanced i.p. tumors, we observed that the ascites contained CD45+ immune infiltrates (figure 1A,B), similar to human ascites.22 We also observed that CD11b+F4/80+ cells accounted for ~30% of all CD45+ immune infiltrates (figure 1B). The CD45+CD11b+ F4/80+ cells were loaded with GFP, as observed by the shift in fluorescence in the ascites from ID8-GFP inoculated mice compared with ascites cells from ID8 (GFP-negative) inoculated mice (online supplementary figure 1A). We further demonstrated that more than 95% of GFP+ cells were F4/80+CD11b+ monocytes and ~15%–20% of them also expressed the CD11c DC marker (figure 1B). Uptake of GFP was confirmed in F4/80+ macrophages using fluorescent microscopy (figure 1C and online supplementary figure 1B). Furthermore, the CD45+CD11b+F4/80+ cells exhibited low CD80, CD86 and CD83 expression, while displaying MHC I and II expression (figure 1D). Thus, mouse OC ascites is rich in phagocytic monocytes with predominant expression of macrophages markers which are laden with tumor protein in vivo but express low levels of maturation markers at the steady state.
Supplemental material

Ascites monocytes are loaded with tumor antigen and exhibit an immature phenotype. (A) 6–8 weeks old C57BL/6 female mice were inoculated with 5×106 ID8 tumor cells expressing or not GFP. Ascites were retrieved when mice reached 30 g and ascites composition evaluated by flow cytometry and microscopy. (B) Representative dotplots showing flow cytometry measurement of CD11b, CD11c and F4/80 expressions on live ascites-derived CD45+ immune cells loaded with GFP. (C) Fluorescent confocal microscopy showing GFP-positivity in CD45+F4/80+ ascites macrophages, indicating the uptake of ID8-GFP tumor in these cells (white arrowheads). (D) Representative histograms showing flow cytometry analysis of costimulatory markers, and MHC Class I and II on CD11b+F4/80+CD45+ macrophages in the ascites of ID8-GFP tumor-bearing mice (blue histograms). Isotype control was presented as gray histograms. (E) IFN-γ expression on coculture of ascites macrophages and T cells from ascites of ID8-VEGF mice. (F) OT-II T cells proliferation on coculture with ascites CD11b+ cells isolated from ID8-OVA inoculated mice or BMDCs pulsed with various concentrations of OVA MHC-II peptide. APCs, antigen-presenting cells; BMDCs, bone-marrow derived dendritic cells; IFN-γ, interferon-γ; OVA, ovalbumin; PMA, phorbol 12-myristate 13-acetate.
Next, we tested whether tumor-laden ascites monocytes could cross-present tumor antigen at the steady state and activate cognate T cells. Ascites-associated monocytes were unable to induce IFN-γ secretion in autologous ascites T cells in coculture experiments (figure 1E). To ensure that this was not due to the inability of ascites T cells to respond to cognate antigen, we isolated ascites monocytes from mice inoculated i.p. with OVA-expressing ID8 tumors (expecting that they would present OVA peptides) and cocultured them with OT-II T cells to assess their ability to activate tumor-specific T cells. Sorted monocytes from ID8-OVA ascites were unable to elicit a proliferation response in OT-II splenocytes, as observed by the lack of proliferation (carboxyfluorescein succinimidyl ester (CFSE) dilution) by the latter (figure 1F). To ensure that this was not due to the inability of monocytes to present antigen, we pulsed ascites monocytes and control canonical BMDCs from C57BL/6 mice with class II OVA (OT-II) peptide and cocultured them with OT-II T cells. At physiologic doses of peptide, ascites monocytes were unable to activate cognate T cells, while BMDC elicited a strong response. Ascites monocytes could activate OT-II cells at supraphysiologic doses of peptide, however, suggesting a tolerogenic phenotype at baseline. In this setup, 100-fold more peptide was required to elicit a comparable proliferation of OT-II T cells relative to peptide-pulsed BMDCs (1 µg/mL vs 0.01 µg/mL). Thus, ascites monocytes are laden with tumor protein, but are unable to activate T cells at the steady state.
Ex vivo TLR activation and IL-10 neutralization enhance the stimulatory activity of ascites monocytes
We hypothesized that ascites CD11b+ monocytes could become effective APCs following ex vivo stimulation with appropriate activation signals (figure 2A). We investigated if TLR agonists could activate ascites monocytes and confer the ability to stimulate tumor-specific T cells. Consistent with their myeloid origin, the combination of TLR4 (LPS) and TLR9 (CpG-oligonucleotides) effectively activated ascites monocytes, resulting in marked upregulation of CD80 and CD86 (figure 2B) as well as a significant increase in the secretion of inflammatory cytokines and T cell-recruiting chemokines (figure 2C and online supplementary figure 2).
