Article Text

Abstract
Background Type 1 conventional dendritic cells (cDC1s) possess efficient antigen presentation and cross-presentation activity, as well as potent T cell priming ability. Tissue-resident cDC1s (CD103+ cDC1s in mice, CD141+ cDC1s in humans) are linked with improved tumor control, yet the efficacy of immunotherapy using this population is understudied.
Methods We generated murine CD103+ cDC1s in vitro and examined their expression of cDC1-related factors, antigen cross-presentation activity, and accumulation in tumor-draining lymph nodes (TdLNs). The antitumor efficacy of the in vitro-generated CD103+ cDC1s was studied in murine melanoma and osteosarcoma models. We evaluated tumor responses on vaccination with CD103+ cDC1s, compared these to vaccination with monocyte-derived DCs (MoDCs), tested CD103+ cDC1 vaccination with checkpoint blockade, and examined the antimetastatic activity of CD103+ cDC1s.
Results In vitro-generated CD103+ cDC1s produced cDC1-associated factors such as interleukin-12p70 and CXCL10, and demonstrated antigen cross-presentation activity on stimulation with the toll-like receptor 3 agonist polyinosinic:polycytidylic acid (poly I:C). In vitro-generated CD103+ cDC1s also migrated to TdLNs following poly I:C treatment and intratumoral delivery. Vaccination with poly I:C-activated and tumor antigen-loaded CD103+ cDC1s enhanced tumor infiltration of tumor antigen-specific and interferon-γ+ CD8+ T cells, and suppressed melanoma and osteosarcoma growth. CD103+ cDC1s showed superior antitumor efficacy compared with MoDC vaccination, and led to complete regression of 100% of osteosarcoma tumors in combination with CTLA-4 antibody-mediated checkpoint blockade. In vitro-generated CD103+ cDC1s effectively protected mice from pulmonary melanoma and osteosarcoma metastases.
Conclusions Our data indicate an in vitro-generated CD103+ cDC1 vaccine elicits systemic and long-lasting tumor-specific T cell-mediated cytotoxicity, which restrains primary and metastatic tumor growth. The CD103+ cDC1 vaccine was superior to MoDCs and enhanced response to immune checkpoint blockade. These results indicate the potential for new immunotherapies based on use of cDC1s alone or in combination with checkpoint blockade.
- CD103+dendritic cell vaccine
- immune checkpoint blockade
- melanoma
- osteosarcoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
T cell-based immunotherapy and antibody-mediated immune checkpoint blockade are among the most exciting advances in cancer therapy over the past decade, eliciting durable control of several cancers and prolonging survival rates.1 2 Nonetheless, limitations exist with current immunotherapies including non-responsiveness or adverse events.3 Thus, approaches to improve the specificity, effectiveness, and safety of cancer immunotherapy across patient populations and cancer types are needed.
Dendritic cells (DCs) are the principal antigen-presenting cells of the immune system and therefore shape adaptive, antitumor immunity.4 These features indicate DCs as a promising tool for anticancer treatment.5–7 The majority of DCs used in clinical trials have been generated from human CD14+ monocytes (MoDCs) or CD34+ progenitors in culture.8 While these DCs can be produced in abundance and are capable of inducing tumor-specific T cells with minimal side effects, their efficacy remains limited.7–9 More recently, specific DC populations including plasmacytoid DCs (pDCs) and type 2 conventional DCs (cDC2s) have yielded clinical responses,10 11 yet these subsets are relatively sparse in vivo. The efficacy or feasibility of current DC vaccines, therefore, may be limited by issues such as use of suboptimal or rare DC subsets.
Type 1 cDCs (cDC1s) exhibit several features that predict important roles in activating antitumor immunity, and abundance of cDC1s within tumors correlates with improved patient outcomes and response to immune checkpoint blockade.12 13 The cDC1 subset possesses antigen uptake, antigen presentation, and antigen cross-presentation abilities. Moreover, migratory CD103+ cDC1s transport tissue or tumor antigens to lymph nodes (LNs) and elicit antigen-specific CD8+ T cell responses.14–18 CD103+ cDC1s can be recruited to tumors by T cell-expressed chemokines including XCL1, where they participate in further T cell recruitment through expression of chemoattractants such as CXCL10.12 19 Consistent with these functions, lymphoid organ-resident CD8α+ cDC1s induced CD8+ T cell responses and protected mice against melanoma engraftment, while treatments to expand and activate locally recruited CD103+ cDC1s increased the efficacy of B-raf kinase (BRAF) inhibition and PD-1 blockade in controlling melanoma.18 20 Collectively, these features suggest cDC1-based vaccines will elicit antitumor activity, yet this concept requires further validation. Moreover, whether cDC1-based vaccines protect from metastatic disease is important to examine, as metastasis is a primary cause of mortality in patients with cancer.
Melanoma and melanoma metastatic disease are responsive to immunotherapies such as checkpoint blockade.2 7 A number of other tumor types, however, remain poorly responsive or refractory. In particular, pediatric solid tumors are frequently non-responsive to immunotherapy. Additionally, these tumors often develop resistance to standard treatments, leaving few clinical options and a need to identify novel approaches for young patients with cancer.
Osteosarcoma is the most common primary malignancy of bone affecting pediatric and adult patients. Chemotherapy and surgery are standard treatments, yet the 5-year survival rate is <20% for osteosarcoma patients who present with metastases or relapse following treatment. Negligible improvements have occurred in osteosarcoma therapeutic options over the past 25 years.21 22 Mifamurtide, a liposome-encapsulated immunotherapy that activates pulmonary macrophages, improved the disease-free and overall survival of patients with osteosarcoma lung metastases.23 By contrast, MoDC vaccines have yielded little to no clinical responsiveness,24–26 while checkpoint blockade with PD-1 or CTLA-4 antibodies led to objective clinical response rates of approximately 5% in osteosarcoma patients.27 28 These results underscore the critical need to improve therapeutic options for osteosarcoma.
