Article Text

Abstract
Cancer immunotherapy with immune-checkpoint blockade has improved the outcomes of patients with various malignancies, yet a majority do not benefit or develop resistance. To address this unmet need, efforts across the field are targeting additional coinhibitory receptors, costimulatory proteins, and intracellular mediators that could prevent or bypass anti-PD1 resistance mechanisms. The CD28 costimulatory pathway is necessary for antigen-specific T cell activation, though prior CD28 agonists did not translate successfully to clinic due to toxicity. Casitas B lymphoma-b (Cbl-b) is a downstream, master regulator of both CD28 and CTLA-4 signaling. This E3 ubiquitin ligase regulates both innate and adaptive immune cells, ultimately promoting an immunosuppressive tumor microenvironment (TME) in the absence of CD28 costimulation. Recent advances in pharmaceutical screening and computational biology have enabled the development of novel platforms to target this once ‘undruggable’ protein. These platforms include DNA encoded library screening, allosteric drug targeting, small-interfering RNA inhibition, CRISPR genome editing, and adoptive cell therapy. Both genetic knock-out models and Cbl-b inhibitors have been shown to reverse immunosuppression in the TME, stimulate cytotoxic T cell activity, and promote tumor regression, findings augmented with PD1 blockade in experimental models. In translating Cbl-b inhibitors to clinic, we propose specific gene expression profiles that may identify patient populations most likely to benefit. Overall, novel Cbl-b inhibitors provide antigen-specific immune stimulation and are a promising therapeutic tool in the field of immuno-oncology.
- CTLA-4 antigen
- drug therapy, combination
- immunotherapy
- review
- therapies, investigational
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The advent of immune checkpoint inhibitors (ICI), specifically cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death (ligand) protein 1 (PD1/PD-L1) blockade, brought immunotherapy into standard treatment paradigms for patients across many advanced cancers. However, the clinical expansion of these agents is plateauing with beneficial outcomes seen in only a minority of patients.1 While efforts are underway to identify more predictive biomarkers of current ICI, the investigation of novel cellular and molecular targets will be necessary to drive more effective and sustained antitumor immunity, reverse immunosuppression in the tumor microenvironment (TME), and further advance immune-based therapies.2
The PD1 and CTLA-4 checkpoints represent only two of many transmembrane receptor-ligand pairs dictating T cell antigenic response. A host of costimulatory and coinhibitory molecular interactions have been characterized between T cells, antigen presenting cells (APCs), and tumor cells.3 While antibody targets have now been developed against some of these interactions, pharmacological intervention against the downstream signaling pathways has been limited.4 These intracellular pathways, ultimately responsible for the transcriptional and post-transcriptional regulation of T cell activation, can serve as resistance and bypass mechanisms against extracellular checkpoint therapy.5 6 Additionally, a paucity of costimulatory receptors or neoantigenicity in the TME of some tumors may limit the use of ICI and therapies targeting these cell-surface interactions altogether.
Success in the next phase of immuno-oncology (IO) will depend on addressing unmet needs and leveraging lessons learned from prior clinical trials.7 Thus future and ongoing goals include: (1) developing biomarker-driven models to predict immunotherapy response based on tumor molecular and genomic characteristics; (2) focusing on the tumor biology of non-responding patients to identify resistance pathways; (3) identifying all potential avenues of immunosuppression in the TME (eg, metabolic insufficiency, hypoxia, adenosine signaling, regulatory cells); (4) facilitating sufficient levels of infiltrating and durably activated T cells; and (5) applying advanced pharmacologic techniques against previously inaccessible proteins and pathways. By addressing this needs assessment, future clinical trials may more effectively combine standard of care ICI with novel immune-based therapies and targeted agents.
Casitas B lymphoma-b (Cbl-b) is an E3 ubiquitin ligase initially characterized as a proto-oncogene but now understood to have a central role in regulating effector T cell function.8 Cbl-b has been identified as a key inhibitor of T cell activation in the absence of CD28 costimulation.9 Through a complex interaction of signal transducers, Cbl-b inhibits T cell transcriptional activity and promotes immune tolerance across innate and adaptive immunity.6 As an intracellular master regulator, Cbl-b inhibition may represent a more specific and efficient route toward broad immune activation regardless of upstream checkpoint signaling (ie, CD28, CTLA-4). Importantly, recent advances in immunology, pharmacology, and data science have enabled the development of novel agents against intracellular proteins, such as Cbl-b.
Here, we will focus on the CD28 pathway, the central role of Cbl-b, and the impact on immune cell subsets by this ubiquitin ligase. We will discuss the preclinical development of Cbl-b inhibition, ranging from small-interfering RNA (siRNA) to allosteric regulatory targets to adoptive T cell therapy. Finally, upcoming clinical trials, combinatorial strategies, relevant biomarkers, and potential toxicities of Cbl-b inhibition will be considered.
T cell activation and regulation: an overview of costimulatory and coinhibitory receptors
Characteristics of the TME associated with an optimal cytotoxic antitumor immune responses include an inflammatory cytokine profile, favorable metabolic milieu, decreased regulatory cell activity, and T cell invasion and activation.10 Dogmatically, CD8+ T cell activation first requires antigen presentation via major histocompatibility complex II (MHC-II) on APCs. Without simultaneous costimulation, or in the presence of coinhibitory competition, however, T cells enter an anergic or apoptotic state. A diverse set of costimulatory and coinhibitory receptors have been characterized that influence this process.5 While evolutionarily conserved to limit T cell overactivation, tumors can evade systemic immunity by ‘hijacking’ this delicate balance of checkpoints in favor of inhibitory pathways.11
Costimulatory receptors
The CD28 costimulatory receptor is an Ig family receptor that plays a central role in canonical T cell activation. Constitutively active in a majority of CD4+ and CD8+ T cells, CD28 binds to the CD80 (B7-1) and CD86 (B7-2) ligands expressed on APCs, ultimately promoting a T cell effector response.12 CD80 and CD86 also compete for the CD28 homolog receptor, CTLA-4, expressed on the surface of T cells.13 CTLA-4 binds these ligands with higher affinity as compared with CD28, maintaining a homeostatic immune response to repeated antigenic presentation. Sequential and differential ligation of CD86 with CD28 is followed by CD80 with CTLA-4—this process initiates cytotoxicity but prevents an unchecked immune response.14 On stimulation, the cytoplasmic domain of CD28 initiates a phosphorylation cascade via PI3K and ZAP70, resulting in proteasomal degradation of Cbl-b, a key inhibitory enzyme, and activation of the NFAT and NF-kB transcription factors (figure 1A).6 15 This multifaceted cascade, activated via TCR/CD3 and CD28 stimulation, ultimately promotes transcriptional upregulation of IL-2, cytoskeletal recruitment, and overall cellular proliferation.16 Unfortunately, early trials with the CD28 ‘superagonist’ TGN1412 lead to severe cytokine release syndrome (CRS), likely due to the constitutive expression of this coreceptor along with the lack of TCR/CD3 primary stimulation.17 These results led to an extended pause in cancer clinical trials addressing the CD28 pathway.