Supplemental material
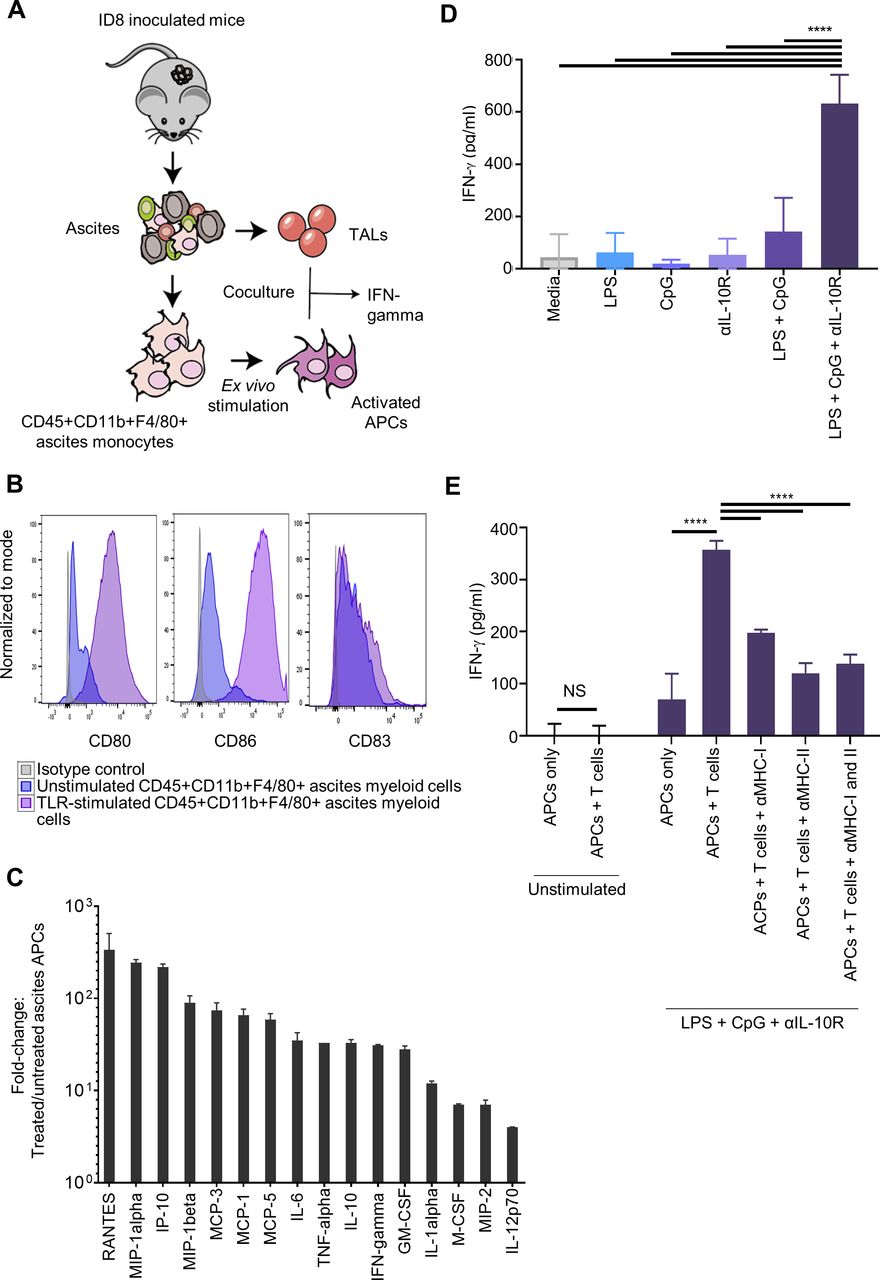
Treatment with toll-like receptor (TLR) agonists, LPS and CpG, in combination with a blocking antibody to the interleukin-10 receptor (IL-10R Ab) restored the immunogenicity of ascites-derived antigen-presenting cells (APCs). (A) Following ID8 tumor cell inoculation, CD45+ APCs and tumor-associated lymphocytes (TALs) were isolated from the ascites of mice. Ascites APCs were stimulated ex vivo for 48 hours with LPS, CpG-oligonucleotides and/or IL-10R Ab, and subsequently, cocultured overnight with autologous TALs. (B) LPS, CpG-oligonucleotides and IL-10R Ab treatment increased CD80 and CD86 expressions on ascites APCs, as assessed by flow cytometry analysis (purple histograms). Isotype control and unstimulated ascites macrophages were shown as gray and blue histograms, respectively. (C) Fold increase in cytokine expressions of ascites APCs after treatment with LPS +CpG-oligonucleotides+IL-10R ab compared with untreated cells. Total n=10 mice per group. (D) Ascites APCs treated with LPS, CpG-oligonucleotides and IL-10R Ab for 48 hours induced significant IFN-γ production from TALs, as measured in cell-free supernatant by ELISA. (E) Ascites APCs treated with LPS, CpG-oligonucleotides and IL-10R Ab were exposed to anti-MHC I and/or anti-MHC II for 30 min prior to coculture with TALs. Decreased IFN-γ production by responding TALs indicated that TCR engagement with MHC-antigen complex was responsible for the response seen in the cells not treated with antibodies blocking MHC molecules. Data were representative of two independent experiments (total n=10 mice per group). Data in C, D and E were presented as mean±SE of the mean (SEM). ****P<0.0001, as highly significant. IFN-γ, interferon-γ; IP, intraperitoneally; LPS, lipopolysaccharide; TCR, T-cell receptor; TNFα, tumor necrosis factor α.