Here, we tested the efficacy of in vitro-generated CD103+ cDC1s in preclinical murine tumor models, using the well-established B16 melanoma as well as the K7M3 osteosarcoma model. We found administration of poly I:C-activated, tumor antigen-loaded CD103+ cDC1s suppressed primary melanoma and osteosarcoma growth, elicited a systemic effect to control untreated bilateral tumors, and restrained distal lung metastasis. These responses were associated with an increase in interferon-γ (IFN-γ)+ CD8+ T cells and tumor antigen-specific CD8+ T cells. Our data suggest an in vitro-generated cDC1-based vaccine offers a novel strategy for cancer treatment including tumors that are refractory to other therapeutic options.
Methods
CD103+ cDC1 vaccination
In vitro-generated CD103+ cDC1s29 were cultured (2.5–4.5×106/mL) in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, Georgia, USA), 1% penicillin-streptomycin, 1 mM sodium pyruvate, and 50 µM β-mercaptoethanol (RPMI 1640 complete medium) ± 20 µg/mL poly I:C (Sigma, St Louis, Missouri, USA), 5% XG-3 supernatant or 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and the surrogate tumor antigens ovalbumin (OVA; 100–400 µg/mL) (Sigma) or K7M3 tumor lysate for 4 hours. Cells were washed twice with phosphate-buffered saline (PBS) and injected (0.5–3×106) into melanoma or osteosarcoma tumors (intratumoral (i.t.)), one or two times 4–7 days following tumor implantation, when tumor sizes reached 10–20 mm2. In bilateral tumor assays, poly I:C-treated and tumor antigen-loaded CD103+ cDC1s (2–3×106 cells) were injected into left-side tumors; right-side tumors remained untreated. For metastasis assays, poly I:C-treated and tumor antigen-loaded CD103+ cDC1s (1–2×106 cells) were injected intravenously (i.v.) approximately 30 days prior to metastasis challenge. In some metastasis assays, mice were injected i.v. with CD45.1+ CD45.2+ OT-I CD8+ T cells (1×106 cells) 1 day prior to CD103+ cDC1 delivery. For combination treatments with checkpoint inhibitors, DC-vaccinated animals were injected intraperitoneally (i.p.) with PD-1 antibody (RMP1-14; 200 µg/mouse) or CTLA-4 antibody (9H10; 200 µg/mouse for the first treatment, 100 µg/mouse for subsequent treatments) 4 days following tumor implantation, with two antibody treatments per week for 3 weeks. PD-1 and CTLA-4 antibodies were purchased from BioXCell company (West Lebanon, New Hampshire, USA).
MoDC vaccination
Murine bone marrow (BM) cultures were initiated at 0.5×106 cells/mL in RPMI 1640 complete medium containing 20 ng/mL GM-CSF. Cells were collected following 6–7 days of culture and MoDCs (CD11c+ cells) were purified using a FACSAria II or III. MoDCs were pretreated with lipopolysaccaride (LPS) (100 ng/mL), OVA (400 µg/mL), and GM-CSF (20 ng/mL) for 4 hours prior to i.t. delivery.
Primary and metastatic melanoma assays
Murine B16-F10 melanoma cells expressing OVA (B16-OVA cells) were cultured in Dulbecco’s modified Eagle medium (DMEM) medium (Gibco, Grand Island, New York, USA) containing 10% FBS and 1% penicillin-streptomycin. For single primary tumors, C57BL/6J mice were shaved on the abdomen and injected subcutaneously (s.c.) with 4×105 melanoma cells on one side. Tumor size (length x width) was measured every 2–3 days using a caliper; mice were euthanized when tumor sizes reached a maximum of 20 mm in any direction or tumor ulceration occurred. To establish bilateral tumors, C57BL/6J mice were injected s.c. with 3×105 melanoma cells on both sides of the abdomen; tumor sizes were monitored every 2–3 days and animals euthanized on one tumor reaching 15 mm in any direction or tumor ulceration. For metastasis assays, C57BL/6J mice were injected with 7.5×105 B16-OVA melanoma cells i.v., approximately 1 month after CD103+ cDC1s were administered. Mice were euthanized 14 d after i.v. melanoma delivery; lung metastases and immune subsets were quantified.
Primary and metastatic osteosarcoma assays
Murine K7M3 cells (a subline of K7M2)30 were cultured in DMEM medium (Gibco) containing 10% FBS, 1% non-essential amino acids, 1% minimal essential medium vitamin solution, 1 mM sodium pyruvate, and 1% penicillin-streptomycin. Balb/c mice were shaved on the abdomen and injected s.c. with 2×106 K7M3 cells. Tumor size (length x width) was measured every 2–3 days using a caliper; mice were euthanized when tumor sizes reached a maximum of 15 mm in any direction or tumor ulceration occurred. For metastasis assays, Balb/c mice were injected with 2×106 K7M3 cells i.v., approximately 1 month after CD103+ cDC1s were administered. Mice were euthanized 28 days later; lung metastases and immune subsets were quantified.
Statistical analyses
Statistical analyses were performed using GraphPad Prism V.7 (La Jolla, California, USA). Data are presented as mean±SEM. Statistical significance was determined by unpaired Student’s t-test, one-way analysis of variance (ANOVA), two-way ANOVA with multiple comparisons, or log-rank (Mantel-Cox) test as indicated in the Figure legends. Differences were considered significant when p=0.05.
Mice, CD103+ cDC1 culture, immune cell profiling, T cell proliferation, DC proliferation and survival, K7M3 tumor lysate preparation, cytokine detection, and quantitative PCR.