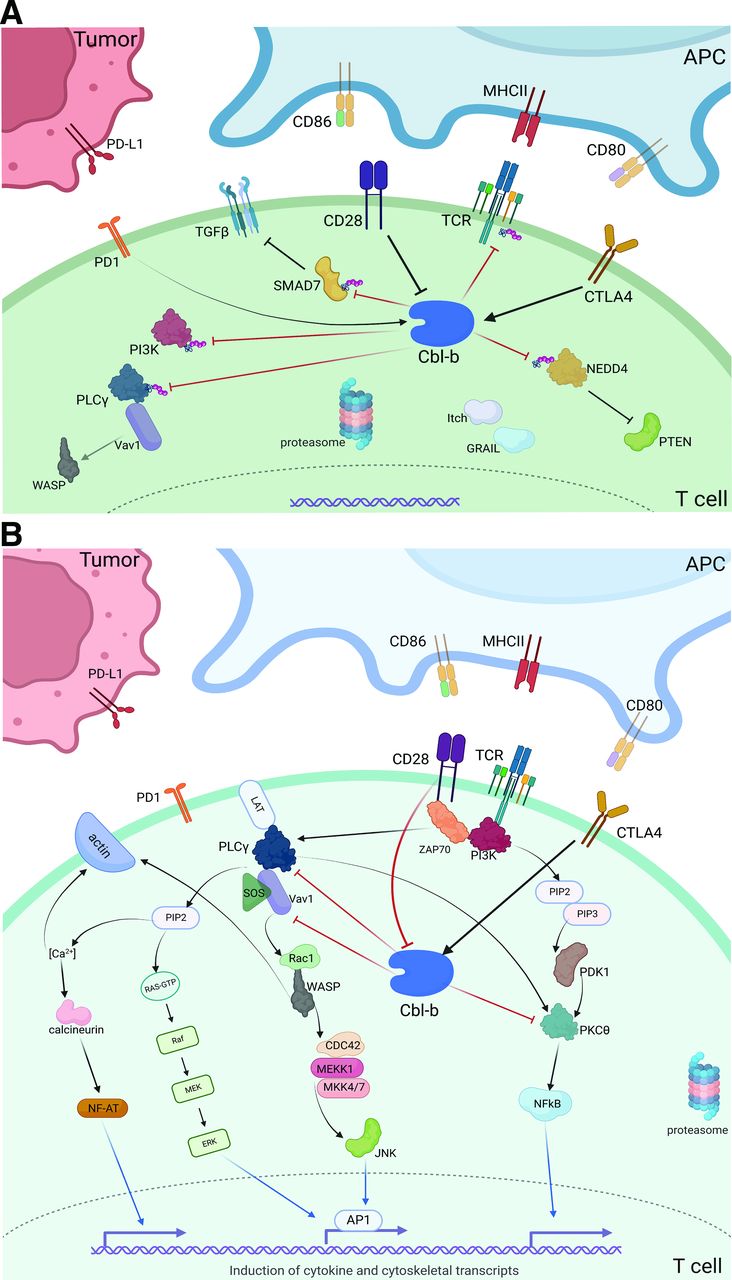
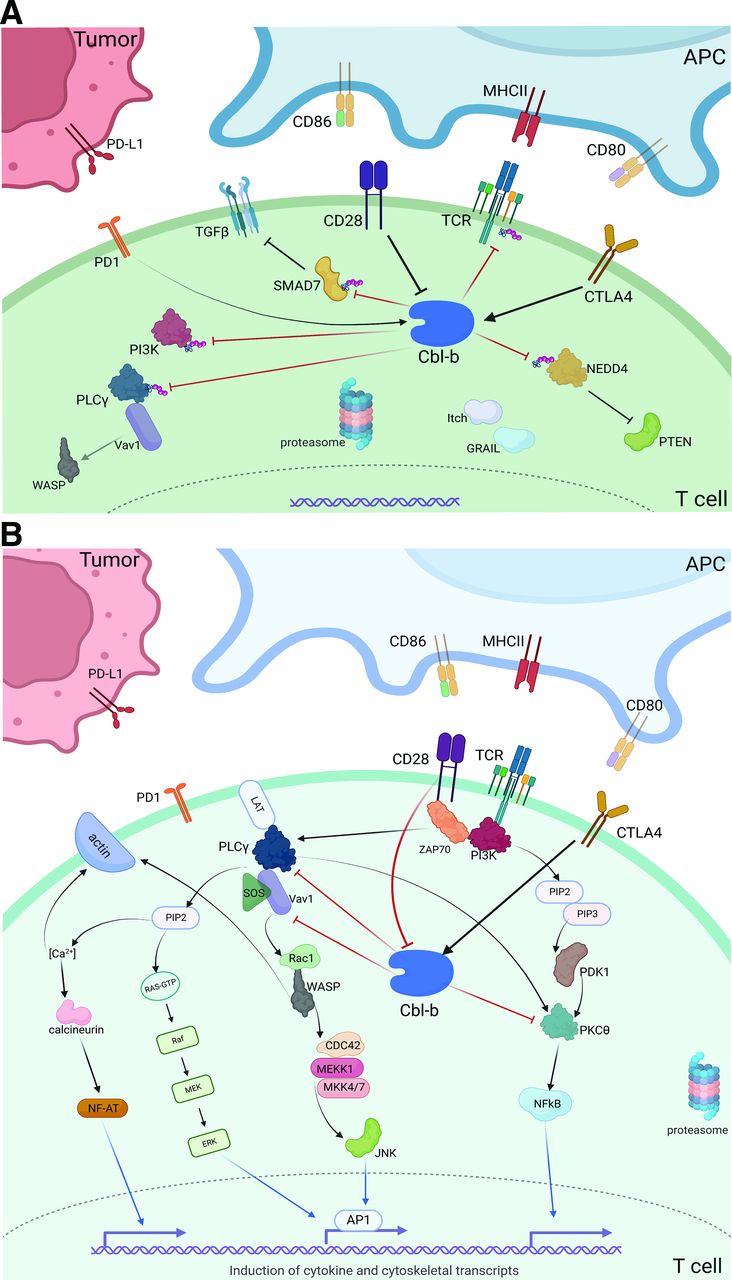
(A) T cell stimulation via CD28 costimulatory signaling cascade. (B) Depiction of intracellular Cbl-b signaling in the tumor microenvironment. CD28 costimulation inhibits downstream Cbl-b signaling, while CTLA4 stimulation promotes Cbl-b activity. On activation, Cbl-b ubiquinates several key proteins that inhibit effector function, promoting an immunosuppressive phenotype. *Red blocked arrows indicate direct ubiquitination. APC, antigen presenting cell; Cbl-b, Casitas B lymphoma-b; MHCII, major histocompatibility complex II.