Ascites-associated T cells in human and mouse OC have been shown to include tumor-reactive T cells.23 To test whether ex vivo activated ascites monocytes could efficiently cross-present autologous tumor antigen previously acquired in vivo, TLR-activated ascites monocytes were cocultured with autologous TALs isolated from the same ascites samples. T cell activation was assessed through IFN-γ secretion. TLR4 stimulation is known to induce IL-10 secretion by monocytes, leading to suppression of APC function of both DCs and macrophages.24 25 Since we indeed observed increased secretion of IL-10 in our ascites monocyte culture on TLR stimulation (figure 2C and online supplementary figure 2), we included in our coculture a neutralizing Ab blocking the IL-10R. Consistent with the results seen above, unstimulated ascites monocytes could not activate autologous TALs (figure 2D). Ascites monocytes treated with either TLR4 or TLR9 agonist alone did not activate autologous TALs, while their combination had a modest effect in enabling ascites monocytes to stimulate IFN-γ production from TALs (figure 2D). Similarly, IL-10R Ab alone did not enable unstimulated monocytes to activate autologous TALs. However, IL-10R Ab in combination with TLR4 and TLR9 agonists significantly increased the ability of ascites monocytes to stimulate IFN-γ production by TALs. Blocking MHC class I and II significantly attenuated the activation of TALs by monocytes, indicating specific TCR engagement and MHC-restricted T cell activation (figure 2E). Altogether, our data demonstrate that while tumor-laden ascites monocytes are unable to elicit a T cell immune response at the steady state, their brief ex vivo exposure to the combination of TLR4 and TLR9 agonists in the presence of IL-10R blockade enables them to activate autologous T cells in an MHC-restricted manner.
TLR-activated ascites monocytes provide effective cancer vaccination
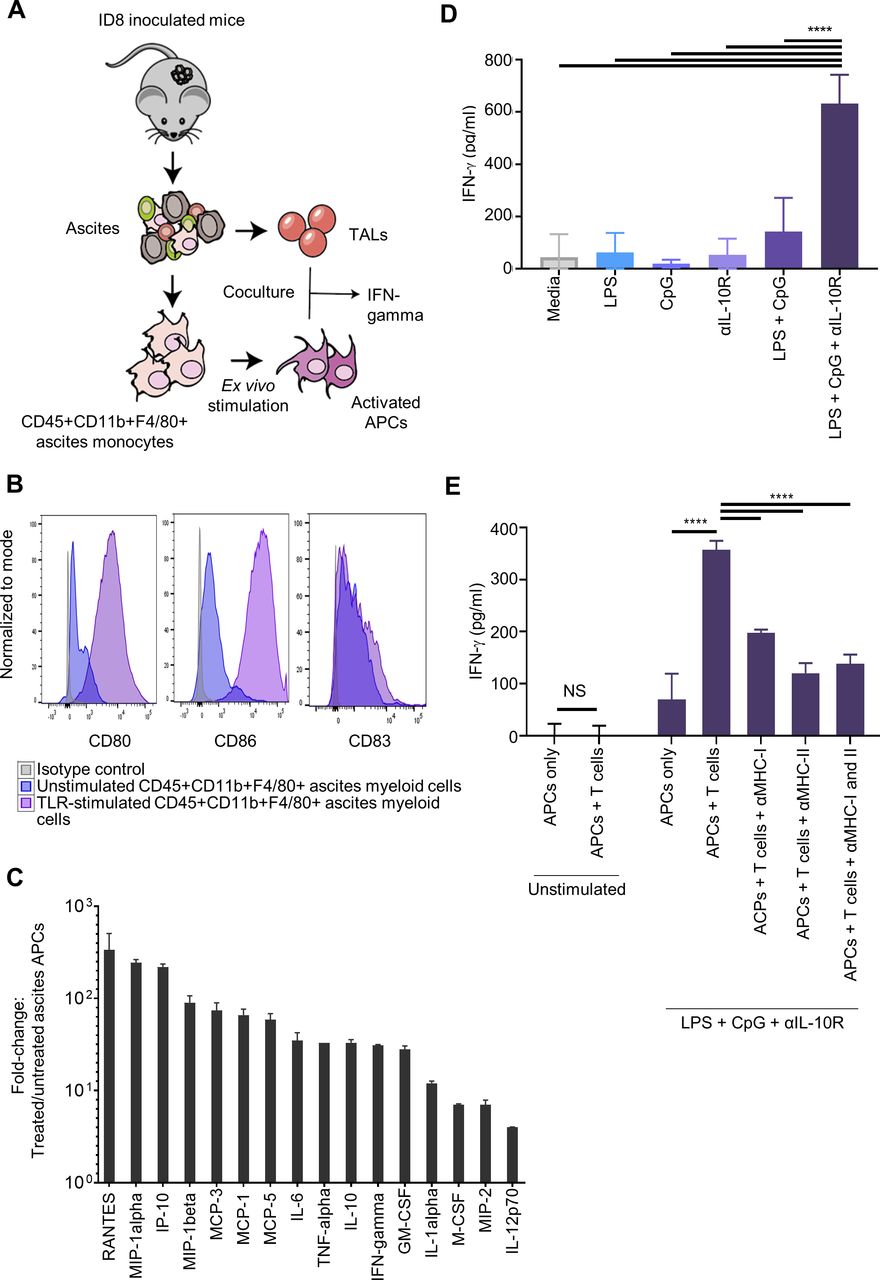
Given the strong immunostimulatory potential of tumor-laden monocytes on short ex vivo stimulation with LPS, CpG-oligonucleotides and IL-10R Ab (henceforth referred to as TLR-activated monocytes or TLRM), we sought to evaluate their potential as a cancer vaccine (figure 3A). In a prophylactic vaccination approach, we injected TLRM or control CD45+ adherent ascites monocytes (incubated in control media for 48 hours) into tumor-naïve mice subcutaneously (s.c.) or i.p. at a first dose of 1×106 cells and followed by two doses of 5×105 cells given over the two subsequent weeks. Mice were then challenged with 5×106 ID8 tumor cells i.p. 2 weeks after the last vaccination, and their survival was followed from the day of tumor challenge and until animals reached 30 g due to ascites accumulation. Intraperitoneal vaccination with TLRMs resulted in significant survival benefit compared with untreated control mice (HR=0.06, p=0.0087, figure 3B). The median survival of mice was 81 days after prophylactic i.p. vaccination with TLRMs and 40 days in untreated control mice. Mice inoculated i.p. with control untreated ascites monocytes experienced more rapid tumor progression with a median survival of 21 days (HR=5.18, p=0.0048), confirming prior observations that tumor-associated myeloid cells could promote tumor growth.26–28 Interestingly, s.c. injection of TLRMs failed to show any beneficial effect (data not shown) and resulted in large acute inflammatory granulomas and severe skin ulceration at the injection sites.