These methods are described in the online supplementary material.
Supplemental material
Results
Local and systemic antitumor efficacy of the CD103+ cDC1 vaccine in melanoma
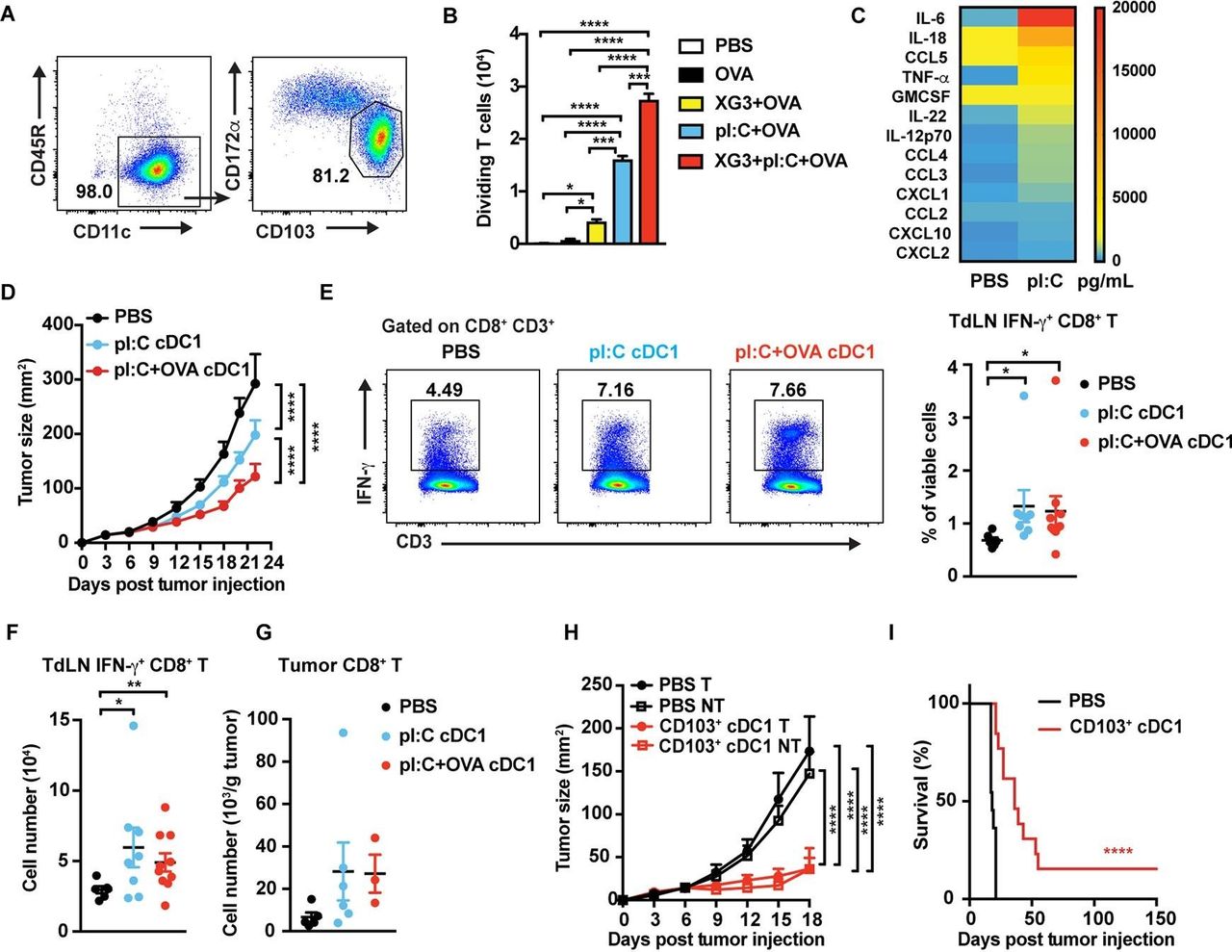
We applied a previously described culture system29 to produce murine nonlymphoid organ CD103+ cDC1s (figure 1A and online supplementary figure S1A). In vitro-generated CD103+ cDC1s expressed cDC1 markers XCR1, CLEC9A, and CD24, while expression of the cDC2 marker CD172α, or myeloid markers Ly6G and Ly6C, was negligible (figure 1A and online supplementary figure S1B). Moreover, the CD103+ cDC1s expressed canonical cDC1 transcriptional regulators including Batf3, Id2, and Irf8. By contrast, expression of pDC (Tcf4, Zeb2) or cDC2 (Irf4) transcription factors was minimal (online supplementary figure S1C).31 In vitro-generated CD103+ cDC1s efficiently cross-presented OVA-derived peptide to activate OT-I CD8+ T cells on stimulation with poly I:C and GM-CSF (figure 1B and online supplementary figure S1D). In addition, poly I:C-treated CD103+ cDC1s produced IL-12p70 and CXCL10, and upregulated MHC-I and cell surface costimulatory molecules CD80, CD86, and CD40 (figure 1C and online supplementary figure S1E,F). MHC-II and toll-like receptor 3 (TLR3) were highly expressed on stimulated and untreated CD103+ cDC1s (online supplementary figure S1F). In vitro-generated CD103+ cDC1s demonstrated the ability to stimulate IFN-γ-producing OT-II CD4+ T cells in culture, suggesting T helper 1 (Th1) induction, but did not promote IL-17+ cell (Th17) generation to a significant degree (online supplementary figure S1G). Collectively, our data indicate in vitro-generated CD103+ cDC1s resemble the nonlymphoid organ cDC1 subset found in vivo in several parameters important for effective Th1 and CD8+ T cell activation.