Given the initial clinical complexity of targeting CD28, a number of costimulatory receptors became high priorities as potential cancer immunotherapy targets, including 4-1BB, OX40, GITR, ICOS, CD27, and CD40 (table 1). In vivo experiments with 4-1BB agonism showed a dramatic increase in CD8+ T cell proliferation, yet overall reduction in B, NK, and CD4+ T cell activity.18 This dichotomous effect, at least in part due to indoleamine 2,3-dioxygenase (IDO) signaling, may partially explain the poor humoral and innate immune response seen with 4-1BB stimulation. A phase I clinical trial with a 4-1BB agonist (urelumab) demonstrated preliminary antitumor activity in advanced cancers,19 although further translation was hampered by hepatic toxicity and lack of target population with clear benefit.20 Multiple next generation approaches targeting 4-1BB are in clinical development, taking advantage of affinity engineered multispecific antibodies (eg, GEN1046, NM21-1480, DSP107).21 22 OX40, part of the TNF receptor family, is a costimulatory molecule with only transient expression in activated T cells.5 Unfortunately, phase I/II trials with the OX40 agonists MEDI0562, BMS-986178+/-checkpoint blockade, and BGB-A445+/-tislelizumab failed to demonstrate clinical efficacy.23 24 This lack of effect could be explained by a complicated kinetic relationship whereby receptor occupancy leads to receptor downregulation.25 Similar to OX40, GITR ligation leads to NF-kB transcriptional activity and an overall decreased threshold of CD8+ T cell activation.26 The safety and efficacy of a GITR agonist, INCAGN-1876, is currently being studied alongside IDO and PD1 blockade in advanced cancers.27 28 Additional GITR agonists under investigation include GWN323+/-spartalizumab, TRX518+/-chemotherapy or PD1 blockade, and BMS-986156+/-nivolumab; though phase I trials have shown minimal single-agent or combined efficacy.29 30 The ICOS costimulatory molecule is also induced on primary T cell activation,31 however, an early phase trial of the ICOS agonist, JTX-2011, alone or in combination with anti-PD1 was discontinued in 2020.32 An additional agonist, feladilimab, has also shown disappointing results in phase II clinical trials alongside anti-PD1 therapy.33 In contrast to most other costimulatory receptors, CD27 is constitutively active in naïve and regulatory T cells but poorly expressed on differentiation.34 This pattern is advantageous for low affinity TCRs and stimulates effector function even in the absence of a strong antigenic presentation. Preliminary results from a phase II trial assessing the CD27 agonist, varlilumab, alongside PD1 blockade showed only marginal response rates in patients with ovarian and colorectal cancer.35
A non-exhaustive list of agents targeting costimulatory receptors with associated clinical trials in the field of immuno-oncology
While initially promising, these costimulatory agonists have yet to demonstrate meaningful benefit in clinical trials. Potential explanations include the dichotomous effects on various immune cell subsets, transient expression of the target receptors, and lack of biomarkers identifying patient populations most likely to benefit. In contrast, the CD28 pathway is constitutively expressed with direct links to the clinically validated CTLA-4 axis.12 14 Furthermore, CD28 signaling is required for response to PD1 therapy.36 Thus, renewed efforts have emerged to safely and more effectively stimulate this coreceptor.
For example, bispecific antibodies have been developed to simultaneously target a tumor-specific antigen (TSA) and CD28 for epithelial tumors, with a PSMAxCD28 agent showing preliminary antitumor activity alongside anti-PD1 (cemiplimab) in a phase I/II trial of patients with metastatic prostate cancer (table 1).37 38 Building off bispecific TSAxCD3 antibodies that initiate primary TCR stimulation,39 the TSAxCD28 platform provides costimulation while maintaining crucial specificity. Despite severe side effects from the original CD28 ‘superagonist’, TGN1412 was rebranded as TAB08 (TheraMAB) and has cautiously re-entered clinical development.40 Additionally, Fc fusion proteins of CD80 (an innate CD28 and CTLA-4 ligand) have also been developed and shown increased T cell effector response in preclinical studies.41 The CD80-Fc fusion protein, FPT155, has shown no evidence of CRS (indicating the requirement of antigenic TCR stimulation), higher affinity for CD28 over CTLA-4, and augmented efficacy alongside anti-PD1 therapy in a CT26 murine model.42 Further, the NEON-1 trial assessing ALPN-202, a PD-L1 dependent CD28 costimulator and CTLA-4 inhibitor, has shown clinical benefit in 25 of 48 patients, though augmentation with pembrolizumab exhibited high-grade AEs (NEON-2).43 44 Finally, given the toxicity of TGN1412, marginal efficacy of costimulatory receptor agonists, and the potential to overcome anti-PD1 resistance, attention has turned to downstream regulators of the CD28 axis (eg, Cbl-b) despite the challenges in targeting these intracellular proteins.17
Coinhibitory receptors: CTLA-4 & PD-1
Antibodies against the CTLA-4 and PD-1 coinhibitory receptors have set a new benchmark for clinical benefit. However, many common malignancies (eg, ovarian, colon, prostate) do not respond and, for relatively responsive cancers (eg, melanoma, non-small cell lung cancer (NSCLC)), a majority of patients develop resistance.45 As mentioned alongside its Ig-receptor homolog, CD28, CTLA-4 exhibits strong affinity to the CD80/CD86 ligands on APCs.46 Downstream signaling ultimately negates actin recruitment, transcription factor activity, and effector response.46 This inhibitory action is also regulated via Cbl-b, the expression of which is significantly elevated on CTLA-4 ligation.16 Unlike CD28, CTLA-4 is not constitutively expressed5; translocation only occurs after TCR stimulation, making it a slightly more focused pharmaceutical target in the TME.