Intraperitoneal transfer of treated ascites APC improves survival in a mouse tumor model. (A) Schedules of ID8 tumor cell inoculation and treated ascites APCs injection for prophylactic, therapeutic and memory studies. (B) Kaplan-Meier survival curve demonstrating a significant disease-specific survival advantage among animals receiving i.p. inoculation of treated ascites APCs compared with control animals or animals receiving untreated ascites APCs (n=10 animals per group unless otherwise stated). Survival was measured from the day of tumor challenge until animals reached a weight of 30 g and were euthanized. Briefly, animals received i.p. injections of 1×106 treated or untreated ascites APC followed by 2 weekly boosters of 5×105 cells prior to tumor challenge with 5×106 tumor cells on day 0. Survival was measured from the day of tumor challenge until animals reached a weight of 30 g and were euthanized. (C) Kaplan-Meier survival curve of animals (n=5 animals per group) receiving 4×106 treated ascites APCs on day 4 after tumor challenge followed by two boosters of 5×105 treated ascites APCS. (D) Animals receiving treated ascites APCs demonstrated evidence of immunologic memory and were able to reject a second tumor challenge. Experimental animals surviving after all control animals succumbed to tumor were injected with an additional 5×106 tumor cells intraperitoneally. At the end of study on day 216, no evidence of tumor was found in seven surviving animals subjected to necropsy. (E) F4/80+ macrophages were responsible for the increased survival of treated animals. F4/80+ cells were FACS-sorted from bulk adherent CD45+ cells after 48 hours in vitro stimulation with LPS +CpG-oligonucleotides+IL-10R Ab, and injected i.p. into mice following a therapeutic setting. (F) CD3+ T cells were sorted from the peritoneal cavity, from mesenteric and retroperitoneal tumor-draining lymph nodes, or from the spleen of vaccinated animals that previously received either prophylactic or therapeutic TLRMs and survived greater than 100 days from tumor inoculation. T cells were coincubated overnight with BMDCs pulsed with ID8 tumor lysate or control non-pulsed DCs and IFN-γ response evaluated by ELISA. (G–I) Splenic CD3+, CD4+ or CD8+ T cells purified from previously vaccinated long-term survivors as above were transferred to tumor-naïve C57BL/6 recipient mice. One day after the T cell transfer, recipient mice were challenged with 5×106 tumor cells i.p. and were monitored for tumor and ascites development. ***Highly significant p value. APC, antigen-presenting cells; BMDC, bone marrow-derived dendritic cells; DC, dendritic cell; IFN-γ, interferon-γ; IL-10R Ab, antibody to interleukin-10 receptor; i.p., intraperitoneally; TLR, Toll-like receptor.