Anti-tumor efficacy of the CD103+ cDC1 vaccine in murine melanoma. (A) Proportion of CD103+ cDC1s 14 days following BM culture initiation. (B) Cross-presentation ability of CD103+ cDC1s in vitro, as assessed by cocultures with CFSE-labeled naïve OT-I CD8+ T cells and flow cytometry. CD103+ cDC1s were stimulated with poly I:C (20 µg/mL), 5% XG-3 supernatant, and OVA (100 ug/mL), as indicated, prior to coculturing with FACS-purified CD8+ T cells, n=2 per group. (C) Cytokine and chemokine expression by activated CD103+ cDC1s in vitro, determined by multiplex analysis of CD103+ cDC1 culture supernatant 18 hours after stimulation with 20 µg/mL poly I:C or PBS, as indicated. n=3 per group. (D) Mice with B16-OVA tumors were vaccinated i.t. on days 4-7 with CD103+ cDC1s or PBS; CD103+ cDC1s were pretreated with poly I:C (pI:C cDC1) or poly I:C+OVA (pI:C+OVA cDC1), as indicated. Tumor size was measured over time. n=11 (PBS), n=19 (pI:C cDC1), n=23 (pI:C+OVA cDC1). (E, F) The percentage (E) and absolute number (F) of IFN-γ+ CD8+ T cells in TdLNs of CD103+ cDC1 vaccinated mice or PBS controls. (E) Representative flow plots (left) and cumulative data (right). Immune profiles were determined from mice euthanized when tumors reached maximum burden. (G) Tumor-infiltrating CD8+ T cells, determined in mice vaccinated with CD103+ cDC1s or PBS at maximum tumor burden. (H) Tumor sizes in mice bearing bilateral B16-OVA tumors, vaccinated i.t. on day 4 with poly I:C activated and OVA-pulsed CD103+ cDC1s or PBS on the left side. Sizes of treated (T) and untreated (NT) tumors were determined, as indicated. n=9 (PBS), n=8 (CD103+ cDC1). (I) Survival of mice bearing bilateral tumors, treated with CD103+ cDC1 vaccine or PBS, as indicated. n=11 (PBS), n=13 (CD103+ cDC1 vaccine). (B, D–H) data shown as mean±SEM. (A-I) Results from 2 to 3 independent experiments. Data were analysed by one-way ANOVA (B, E-G), two-way ANOVA (D, H), and log-rank (Mantel-Cox) test (I). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANOVA, analysis of variance; BM, bone marrow; CFSE, carboxyfluorescein succinimidyl ester; cDC1, type 1 conventional dendritic cell; FACS, fluorescence-activated cell sorting; IFN-γ, interferon-γ; TdLNs, tumor-draining lymph nodes; OVA, ovalbumin.
On i.t. vaccination, poly I:C-activated, OVA-loaded CD103+ cDC1s restrained melanoma growth and prolonged mouse survival more effectively than controls (PBS treatment) or CD103+ cDC1s stimulated with poly I:C alone (figure 1D and online supplementary figure S2A). Furthermore, i.t. CD103+ cDC1 delivery associated with increased amounts of IFN-γ-producing CD8+ T cells in tumor-draining LNs (TdLNs) at maximum tumor burden (figure 1E,F). Tumor-infiltrating CD8+ T cells were also enhanced by CD103+ cDC1 vaccination, although the difference from controls was not significant (figure 1G). CD103+ cDC1 vaccination increased CD4+ CD25+ Foxp3+ T regulatory cells (Tregs) in TdLNs, while TdLN pDCs were significantly reduced on poly I:C and OVA-activated CD103+ cDC1 treatment (online supplementary figure S2B,C). Other TdLN immune subsets analyzed were unaffected (online supplementary figure S2B,C). These results suggest CD103+ cDC1 vaccination inhibits melanoma growth by promoting CD8+ T cell responses in vivo.
To address whether the CD103+ cDC1 vaccine elicited systemic tumor immunity, mice bearing bilateral melanoma tumors were treated with poly I:C-stimulated, OVA-pulsed CD103+ cDC1s i.t. on the left-side, while right-side tumors remained untreated. Growth of both tumors was restrained by unilateral CD103+ cDC1 vaccination, and survival of vaccinated mice was prolonged over controls (figure 1H,I). Earlier vaccination of smaller tumors also correlated with improved tumor control (figure 1D,H). Together, our data indicate immunization with in vitro-generated CD103+ cDC1s drives systemic immunity and controls local and distal melanoma growth effectively.
Comparison of CD103+ cDC1- and MoDC-mediated antitumor immunity
Since MoDC-based vaccines have been used clinically, we compared the antitumor efficacy of in vitro-generated CD103+ cDC1s with MoDCs. Mice bearing bilateral melanoma tumors were injected i.t. with OVA-loaded and poly I:C-treated CD103+ cDC1s, or OVA-loaded and LPS-treated MoDCs, on the left-side while right-side tumors remained untreated (figure 2A). We used distinct TLR agonists to activate each DC population, as expression of poly I:C-responsive TLR3 is enriched in CD103+ cDC1s, while LPS-responsive TLR4 is abundantly expressed by MoDCs.4 32 Vaccination with MoDCs induced systemic antitumor responses, however, CD103+ cDC1s showed superior activity in suppressing bilateral tumor growth (figure 2B). In addition, survival of CD103+ cDC1 vaccinated mice was prolonged significantly compared with MoDC- or PBS-treated animals (figure 2C), indicating CD103+ cDC1s elicit more effective antitumor immunity versus MoDCs.