PD1, perhaps the most well-recognized co-inhibitory receptor, is another homolog to CD28. Ligation with PD-L1 leads to inhibition of CD28-mediated PI3K activity, promoting T cell anergy.47 Clinical trials have demonstrated superior outcomes with anti-PD1 versus anti-CTLA-4 therapy in many cancers, though precise mechanisms behind these differential outcomes are less clear. While gene expression analyses have identified certain genes (eg, Bcl-xL, JAK2) as potential mediators of both anti-PD1 efficacy and resistance,48 functional assays in Cbl-b knockout (KO) mice showed a complete lack of tumor regression with PD-L1 blockade as compared with wild-type controls. These results in syngeneic melanoma murine models highlight Cbl-b as a critical, downstream mediator of PD(L)1 signaling, a finding validated by multiple groups.49 Additionally, PD-L1 silencing in murine dendritic cells (DCs) was shown to reduce Cbl-b expression in CD8+ T cells,50 providing additional data linking these proteins to similar immune pathways. Overall, Cbl-b not only mediates CTLA-4 signaling but also PD1-induced immunosuppression in the TME.
Intracellular signaling: the central role of Cbl-b, an E3 ubiquitin ligase
Despite the promise of coinhibitory receptor blockade (ie, ICI), the most potent antitumor immune responses may yet be harnessed via direct augmentation of T cell activation machinery. The costimulatory receptor, CD28, provides a critical secondary signal to promote T cell activation and prevent anergy on TCR/MHC-II ligation. Following T cell activation, CTLA-4 expression escalates to outcompete the binding of CD28 to its APC ligands (CD80, CD86),4 reducing the risk of unnecessary T cell proliferation under physiologic conditions. Cbl-b, a RING finger E3 ubiquitin ligase, is a key intracellular mediator between CD28 and CTLA-4 receptor signaling (figure 1B). Without CD28 stimulation, Cbl-b, along with the ligases Itch and GRAIL, lead to ubiquitination of several key proteins—namely, PI3K and PLCγ, the zeta-subunit of TCR, and NEDD4.15 16 Ultimately, this ubiquitination prevents Vav1-mediated cytoskeletal activation, TCR phosphorylation, and PTEN inactivation, respectively. Cbl-b also directly ubiquitinates SMAD7, promoting TGF-ß signaling and an overall immunosuppressive phenotype.8 In the event of successful CD28 ligation, however, Cbl-b itself is ubiquitinated and tagged for lysosomal degradation to prevent this anergic response.
The precise mechanism by which CTLA-4 stimulation leads to Cbl-b activation has not yet been elucidated, but several studies have shown a strong correlation between CTLA-4 ligation and both the function and expression of Cbl-b.51 Furthermore, CTLA-4 signaling leads to increased expression of both Itch and GRAIL, initiating a host of ligase-mediated inhibitory activity.4 In addition to CD8+ T cell effects, CTLA-4 receptor ligation has been associated with Treg activation—preliminary studies show that GRAIL and associated ligases may be key, downstream mediators of Treg development.52 Overall, given its critical role in the CD28, CTLA-4, and PD(L)1 pathways, Cbl-b may be tied to both the success of checkpoint blockade along with a possible source of resistance.
The effect of Cbl-b on immune cells
T cells
A number of KO models have been developed to elucidate the effect of Cbl-b signaling in innate and adaptive immune cells. Starting with T cell differentiation, Cbl-b-deficient mice demonstrate increased Th2, Th9, and Th17 cellular differentiation.53 Phenotypic traits related to this differentiation included a hyperacute immune response secondary to increased JAK/Stat6 signaling in the absence of Cbl-b mediated ubiquitination.53
Cbl-b-/- murine models demonstrate a stark response to both infections and malignancy. For example, in the context of lymphocytic choriomeningitis virus (LCMV) infection in Cbl-b KO mice, CD8+ TCR downregulation is limited with increased levels of IFN-γproduction.54 While Cbl-b inhibition has been shown to be an effective adjunct for inactivated vaccines, complete deficiency has also led to cytokine-storm mediated fatalities in certain murine KO models.55 Notably, however, Cbl-b knockout models only develop detrimental autoimmune effects on stimulation or after a significant time period, whereas CTLA-4-/- murine models often develop spontaneous, lethal autoimmunity at an early age.56
The response of CD8+ T cells in Cbl-b-/- mice exposed to cancer is also enhanced. In a UVB-induced tumor model, over 80% of Cbl-b-/- mice spontaneously rejected tumors as compared with wild-type models.55 Further murine experiments also showed that adoptively transferred Cbl-b-/- CD8+ T cells could eradicate TC-1 injected tumor cells while promoting ongoing CD8+ T cell proliferation.55 Similarly, Cbl-b-deficient CAR T cells engineered against carcinoembryonic antigen demonstrated reduced exhaustion markers (PD1+TIM3+) in MC38 syngeneic mice.57 Additional mechanistic studies have identified IL-9 and Th9 cells as key mediators of antitumor efficacy in Cbl-b-/- models.58 These results not only reveal the unique role of Cbl-b in defining the cytokine profile and cellular differentiation of the TME, but also support the translational potential of Cbl-b inhibition with cellular therapies.