The vaccination effect of TLRMs was further evaluated in the therapeutic setting. Tumor-bearing mice received an i.p. injection of TLRMs (4×106 cells followed by 2 weekly doses of 5×105 cells), starting 4 days after i.p. tumor inoculation (5×106 tumor cells). Tumor-bearing recipients that received TLRMs demonstrated a statistically significant survival advantage (p=0.0198), and 60% of the treated animals were alive at the end of study on day 135 (figure 3C).
Next, we investigated whether the therapeutic effect of TLRMs was mediated by T cells, that is, whether TLRMs could induce protective immunological memory. Tumor-bearing mice previously vaccinated i.p. with TLRMs and surviving with no evidence of tumor, were rechallenged on day 49 (after all control animals succumbed to disease) with 5×106 ID8 tumor cells i.p. (figure 3D). Significant long-term protection was seen in these mice (p<0.00018); in a representative experiment, one of nine evaluable mice developed tumor by day 111, while the remaining mice (8/9) were disease-free at gross necropsy and at histologic examination of the peritoneal surfaces and key intra-abdominal organs and lungs at the their exit from the study (figure 3D).
Finally, we tested which monocyte population was responsible for the vaccination effect observed above. We FACS sorted F4/80+ macrophages and F4/80- monocytes from ID8 ascites, treated them ex vivo with TLR agonists and IL-10R Ab as above and administered them to ID8 tumor-bearing mice i.p. using the same therapeutic regimen as above. We observed that the therapeutic effect was largely mediated by the activated macrophage subset while the F4/80- monocytes provided no protective effect (figure 3E).
TLRM vaccination elicits antitumor T cell-mediated immune responses in vivo
We asked whether the observed therapeutic benefit was associated with the induction of tumor antigen-specific T cells, and in which compartment such T cells could localize. We sorted CD3+ T cells from the peritoneal cavity, from mesenteric and retroperitoneal tumor-draining lymph nodes, or from the spleens of vaccinated animals that received either prophylactic or therapeutic TLRMs and which survived greater than 100 days from tumor inoculation. Control T cells were sorted from the same compartments of non-vaccinated animals bearing intraperitoneal tumor. To identify tumor-specific lymphocytes, we coincubated T cells from the above compartments overnight with BMDCs pulsed with ID8 tumor lysate or control unpulsed DCs (figure 3F). T cells from control untreated tumor-bearing mice produced no IFN-γ in response to lysate-pulsed BMDCs. In contrast, T cells harvested from all the compartments of mice vaccinated either prophylactically or therapeutically with TLRMs produced strong IFN-γ specifically in recognition of lysate-pulsed BMDCs. T cell activation was MHC-mediated, as the addition of anti-MHC-I and/or anti-MHC-II antibodies reduced IFN-γ production. Taken together, these data suggest that i.p. vaccination using TLRMs can induce a strong T cell response which is associated with expansion of tumor-specific T cells that are present in draining lymph nodes, and circulate through ascites and spleen affording immune protection and long-term survival.
To determine whether such T cells mediated the protective effect of TLRMs in vivo, whole CD3+ splenocytes purified from previously vaccinated long-term survivors as above were transferred into tumor-naïve C57BL/6 recipient mice. To dissect the contribution of CD8+ versus CD4+ T cells, we also sorted each population separately from spleens of vaccinated long-term survivors and transferred them to tumor-naïve recipient mice as above. Recipients were subjected to whole-body irradiation (400 rad) 24 hours before T cell transfer to maximize T cell engraftment. Recipient mice were challenged with 5×106 ID8 tumor cells i.p. a day after the T cell transfer and monitored for tumor and ascites development. Untreated control mice developed tumor and ascites rapidly after tumor inoculation and were euthanized by day 44 (figure 3G–I). In contrast, the transfer of total CD3+, or purified CD4+ or CD8+ splenocytes from vaccinated donors demonstrated a marked protective effect with at least 80% survival in each group (figure 3G–I). Taken together, our data show that TLR-activated ascites macrophages function as bona fide APCs which can prime effector T cells against tumor antigen in vivo and result in immune-mediated tumor clearance. These data suggest that macrophages isolated from ascites could be used directly as APCs without further antigen loading, provided a short activation is performed ex vivo.