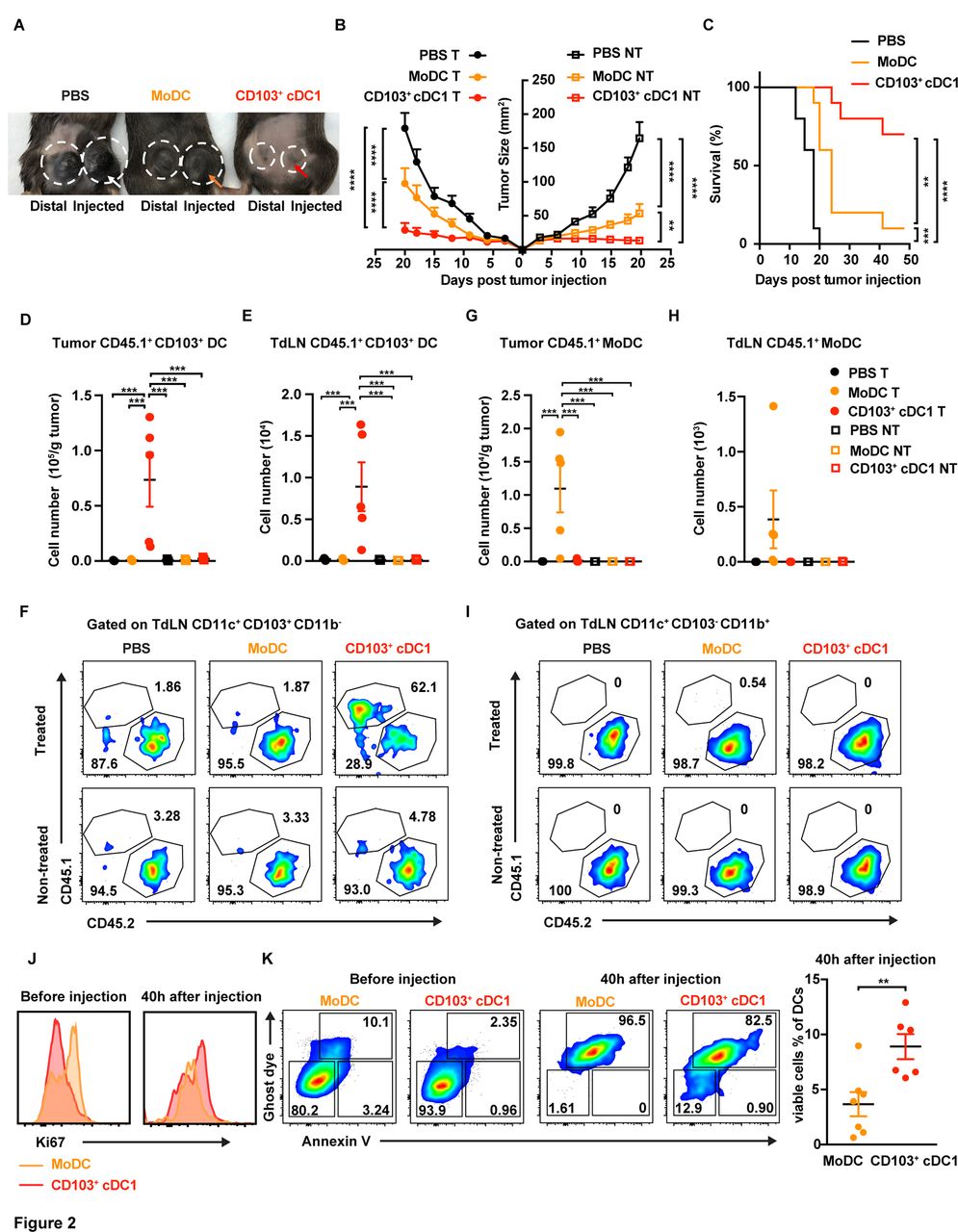
Comparison of CD103+ cDC1 and MoDC vaccine activity in melanoma. (A–C) Animals with bilateral B16-OVA tumors were vaccinated with CD103+ cDC1s or MoDCs at 4 and 7 days following tumor establishment. Prior to DC injection, cells were incubated with GM-CSF (20 ng/mL) and OVA (400 µg/mL). CD103+ cDC1s were stimulated with poly I:C (20 µg/mL) and MoDCs were stimulated with LPS (100 ng/mL). Animals were vaccinated on the left-side tumor (circle with arrow); the distal right-side tumor was untreated (circle without arrow), as indicated (A). Sizes of vaccinated B16-OVA (treated, (T)) tumors and distal untreated (NT) tumors over time, as indicated (B). Survival of mice bearing bilateral tumors, following treatment with CD103+ cDC1 vaccine, MoDC vaccine, or PBS on one side, as indicated (C). n=10 for each group. (D–I) CD45.1+ CD103+ cDC1s or CD45.1+ MoDCs were injected into the left-side tumor of CD45.2+ mice bearing bilateral B16-OVA tumors, using 1×107 DCs/animal, 4 days following tumor implantation. After 40 hours, the amounts of CD45.1+ CD11c+ CD103+ CD11b- cDC1s (CD103+ cDC1s) (D–F) and CD45.1+ CD11c+ CD103- CD11b+ MoDCs (G–I) were determined in tumors and TdLNs; n=5 per group. (J, K) Proliferation (J) and survival (K) of DC vaccines prior to i.t. delivery, or 40 hours following vaccination (n=6–7), as indicated. (B, D, E, G, H, K) Data shown as mean±SEM. (A–K) Results from two independent experiments. Data were analysed by unpaired Student’s t-test (K), one-way ANOVA (D, E, G, H), two-way ANOVA (B), or log-rank (Mantel-Cox) test (C). **P<0.01, ***P<0.001, ****P<0.0001. ANOVA, analysis of variance; cDC1, type 1 conventional dendritic cell; DC, dendritic cell; i.t., intratumoral; MoDC, monocyte-derived dendritic cell; TdLNs, tumor-draining lymph nodes; OVA, ovalbumin.