Treg and myeloid-derived suppressor cells
Treg and myeloid-derived suppressor cells (MDSC) protect against autoimmunity in the setting of chronic antigenic presentation, but in the TME, these cells can lead to anergy, ICI resistance, and increased tumor proliferation. Cbl-b promotes peripheral Treg development via Akt2, converting CD4+CD25- cells into CD4+CD25+FoxP3+ T cells.59 In contrast, Cbl-b deficient CD8+ T cells are resistant to Treg or TGF-β-mediated suppression.59 While the immunosuppressive effects of MDSCs are less well characterized, connections have been identified between the glycolipid, alpha-galactosylceramide (aGalCer), with both Cbl-b activation and MDSC induction.60 aGalCer has been shown to upregulate Erg2/3 with subsequent induction of PD1 and Cbl-b in NKT cells along with MDSC stimulation.60
Natural killer cells
Natural killer (NK) cells directly promote cytotoxicity against stressed and foreign cells in the absence of MHC-II or antibody signaling.61 Unlike T cells, Cbl-b deficiency does not impact NK differentiation, however Cbl-b-/- mice display markedly enhanced NK proliferation and IFN-γ production.62 This may be regulated by TAM kinases receptors, ubiquitylation targets of Cbl-b. A small molecule TAM modulator has been shown to significantly reduce metastatic tumor growth in a melanoma murine model, a finding reversed on NK cell depletion.62 Based on this model, a TAM/Cbl-b inhibitory pathway rendering NK cells dysfunctional has been proposed. Additionally, elucidation of intracellular signaling revealed that NK cell anergy can be induced by Cbl-b mediated ubiquitination of CARMA1.63 On targeting CARMA1, a component of the CBM complex, Cbl-b ultimately blocks NF-kB transcriptional activity leading to NK cell suppression. Given these data, and the importance of NK-mediated cytotoxicity in the TME, therapeutic Cbl-b inhibition may offer a multicellular approach toward antitumor immunity.
B cells
While humoral-mediated adaptative immunity is not the primary defense against tumor invasion, B cells play a critical role via antigen presentation and cytokine signaling. CD40 is a key costimulatory receptor that leads to isotype switching, germinal cell formation, and NF-kB-mediated proliferation in B cells.64 Cbl-b induced TRAF signaling inhibits B cell activation in the absence of CD40 activation.65 Unsurprisingly then, Cbl-b deficiency can significantly enhance CD40-mediated B cell proliferation.66 Akin to the TCR and CD28 coreceptor on T cells, the simultaneous activation of the BCR and CD40 on B cells leads to a high-affinity, antigen-specific clonal expansion.6 Cbl-b inhibition lowers the threshold of BCR induced activation regardless of CD40 costimulation.
Dendritic cells
Macrophages and DCs preferentially target and phagocytose microbial antigens or stressed cells via generalized danger sensing receptors (eg, Toll-like receptors, TLR).67 This rapid but non-specific process recruits additional immune cells and activates the adaptive immune system via antigen presentation using MHC-II receptors.68 Similar to NK cells, Cbl-b deficiency does not affect DC differentiation. However, Cbl-b-/- mice produced significantly increased levels of TNF-α and IL-6 on LPS-induced TLR4 stimulation as compared with wild-type mice.8 As opposed to Cbl-b deficient T cells, the enhanced cytokine production of Cbl-b-/- DCs alone does not suffice to prime antigen-specific CD8+ T cell expansion. Additionally, Cbl-b-deficient mice exposed to LPS, an innate ligand to TLRs, were shown to be highly susceptible to septic shock.69 Overall, these results clarify the lack of benefit for systemic Cbl-b blockade in the absence of a primary antigenic presentation to adaptive lymphocytes.
Targeting CD28 and Cbl-b in the preclinical setting
The CD28 pathway remains a high priority target for the next generation of IO therapies for several reasons: (1) CD28 ligation is the primary costimulatory signal required for effector T cell response70; (2) CD28 agonists have been shown to lower the threshold for T cell stimulation17; (3) CD28 receptors are constitutively active in a majority of CD4+ and CD8+ T cells, as opposed to other costimulatory receptors14; (4) CTLA-4 inhibitors have been shown to boost CD8+ T cell activity and promote clinical response71; and (5) a critical downstream protein, Cbl-b, has been shown to regulate multiple costimulatory and coinhibitory pathways, T cell transcriptional activity, and ultimate immune cell response.16
Though certainly promising, numerous challenges have surfaced in the effort to target the CD28 pathway. First, early clinical data demonstrated the risk of highly morbid, autoimmune effects following non-specific agonism of this constitutively active coreceptor.17 Additionally, extracellular blockade of CTLA-4 alone has had limited success, likely secondary to downstream resistance or bypass mechanisms.72 Further, T cell infiltration itself is limited by immunosuppressive cytokine (eg, TGF-ß) and cellular (eg, Treg, MDSC) profiles along with poor neoantigenicity.73 Finally, many downstream molecular mediators (ie, Cbl-b) have been labeled ‘undruggable’ targets due to unfavorable chemical structures, competitive active sites, and elusive allosteric pockets.16 55 74
Because Cbl-b-/- CD8+ T cells have shown resistance to Treg and TGF-β mediated suppression, all while preserving antigenic specificity,75 multiple groups have pursued novel platforms with the intent of developing clinical therapeutics that inhibit Cbl-b (table 2). Pharmaceutical approaches include DNA encoded libraries and machine learning algorithms, CRISPR genome editing combined with adoptive cell therapy, and siRNA knockdown, among others.
Platforms targeting Cbl-b currently in development or under investigation
The most advanced Cbl-b targeting program has been developed by Nurix, utilizing a priority DNA encoded library screening platform toward identifying compounds that can link E3 ligases with proteins of interest.76 This process can target proteins for ligase mediated ubiquitination and ultimately proteasomal destruction. NX-1607 is an oral small molecule that significantly reduces tumor growth in colon and triple negative breast cancer murine models.77 Cellular and molecular correlates of this inhibition demonstrate enhanced NK and CD8+ T cell activity in the TME; additionally, NX-1607 mediated tumor inhibition was reversed on NK or CD8+ T cell depletion in these models.77 Given a reduction in TCR diversity and tumor reactive TIL following checkpoint blockade, Nurix has also developed an ex vivo Cbl-b inhibitor (NX-0255) to be used alongside adoptive T cell therapy. This platform, termed drug enhanced tumor infiltrating lymphocyte (DeTIL) therapy, cultures patient-derived tumor fragments with IL-2 and NX-0255 to further stimulate antigen-specific CD8+ T cells. These DeTILs are then reinfused back into the patient. Both NX-1607 (NCT05107674) and DeTIL-0255 (NCT05107739) have started recruitment efforts for phase I clinical trials in advanced malignancies.