Human OC ascites monocytes present TAA peptides in vivo
To test the translational potential of this vaccine, we collected ascitic fluid via paracentesis from four chemotherapy-naïve patients with stage III/IV epithelial OC prior to primary debulking surgery (figure 4A). Ascites cellular content was heterogeneous among patients; tumor cells (CD45-Epcam+) and myeloid cell populations accounted for 1%–85% and 10%–30% of the total cells isolated from OC ascites, respectively (figure 4B). CD14+ peritoneal monocytes coexpressed CD11b and CD11c, with a fraction also expressing CD16, CD303 or CD141 (figure 4C). Consistent with our results in the ID8 ovarian model, CD14+ cells from human ascites lacked costimulatory CD80 and CD83 expression and were low in CD86 and CD40 expression (figure 4D). Ascites monocytes from OC patients exhibited transcriptome profiles of protumorigenic macrophages,29 30 which in principal component analyzes were closer to cancer macrophages than to normal circulating monocytes or normal tissue macrophages31 (figure 4E).

CD14+ cells retrieved from human OC ascites express low levels of costimulatory and antigen-presentation markers and present TAAs in situ. (A) Malignant ascites were collected via paracentesis from chemotherapy-naïve patients with stage III or IV epithelial OC prior to primary debulking surgery. (B) Flow cytometry characterization revealed different percentages of immune cells representing CD14+ cells, T, B, natural killer and tumor cells in the malignant ascites of four OC patients’ samples. (C) Representative dotplots of ascites-derived CD14+ cells showing the expression of myeloid-lineage markers and D) costimulatory markers. (E) Principal component analysis showing clustering of ascites CD14+ cells with endometrial cancer macrophages. (F) Left panel in blue and white, immunopeptidomic analysis with mass spectrophotometry highlighting the MHC Class I and II presentation of TAAs on CD14+ cells and tumor cells from two OC patients. The data was presented as relative MS signal (log2). Right panel with red and light pink, comparing the transcriptional levels of expression of TAAs in CD14+ cells and tumor cells from the same two OC patients. The data were presented as relative expression (transcripts per million (TPM)/TPM average). OC, ovarian cancer; TAAs, tumor-associated antigens.
To determine if human ascites monocytes are loaded and present tumor antigens in situ, we purified CD14+ cells from ascites by FACS sorting (purity ~99%; online supplementary figure 3A and B) and analyzed the immunopeptides bound to surface HLA molecules by MS/MS profiling of peptides eluted from class I and II HLA molecules. We identified in two out of three patients several TAA candidates presented on HLA class I and II molecules of ascites CD14+ cells, which were also presented by autologous tumor cells isolated from the same ascites. These TAAs included cancer-testis antigens (CT55, MAGEs, PRAME, SAGE1); known OC-associated antigens (MUC1, ERBB2, mesothelin, TP53); and other TAAs (GPC3, PMEL, TYRP1, ODF2) (figure 4F and online supplementary file 1). We demonstrated by RNAseq analysis the selective expression of some of the presented TAAs in tumor cells but not in CD14+ cells at the mRNA level, confirming that these immunopeptides presented by CD14+ cells had originated from tumor protein uptake (figure 4F and online supplementary file 2). Altogether, these data suggest that CD14+ monocytes from OC ascites have a tumor-associated macrophage phenotype with reduced expression of costimulatory molecules and present TAAs in situ.
Supplemental material
Supplemental material
Supplemental material
Human monocytes from ascites can be stimulated by LPS, CpG-oligonucleotides and IL-10R Ab and subsequently activate autologous TALs upon ex vivo coculture
Consistent with our mouse results, human CD14+ cells from OC ascites responded potently to a short (48 hours) ex vivo stimulation with TLR agonists. LPS stimulation induced significant cytokine and chemokine secretion in human ascites monocytes (figure 5A and B), and upregulated CD40, CD80, CD83 and CD86 expression (figure 5C). As expected by the reduced/lack of TLR9 expression in human peritoneal macrophages (online supplementary figure 4), CpG-oligonucleotides did not impact nor add to the activation induced by LPS.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD14+ cells retrieved from human ascites can be matured ex vivo on stimulation with LPS, CpG and IL-10RAb and are capable of activating autologous TALs. (A) CD14+ cells from ascites were isolated by magnetic cell sorting and stimulated in vitro with LPS, CpG and/or IL-10R Ab for 48 hours. Activated CD14+ cells were subsequently cocultured with autologous TALs for 72 hours to assess T cell activation, and TALs were further expanded to assess proliferation and reactivity. (B) Fold-increase in cytokine secretion on TLR stimulation and IL-10R Ab treatment, and relative to untreated ascites monocytes as measured by multiparameter analyte profiling (Myriad Rules Based Medicine Luminex multianalyte profile). (C) Flow cytometry analysis depicting upregulation of maturation markers expression on CD14+ cells on TLR agonist stimulation and/or IL-10R Ab treatment. (D) activated CD14+ cells were cocultured with autologous TALs, and IFN-γ, granzyme A (GzmA) and B (GzmB) levels in the supernatant were measured after 72 hours coculture by CBA. (E) After 72 hours of coculture, media was changed and IL-2 (3000 U/mL) added for TAL expansion over 21 days. The expansion was compared with that of TALs expanded in the absence of DCs or following conventional expansion protocol. (F) T cell repertoire analysis at the end of the 21 days expansion. (G) IFN-γ ELISPOT assay was used to evaluate tumor-associated antigens TALs reactivity following 21 days of expansion. Data was representative of three independent experiments and were presented as mean±SEM *P<0.05, **P<0.001, ****P<0.0001. CBA, Cytometric Bead Array; DC, dendritic cell; IFN-γ, interferon-γ; IL-10R Ab, antibody to interleukin-10 receptor; LPS, lipopolysaccharide; TAA, tumor-associated antigens; TALs, tumor-associated lymphocytes; TLR, Toll-like receptor.