To evaluate potential mechanisms for the improved response to CD103+ cDC1 vaccination, we compared DC amounts in vaccinated and unvaccinated tumors and corresponding TdLNs 40 hours following i.t. delivery. Congenic CD45.1+ CD103+ cDC1s or CD45.1+ MoDCs were used in these assays to distinguish from endogenous populations. CD45.1+ CD103+ cDC1s were detected in tumors and TdLNs on the side of i.t. injection only (figure 2D–F). In addition, CD45.1+ CD103+ cDC1s were more abundant in TdLNs compared with endogenous cells, as judged by relative frequencies of CD45.1+ versus CD45.2+ CD103+ cDC1s on the treatment side (figure 2F). By contrast, the majority of CD45.1+ MoDCs remained within injected tumors; CD45.1+ MoDCs were rarely observed in TdLNs (figure 2G–I). The chemokine receptor CCR7, which mediates DC migration to LNs, was expressed at higher amounts on tumor-associated CD45.1+ CD103+ cDC1s compared with CD45.1+ MoDCs, while TdLN-associated populations expressed similar CCR7 amounts (online supplementary figure S2D). In addition, despite i.t. injection of equivalent DC numbers, CD45.1+ MoDC amounts were approximately 10% of CD45.1+ CD103+ cDC1s within tumors (figure 2D,G). These results suggest CD103+ cDC1s persist in tumors longer, express greater amounts of CCR7, and have superior TdLN accumulation versus MoDCs on vaccination.
We analyzed DC proliferation and survival in vaccine conditions, to further understand the improved efficacy of CD103+ cDC1s. These assays revealed modest but non-significant differences in Ki67 status prior to vaccination, and no detectable differences 40 hours following i.t. delivery, suggesting similar proliferation rates (figure 2J). By contrast, the activated CD103+ cDC1 vaccine contained fewer apoptotic and dead cells, and a greater proportion of viable cells prior to i.t. delivery, relative to the MoDC vaccine (figure 2K). In addition, CD103+ cDC1s showed enhanced viability following delivery to melanoma tumors versus MoDCs, although the viability of both populations 40 hours after vaccination was low (figure 2K and online supplementary figure S2E). These results indicate CD103+ cDC1s have improved survival following antigen and TLR agonist stimulation, as well as on exposure to melanoma tumors, compared with MoDCs, suggesting enhanced CD103+ cDC1 viability contributes to the efficacy of this population as a tumor vaccine.
Immune responses associated with CD103+ cDC1 and MoDC vaccination
To further examine mechanisms by which the CD103+ cDC1- and MoDC-based vaccines mediate distinct antitumor efficacies, we evaluated their ability to stimulate T cell proliferation in vitro. Poly I:C-activated CD103+ cDC1s showed modestly enhanced ability to induce CD8+ T cell proliferation versus poly I:C-stimulated MoDCs, while LPS-treated DCs demonstrated indistinguishable activity (online supplementary figure S3A). CD103+ cDC1s, however, were significantly inferior to MoDCs in their ability to stimulate CD4+ T cell proliferation in vitro (online supplementary figure S3A).
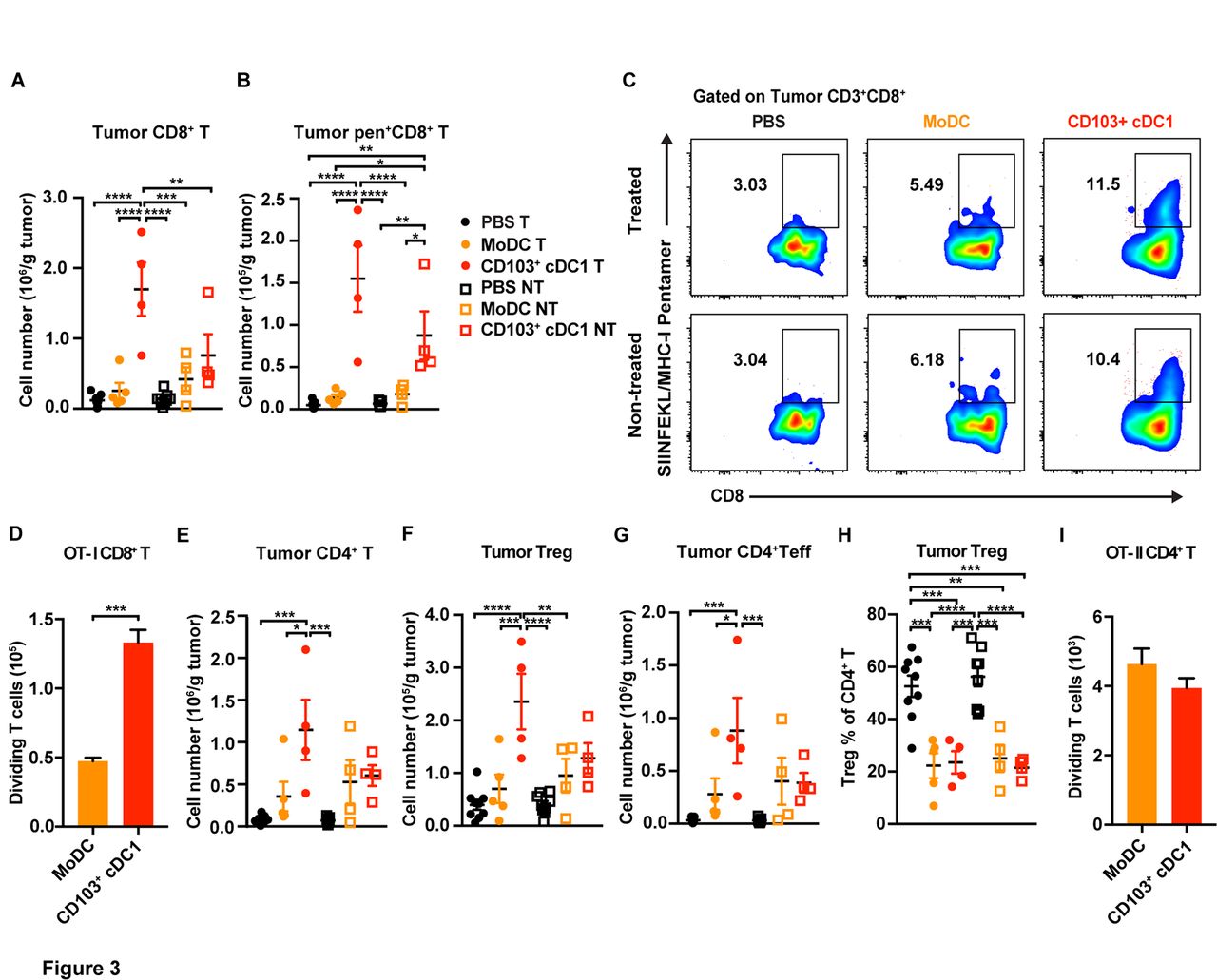