Applying a siRNA platform, Apeiron is combining adoptive T cell therapy with novel Cbl-b inhibition. The APN401 approach uses electroporation to transfect peripheral leukocytes with Cbl-b specific siRNA, and reinfuses these transfected cells back into the patient. While not as tumor specific as DeTIL therapy, APN401 forfeits cellular specificity for the benefits of a completely ambulatory treatment model. Preclinical data assessing siRNA transfected CD8+ T cells alongside a DC-based tumor vaccine showed enhanced intratumoral inflammatory cytokine signaling along with augmented tumor inhibition in a B16 murine model.78 Preliminary data from a phase I clinical trial, however, showed only marginal clinical improvements—best tumor response entailed four of sixteen patients with stable disease.79 Regardless, a lack of immune-related adverse events (irAE) has encouraged ongoing translational work with this platform.
Using allosteric chemistry, HotSpot Therapeutics has developed a series of small molecule Cbl-b inhibitors.80 HotSpot has described a novel regulatory pocket on Cbl-b and developed an inhibitor (HOT-A) that prevents Cbl-b phosphorylation and functional activity.81 In a series of stepwise experiments, HOT-A was shown to increase IFNγ secretion and CD8+ T cell proliferation using human-derived peripheral blood mononuclear cells. Additionally, K562 tumor cells were killed on coadministration of HOT-A with NK cells. Finally, anti-CD3 treated mice showed enhanced IL-2 secretion and increased differentiation of activated (CD3+/CD69+) T cells on HOT-A delivery.81 These results, though preliminary, provide a promising foundation for upcoming clinical trials (NCT05662397).
Clinical implications and next steps
The data presented above suggest that Cbl-b, part of the CD28 pathway, is a key mediator of immunosuppression in the TME. Thus, this intracellular ligase may represent a novel and important target for the next phase of IO. To effectively translate the known biology of Cbl-b into clinic, however, focus should be placed on unexplored spaces based on the development of anti-CTLA-4 therapy, identifying biomarkers for optimal patient selection, assessing appropriate combinatorial strategies, and anticipating irAEs.
CD28 and CTLA-4 control Cbl-b expression and activity in a potent but inverse fashion.51 While CD28 activation inhibits Cbl-b-mediated immunosuppression, CTLA-4 ligation promotes Cbl-b activity, TGF-β signaling, loss of PTEN inactivation, and ultimate T cell anergy (figure 1B).82 The efficacy of CTLA-4 blockade is dependent on these downstream effects, however, until recently, targeting these intracellular mediators for ICI augmentation has been limited. While anti-CTLA-4 therapy quickly revolutionized the treatment of some advanced cancers after the Food and Drug Administration (FDA) approval of ipilimumab in 2011, CTLA-4 blockade was soon overshadowed by PD(L)1 inhibitors given significantly improved survival and less toxicity with anti-PD1 versus anti-CTLA-4 monotherapy.83 Currently, ipilimumab is primarily used in combination with PD1 blockade as induction therapy in select cancers, while anti-PD1 therapy is approved in an ever-growing list of advanced malignancies.84
Whereas the differential clinical outcomes between CTLA-4 and PD1 blockade are relatively clear, the cellular and molecular mechanisms explaining this distinction are less well understood. This lack of understanding in pharmacological variation, along with favorable toxicity profile of PD1 monotherapy, may have led to a premature dismissal in targeting the CTLA-4 pathway. Tumors exposed to PD1 antibodies exhibit increased levels of cytolytic transcription factors along with decreased Treg infiltration as compared with CTLA-4 blockade.85 Additional cellular correlates of therapeutic benefit in patients with melanoma suggest that changes in NK cell subsets may differentiate response to anti-PD1 therapy whereas anti-CLTA-4 response rely primarily on CD4+ and CD8+ memory T-cell subsets.86 Though further characterization of these independent pathways is still needed, dual checkpoint blockade has been shown to prevent resistance to anti-PD1 therapy caused by compensatory upregulation of secondary inhibitory receptors, a mechanistic explanation behind both augmented clinical benefit as well as increased irAEs.87 Despite focus on anti-PD1(L)1, several clinical trials have suggested an ongoing role for CTLA-4 inhibitors in multiple cancers, particularly as an immunologic primer alongside PD1 therapy. For example, ipilimumab was shown to improve PFS (4.0 vs 3.1 months, p<0.01) but fell short demonstrating an improvement in OS (11.2 vs 10.0 months, p=0.053) in castration-resistant prostate cancer.88 In a phase II trial of recurrent ovarian cancer, the addition of ipilimumab to nivolumab resulted in improved and clinically relevant 6 month PFS relative to PD1 monotherapy (25.5% vs 16.3%, p=0.13), though the study was not powered to confirm this difference.89 Results from CheckMate 227 confirmed OS advantage with ipilimumab plus nivolumab versus chemotherapy as first-line treatment for advanced NSCLC, regardless of PD-L1 expression (median OS 17.1 vs 13.9 mos, HR 0.73, 95% CI 0.64 to 0.84). Notably, the phase III HIMALAYA trial led the FDA to accept tremelimumab under priority review for the treatment of unresectable HCC alongside durvalumab (HR 0.78, p=0.0035, vs sorafenib alone). Finally, ipilimumab has been shown to have some salvage treatment potential in PD1-refractory advanced RCC, providing an immunotherapeutic ‘boost’ on progression with nivolumab monotherapy.90 Overall, further investigation and targeting of the CD28, CTLA-4, and Cbl-b pathways may provide a key route toward effective treatment of PD1-resistant tumors.