To assess their T cell immunostimulatory potential, we cocultured human TLRMs with TALs purified from autologous ascites specimens. Confirming their anergic state, untreated ascites monocytes were unable to elicit any T cell activation (figure 5D). While ascites monocytes treated with LPS or IL-10R Ab alone showed minimal effects on autologous TAL activation, stimulating ascites monocytes with LPS +IL-10R Ab efficiently primed TALs as measured by the strong granzyme (Gzm) A, GzmB and IFN-γ secretions (figure 5D). TALs activation in this context was mediated by TCR-MHC peptide recognition, as evidenced by the reduced expression of these cytokines on HLA-blockade (figure 5D). Importantly, TLRMs retained their ability to elicit activation of autologous TALs following re-exposure to the peritoneal environment of OC ascites as observed by their persistent T cell stimulatory potential in the presence of ascitic fluid (online supplementary figure 5).
Supplemental material
Following coincubation with autologous TLRMs, TALs were expanded for 21 days with IL-2. TALs pre-exposed to autologous TLRMs activated with LPS or LPS +IL-10R Ab exhibited a fourfold higher expansion after coculture relative to TALs not exposed to ascites CD14+ cells or exposed to untreated ascites CD14+ cells (figure 5E). We analyzed the T cell repertoire by deep TCRVβ-sequencing to assess the clonal diversity of output cells from these cultures. The output expanded TALs that had been initially exposed to TLRMs were less diverse compared with TALs expanded without such priming, suggesting a clonal expansion elicited by human TLRMs (figure 5F). Finally, we assessed if these expanded TALs were tumor reactive. TAA peptides discovered by immunopeptidomic analysis of ascites CD14+ cells were synthesized, and TAL reactivity assessed by ELISPOT. TALs stimulated with autologous ascites TLRMs were able to subsequently react against autologous tumor peptides, while TALs expanded without exposure to TLRMs or exposed to untreated ascites CD14+ monocytes (figure 5G). Thus, human ascites monocytes can be reprogrammed to immunogenic APCs, leading to subsequent T cell activation that enhances tumor-reactive T cell expansion.
Discussion
Our results demonstrate that the rapid ex vivo treatment of ascites-derived monocytes with TLR-agonists plus a blocking Ab to the IL-10R induces phenotypic and functional changes in these cells. These activated APCs promote T cell activation and immune-mediated tumor clearance when returned to the peritoneal cavity as a cellular vaccine in an OC mouse model. This strategy resulted in tumor rejection in both prophylactic and therapeutic settings. We observed similar changes in the phenotype, functional status and cytokine profile of monocytes from human ascites following treatment with TLR agonists plus IL-10R Ab. Immunopeptidomic profiling demonstrated in situ TAA presentation on human ascites CD14+ cells, and coculture of TLRMs with autologous TALs led to enhanced expansion of TALs on high-dose IL-2 stimulation. These observations suggest that the results obtained in our mouse OC model are relevant to human disease and have translational potential as the basis for novel immunotherapeutic strategies.
The ID8 model used in this study closely approximates clinical disease in patients with disseminated peritoneal implants. Consequently, we would predict that adoptive transfer of TLRMs would be most effective as a consolidation therapy following surgical cytoreduction and primary adjuvant therapy in the setting of minimal residual disease, when tumor-induced suppressive effects would be minimized. Additionally, it is important to note that in the mouse studies, the approach of CD45 magnetic bead-based purification of ascites APCs did not completely eliminate tumor cells. We were able to confirm, however, that injection of TLR agonist-treated tumor cells, in lieu of ascites APCs, did not hasten tumor development in the prophylactic model (data not shown), indicating that the presence of a tumor fraction likely did not reduce the survival benefit associated with vaccination.