To evaluate whether differential T cell responses were elicited by each DC population in vivo, we investigated immune responses in tumors and TdLNs during the predicted T cell expansion phase,17 14 days after i.t. vaccination. CD8+ T cell amounts were increased in tumors vaccinated with CD103+ cDC1s, compared with MoDC-vaccinated tumors, PBS-treated controls and distal untreated tumors (figure 3A). Moreover, OVA-specific CD8+ T cells within treated as well as distal untreated tumors were increased significantly on vaccination with CD103+ cDC1s, as judged by analysis of SIINFEKL/H-2Kb pentamer+ CD8+ T cells (figure 3B,C). By contrast, tumors in MoDC-vaccinated mice or controls did not accumulate OVA-specific CD8+ T cells (figure 3B,C). In addition, vaccine-origin CD45.1+ CD103+ cDC1s isolated 40 hours following i.t. delivery showed improved ability to stimulate OT-I CD8+ T cell proliferation in vitro, versus vaccine-origin CD45.1+ MoDCs (figure 3D).

Immune responses elicited by CD103+ cDC1 and MoDC vaccines in melanoma. Mice bearing bilateral B16-OVA tumors were vaccinated on one side with CD103+ cDC1s, MoDCs, or PBS as described in the legend to figure 2. (A–C) The number of tumor-infiltrating CD8+ T cells (A), and numbers and proportions of OVA-specific CD8+ T cells in tumors (B, C) were determined 14 days following vaccination in tumors that received the DC vaccine (treated, (T)) and in distal untreated (NT) tumors, as indicated. (D) The ability of vaccine-derived CD45.1+ CD103+ cDC1s or CD45.1+ MoDCs, purified 40 hours following i.t. delivery, to stimulate OT-I CD8+ T cell proliferation was determined in coculture assays in vitro. Proliferating OT-I CD8+ T cells were measured at 72 hours; assays were performed in the presence of 20 ng/mL GM-CSF. (E–G) The numbers of CD4+ T cells (E), CD4+ CD25+ Foxp3+ Treg (F), and CD4+ Foxp3- effector T cells (Teff) (G) were determined 14 days following vaccination in tumors that received the DC vaccine (treated, (T)) and in distal untreated (NT) tumors, as indicated. (H) The percentage of Tregs within the total CD4+ T cell population in tumors from mice vaccinated as indicated, 14 days following vaccination. (I) The ability of vaccine-derived CD45.1+ CD103+ cDC1s or CD45.1+ MoDCs, purified from tumors 40 hours following i.t. delivery, to stimulate OT-II CD4+ T cell proliferation was determined in coculture assays in vitro. Proliferating OT-II CD4+ T cells were measured at 72 hours; assays were performed in the presence of 20 ng/mL GM-CSF. (A, B, D–I) Data shown as mean±SEM. (A–I) Representative (C) or cumulative (A, B, D–I) results from two independent experiments; n=9 (PBS), n=5 (MoDC), n=4 (CD103+ cDC1). Results were analysed by one-way ANOVA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANOVA, analysis of variance; cDC1, type 1 conventional dendritic cells; i.t., intratumoral; MoDC, monocyte-derived dendritic cell; OVA, ovalbumin.
The CD103+ cDC1 vaccine also promoted an increase in tumor-infiltrating CD4+ T cells, including Treg and T effector subsets, in vaccinated tumors but not distal untreated tumors, compared with MoDC vaccination or PBS treatment (figure 3E–G). The relative frequency of Tregs within the tumor-infiltrating CD4+ T cell population was reduced in DC vaccine-treated mice, however, compared with controls (figure 3H). Furthermore, vaccine-origin CD45.1+ CD103+ cDC1s and CD45.1+ MoDCs isolated from tumors were equivalently effective at stimulating CD4+ T cell proliferation in vitro, although CD4+ T cell proliferation was notably reduced compared with CD8+ T cell proliferation in these conditions (figure 3D,I). Tumor-infiltrating B cells, monocytes, macrophages, cDC1s, and cDC2s were not significantly different in treated or untreated tumors among the three groups, while neutrophils were induced in tumors on CD103+ cDC1 but not MoDC vaccination (online supplementary figure S3B).