Biomarkers for Cbl-b therapeutics
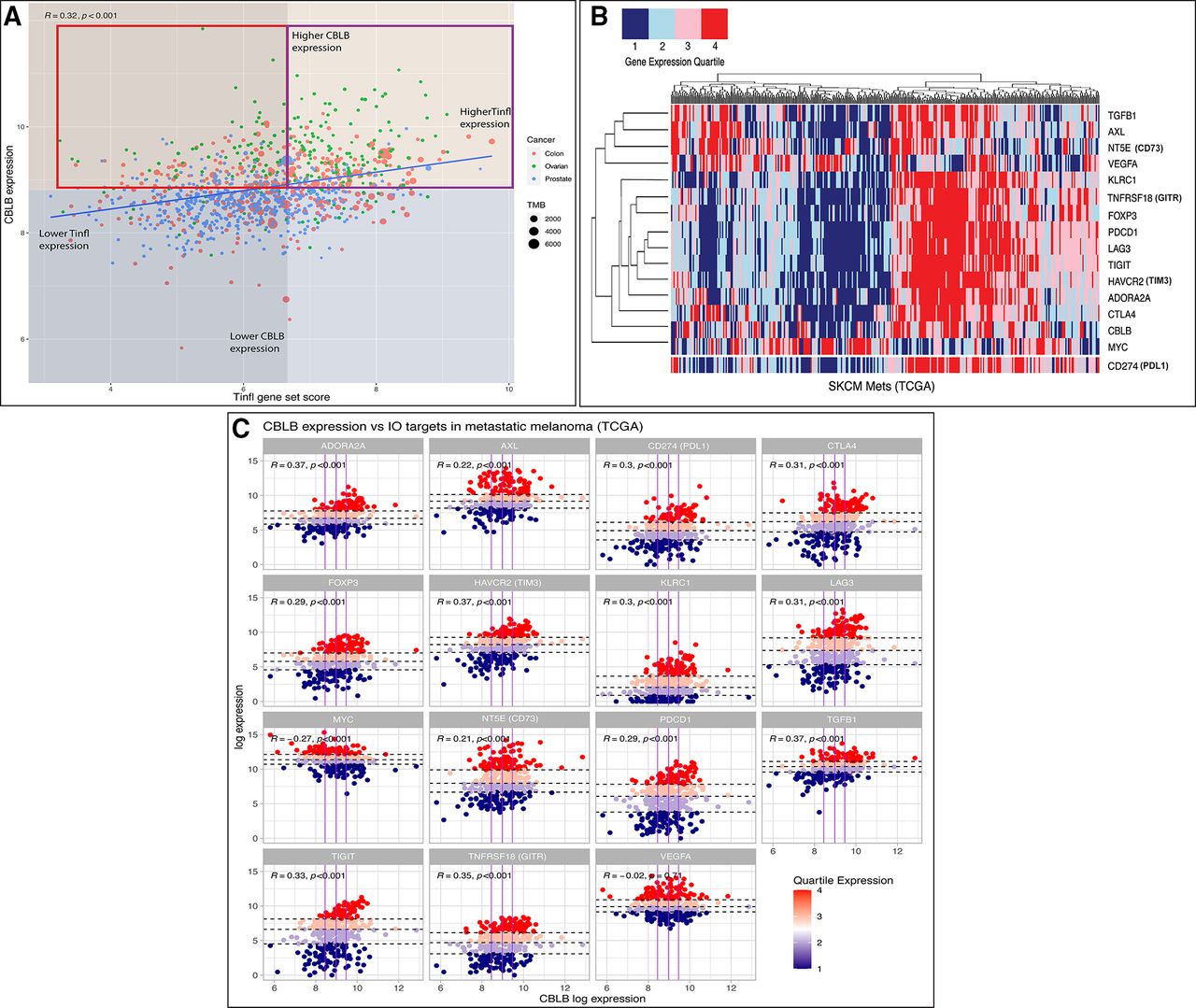
Biomarker development remains an area of unmet need in IO and may be of particular relevance surrounding therapeutic targeting of Cbl-b. PD-L1 expression, tumor mutational burden (TMB), and CD8+ T cell infiltration are widely used clinical biomarkers for ICI efficacy, though predictive utility has been inconsistent in many cancers.91 Gene signatures have been developed to characterize the transcriptional landscape of responding versus non-responding tumors. For example, a T cell-inflamed signature (Tinfl) containing IFN-γ responsive genes has shown predictive value in multiple cancers treated with anti-PD1 therapy.92 Additionally, an adenosine signature (AdenoSig) containing myeloid and inflammatory related genes was found to predict tumor regression on anti-adenosine therapy.93 Thus, pretreatment gene expression profiles might be useful in identifying patients who could benefit from anti-Cbl-b therapy. As a preliminary assessment, we postulate that patient tumors with low/moderate Tinfl expression, low/moderate TMB, and high CBLB expression might be a high unmet need population who could benefit from Cbl-b inhibition. Cancers historically unresponsive to ICI, for example, CRC, ovarian, and prostate, can be classified into Tinfllo/CBLBhi tumors based on expression data (figure 2A). Notably, the pharmacokinetic penetration of small molecule Cbl-b inhibitors into the TME may increase their effectiveness in TMBlo/TILlo tumors as compared with traditional checkpoint antibodies. Overall, the immunostimulatory properties of this therapy, IO non-responsive nature of these tumors, and lack of effective options for this cohort could provide useful guidance for clinical application. Integrating additional biomarkers, including microsatellite instability, mismatch repair deficiency, and TCR diversity, may offer further predictive power in the future.

{kind=link}
{kind=link}
(A) Comparison of T cell-inflamed gene set enrichment score (Tinfl)100 versus CBLB gene expression across select TCGA tumor samples. Red box highlights tumor samples postulated to receive greatest benefit from Cbl-b inhibition versus ICI therapy±Cbl-b inhibition (purple box). Median expression values denoted by orange (higher CBLB), blue (lower CBLB), gray (lower Tinfl), and transparent (higher Tinfl). TMB, tumor mutational burden. Spearman’s correlation coefficient and p value are shown at the left corner of the figure. Methods: Gene set enrichment analysis (GSEA) was performed using normalized and log2-transformed RNAseq gene expression data from the TCGA. All analyses were performed using Bioconductor packages in R (V.4.0.3).100 (B) Heatmap of various IO targets (right) based on quartile expression of TCGA metastatic melanoma samples. CD274 (PDL1) data were added to bottom of heatmap for visual reference. Methods: Unsupervised hierarchical clustering performed across tumor samples; quartile expression calculated across all samples per gene. All analyses were performed using R (V.4.0.3).100 (C) Expression of fifteen IO targets versus CBLB expression in TCGA metastatic melanoma samples. Vertical (purple) lines denote CBLB quartiles; horizontal (dashed) lines denote IO target quartiles per facet. Methods: Faceted scatterplot with colors denoting quartile expression of each gene in R (V.4.0.3). Cbl-b, Casitas B lymphoma-b; ICI, immune checkpoint inhibitor; IO, immuno-oncology.