In mouse models, Chu et al found that bona fide DC could be derived from ascites macrophages cultured in granulocyte-macrophage colony-stimulating factor and IL-4.15 When these cells were pulsed with tumor antigen and activated with tumor necrosis factor-α, they induced cytotoxic T lymphocytes to lyse tumor cells in vitro.15 In addition, Scarlett et al demonstrated that direct i.p. treatment with a TLR3 agonist plus CD40 activation enhanced T cell-mediated antitumor immunity and improved survival in a mouse OC model.16 Taken together, these studies suggest that peritoneal leucocytes can be modulated to promote immune-mediated clearance of OC. Our results are in keeping with prior work demonstrating that changes in the phenotypic and functional status of leucocytes in the OC microenvironment could significantly impact tumor clearance and ultimately survival.16 32–35 Importantly, we show that interventions that restore the immunogenic status of ascites monocytes can induce T cell-mediated rejection of peritoneal carcinomatosis and we demonstrate the impact of i.p. administration on vaccine efficacy in a model of HGSOC. Given that OC characteristically recurs in the peritoneal cavity, interventions that block intraperitoneal tumor development would be expected to have a significant impact on the morbidity and mortality of this disease. Fortunately, intraperitoneal ports, which are used to administer chemotherapy to advanced OC patients, provide the opportunity to access the peritoneal cavity for treatment and immune monitoring. Thus, vaccine strategies such as the one we describe could be administered directly i.p. to maximize therapeutic impact while limiting toxicity.
Importantly, macrophage plasticity must be considered when designing a TLRM vaccine.36 Indeed, OC could turn macrophages into an M2-like phenotype, a switch which would dramatically impact the therapeutic efficacy of this intervention.26 28 37 38 Macrophage versatility could explain why prior clinical trials using macrophage-based vaccines failed to demonstrate clinical benefits.39 Several molecules are currently being developed to reverse this immunosuppressive state and reeducate tumor-associated macrophages from an M2 to a M1 phenotype,40 41 and could potentially be used in combination with APC vaccine to enhance therapeutic efficacy.
In preparation for clinical trials, we are currently evaluating safety measures to preserve the immunogenicity of this strategy without exposing patients to potentially viable tumor cells. Additional advantages of this protocol compared with conventional cellular immunotherapeutic strategies include the short duration of ascites APC treatment ex vivo and the exclusion of exogenous antigen pulsing, which would eliminate the production requirement for specialize cell culture techniques. Because large volumes of ascites can be retrieved from OC patients undergoing treatment even in the outpatient setting, this protocol would facilitate the development of a rapid and cost-effective approach to OC immunotherapy, and improve access to individualized immune-based treatments for more OC patients.42
Acknowledgments
We thank the staff of Tumor Tissue and Biospecimen Bank (TTAB) at the University of Pennsylvania Health System as well as the Biobank of CHUV for providing ovarian tumors ascites. We thank the staff from the Microscopy facility, flow cytometry facility and Mouse facility for their help. We thank Paul Terranova for providing the ID8 cells and Alexandre Harari for the helpful discussions. Library preparation and RNA-sequencing was performed at the Lausanne Genomic Technologies Facility, University of Lausanne, Switzerland (https://www.unil.ch/gtf/en/home.html).
References
Footnotes
Twitter @ChiangCheryl1
SFA and AJG contributed equally.
Contributors LEK and GC directed the project. SFA and AJG planned and conducted experiments, analyzed data and wrote the manuscript with the assistance of the other authors. AM, DF, SJ and GAM conducted experiments and analyzed data. AF analyzed data, PAG designed experiments and performed statistical analyses. MB-S supervised the immunopeptidomics antigen discovery analyses; JM performed extraction of RNA and HLA peptides and HP was responsible for MS analyses. FH performed RNA seq data analysis associated to the immunopeptidomic study. CL-LC, CN and AJG performed additional mouse experiments. CL-LC and LEK revised and finalized the paper. SR and DD helped with experimental designs and discussions. All authors read and approved the final manuscript.
Funding This work was funded by grants from the Gynecologic Cancer Foundation, Sandy Rollman Foundation, American Cancer Society, the Kaliedoscope of Hope Foundation and the Ludwig Institute for Cancer Research.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All human ascites, blood and tissue samples were obtained through protocols approved by the local Ethics Committee of Lausanne University Hospital, Switzerland (CHUV-DO-pre-IT; Ethics Committee number (BASEC): 2016-02094, TRP 1 BASEC 2017-00359, TRP 2 BASEC: 2017-00305 and TRP 3 BASEC: 2017-00490), and University of Pennsylvania’s Institutional Review Board (IRB) [protocol number 702679]. Written informed consent was obtained from all donors.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplementary information. The list of identified HLA-I and HLA-II binding peptides in two OC patients can be found in online supplementary file 1. The expression values of genes expressed in the same two OC patients can be found in online supplementary file 2.