Further analyzing the expression data of melanoma samples reveals that CBLB clusters with other IO targets and associates with PDL1 expression, indicating a unified phenotype among T cell-inflamed tumors (figure 2B).94 As shown in figure 2C, this pattern offers potential strategies for combinatorial therapies. For example, TGFβ1 expression is correlated with both CBLB and PDL1, denoting a similar profile to the aggressive CRC mesenchymal subtype (CMS4).95 This molecular subtype is characterized by increased stromal invasion, moderate immune infiltration scores, and upregulation of TGF-β, prompting novel drug development and ongoing clinical trials targeting the TGF-β pathway.96 The exceptions to this pattern, however, suggest other potential strategies for treating non-T cell-inflamed tumors. Figure 2B,C shows that VEGFA and MYC do not correlate with either PD-L1 or CBLB. Robust preclinical work has led to numerous clinical trials aiming to ‘normalize’ the chaotic vasculature of the TME and promote immune cell infiltration using anti-VEGF priming plus checkpoint blockade.97 Similarly, the MYC family of transcription factors has long been implicated in decreasing the immunogenicity of the TME, yet targeting this master regulator and its downstream processes has remained elusive due to its pharmacologic challenges, not unlike Cbl-b.98 Overall, further in silico studies of gene signatures will continue to offer strategies to augment existing ICI, reverse the immunosuppressive TME, and expose novel IO targets.
While Cbl-b has been most frequently studied in the context of CD28 and CTLA-4, other relevant associations have been identified with pathways such as PD1/PD-L1 signaling.99 A B16 melanoma model resistant to PD-L1 mediated suppression showed significant tumor regression with Cbl-b-/- T cells.99 These results highlight Cbl-b as a key downstream mediator of PD1 signaling that could also serve as a potential source of anti-PD1 resistance and emphasize the potential for an augmented benefit of combinatorial anti-PD1 plus Cbl-b blockade. Future studies comparing the mutations, expression, and functional levels of Cbl-b in anti-PD1 responsive versus non-responsive tumors would provide further clarification. Additional elements leading to immunosuppression in the TME include certain metabolites, vascular components, and other T cell coreceptors. Ongoing trials targeting these immunological mediators alongside ICI may help identify unmet needs in non-responding cohorts, predict the utility of combinatorial strategies with Cbl-b blockade, and highlight potential patient populations most likely to benefit.
As described with the novel treatment platforms, combining Cbl-b inhibition with tumor specific T cells will be a key element in the effort to maximize site specific immune stimulation and limit irAEs. Adoptive T cell transfer and antibody-drug conjugates provide routes for tumor specific drug delivery. While the former specifically addresses T cell infiltration, the latter may provide a more feasible and less intensive treatment algorithm for delivering Cbl-b inhibitors to the TME. Regardless, the dichotomy between tumor-specific targets and systemic immune activation is not a new IO-related principle, but one that remains relevant for achieving clinical benefit while minimizing dose-related toxicities.
Cbl-b is a downstream, master regulator of both costimulatory and coinhibitory pathways of T cell activation—thus representing a prime target to reverse immune suppression in the TME. With the aid of novel biopharmaceutical platforms, Cbl-b inhibitors can now target this once ‘undruggable’ downstream mediator of CD28/CTLA-4 signaling, provide immune stimulation in the context of antigen specificity, offer antitumor activity in cancers unresponsive to established checkpoint therapy, and could soon be added to the armamentarium of IO.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @RiyueSunnyBao, @jasonlukemd
Contributors Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work: RCA, RB and JJL. Drafting the work or revising it critically for important intellectual content: RCA, RB and JJL. Final approval of the version to be published: RCA, RB and JJL. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: RCA, RB and JJL.
Funding JJL acknowledges NIH UM1CA186690-06, P50CA254865-01A1, P30CA047904-32, R01DE031729-01A1. RB acknowledges NIH P30CA047904, P50CA254865-01A1, P50CA097190, R01DE031729-01A1.
Competing interests RB: (all provisional) PCT/US15/612657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof), PCT/US63/055227 (Methods and Compositions for Treating Autoimmune and Allergic Disorders). JJL: DSMB: Abbvie, Immutep; Scientific Advisory Board: (no stock) 7 Hills, Fstar, Inzen, RefleXion, Xilio (stock) Actym, Alphamab Oncology, Arch Oncology, Kanaph, Mavu, Onc.AI, Pyxis, Tempest; Consultancy with compensation: Abbvie, Alnylam, Avillion, Bayer, Bristol-Myers Squibb, Checkmate, Codiak, Crown, Day One, Eisai, EMD Serono, Flame, Genentech, Gilead, HotSpot, Kadmon, KSQ, Janssen, Ikena, Immunocore, Incyte, Macrogenics, Merck, Mersana, Nektar, Novartis, Pfizer, Regeneron, Ribon, Rubius, Silicon, Synlogic, Synthekine, TRex, Werewolf, Xencor; Research Support: (all to institution for clinical trials unless noted) AbbVie, Agios (IIT), Astellas, Astrazeneca, Bristol-Myers Squibb (IIT & industry), Corvus, Day One, EMD Serono, Fstar, Genmab, Ikena, Immatics, Incyte, Kadmon, KAHR, Macrogenics, Merck, Moderna, Nektar, Next Cure, Numab, Pfizer (IIT & industry) Replimmune, Rubius, Scholar Rock, Synlogic, Takeda, Trishula, Tizona, Xencor; Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof).
Provenance and peer review Commissioned; externally peer reviewed.