Within TdLNs from the vaccine-treated side, MoDC vaccination was associated with induction of CD4+ and CD8+ T cells, as well as IFN-γ+ CD4+ Th1 and IL-4+ CD4+ Th2 subsets, compared with PBS or CD103+ cDC1 vaccination (online supplementary figure S3C). TdLNs on the untreated side did not show appreciable changes in T cell amounts on DC vaccination (online supplementary figure S3C). Furthermore, we did not detect differences in SIINFEKL/H2Kb pentamer+ CD8+ T cells, Tregs, B cells, myeloid subsets or DC populations in TdLNs among the three treatment groups (online supplementary figure S3C–F). Vaccine-origin CD45.1+ MoDCs isolated from TdLNs, however, were superior to TdLN-derived vaccine CD45.1+ CD103+ cDC1s in eliciting CD4+ T cell proliferation in vitro, while both DC populations from TdLNs stimulated CD8+ T cell proliferation in vitro similarly (online supplementary figure S3G). Collectively, our data suggest a propensity for CD103+ cDC1s to elicit CD8+ T cell responses, while MoDCs promote greater CD4+ T cell proliferation in vitro. Moreover, our results indicate in vitro-generated CD103+ cDC1s enhance systemic immunity, as well as i.t. accumulation of total CD8+ and tumor antigen-specific CD8+ T cells, more effectively than MoDC vaccination.
Activity of the CD103+ cDC1 vaccine toward melanoma lung metastases
Metastasis treatment remains a major clinical challenge33; thus, we investigated whether the CD103+ cDC1 vaccine would control metastatic tumors. We transferred tumor antigen (OVA)-specific CD45.1+ CD45.2+ OT-I CD8+ T cells to C57BL/6J mice (CD45.2+) and vaccinated animals with poly I:C activated and OVA-pulsed CD103+ cDC1s 1 day later by i.v. delivery. After approximately 1 month, to encompass initial T cell expansion and memory generation phases,34 animals were challenged i.v. with B16-OVA cells to establish experimental lung metastasis (figure 4A). This strategy allowed us to examine whether the in vitro-generated CD103+ cDC1 vaccine elicits long-lasting antitumor immunity on combination treatment with adoptive T cell transfer. Our experiments revealed significant restraint of lung metastases in CD103+ cDC1 vaccine-treated animals versus controls (figure 4B,C), indicating CD103+ cDC1s delivered i.v. stimulate a durable antitumor immune response that suppresses pulmonary metastases.

Efficacy of the CD103+ cDC1 vaccine in metastatic melanoma. (A) Schematic diagram of the experimental approach. (B, C) Representative image (B) and quantification (C) of lung melanoma foci following treatment with poly I:C+OVA-stimulated CD103+ cDC1 vaccine or PBS and challenge with B16-OVA cells i.v., as indicated. n=5 (PBS), n=6 (CD103+ cDC1 vaccine). (D–F) Percentages (D) and absolute numbers (E and F) of donor CD45.1+ IFN-γ+ CD8+ T cells and endogenous CD45.1- IFN-γ+ CD8+ T cells in the spleen of mice treated with CD103+ cDC1 vaccine or PBS. (G, H) Percentages (G) and absolute numbers (H) of splenic Th1, Th2, Th17, and Treg subsets. (B–H) Results from animals 14 days after challenge with B16-OVA cells i.v. Data shown as mean±SEM, from two independent experiments. Results were analysed by unpaired Student’s t-test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. cDC1, type 1 conventional dendritic cells; IFN-γ, interferon-γ; OVA, ovalbumin; Th1, T helper 1; Th2, T helper 2; Th17, interleukin-17-positive T cells; Treg, T regulatory cells.
To evaluate immune responses induced on T cell transfer and i.v. CD103+ cDC1 vaccination, we measured IFN-γ-producing CD8+ T cells in lymphoid organs 14 days following i.v. challenge with B16-OVA. These assays revealed increased amounts of CD45.1+ IFN-γ+ OT-I CD8+ T cells in spleen and inguinal LNs relative to controls (figure 4D,E; online supplementary figure S4A). Importantly, CD45.1+ IFN-γ+ OT-I CD8+ T cells were elevated approximately 6 weeks after OT-I T cell transfer and CD103+ cDC1 vaccination, suggesting long-term maintenance of the OVA antigen-specific T cell response. CD103+ cDC1 vaccination also enhanced endogenous CD45.1- IFN-γ+ CD8+ T cell amounts in spleen, while their amounts in inguinal and lung-draining LNs showed an increased trend (figure 4D,F and online supplementary figure S4A,B). Furthermore, IFN-γ+ CD4+ Th1 cells were increased in spleen, and inguinal and lung-draining LNs, following CD103+ cDC1 vaccination (figure 4G,H and online supplementary figure S4C,D). CD103+ cDC1 vaccination did not affect other CD4+ T cell subsets, B cells, or myeloid cells in lymphoid organs appreciably (figure 4H; online supplementary figure S4C–G). These results suggest CD103+ cDC1 vaccination induces CD8+ T cell and Th1 responses without eliciting global inflammatory or immune system activity.
We next treated mice by i.v. CD103+ cDC1 delivery alone, and challenged animals approximately 1 month later with melanoma cells i.v., to examine whether cotransfer with antigen-specific OT-I CD8+ T cells was required for metastasis suppression. These assays revealed CD103+ cDC1 vaccination alone controlled lung metastases (online supplementary figure S4H,I), suggesting activation of endogenous immune responses by the CD103+ cDC1 vaccine is sufficient to inhibit metastatic tumors. Collectively, our data imply CD103+ cDC1 vaccination confers effective protection against primary melanoma tumors as well as melanoma metastases by eliciting long-lasting, tumor antigen-specific CD8+ T cell-mediated and Th1-mediated immunity.
Efficacy of the CD103+ cDC1 vaccine in primary and metastatic osteosarcoma
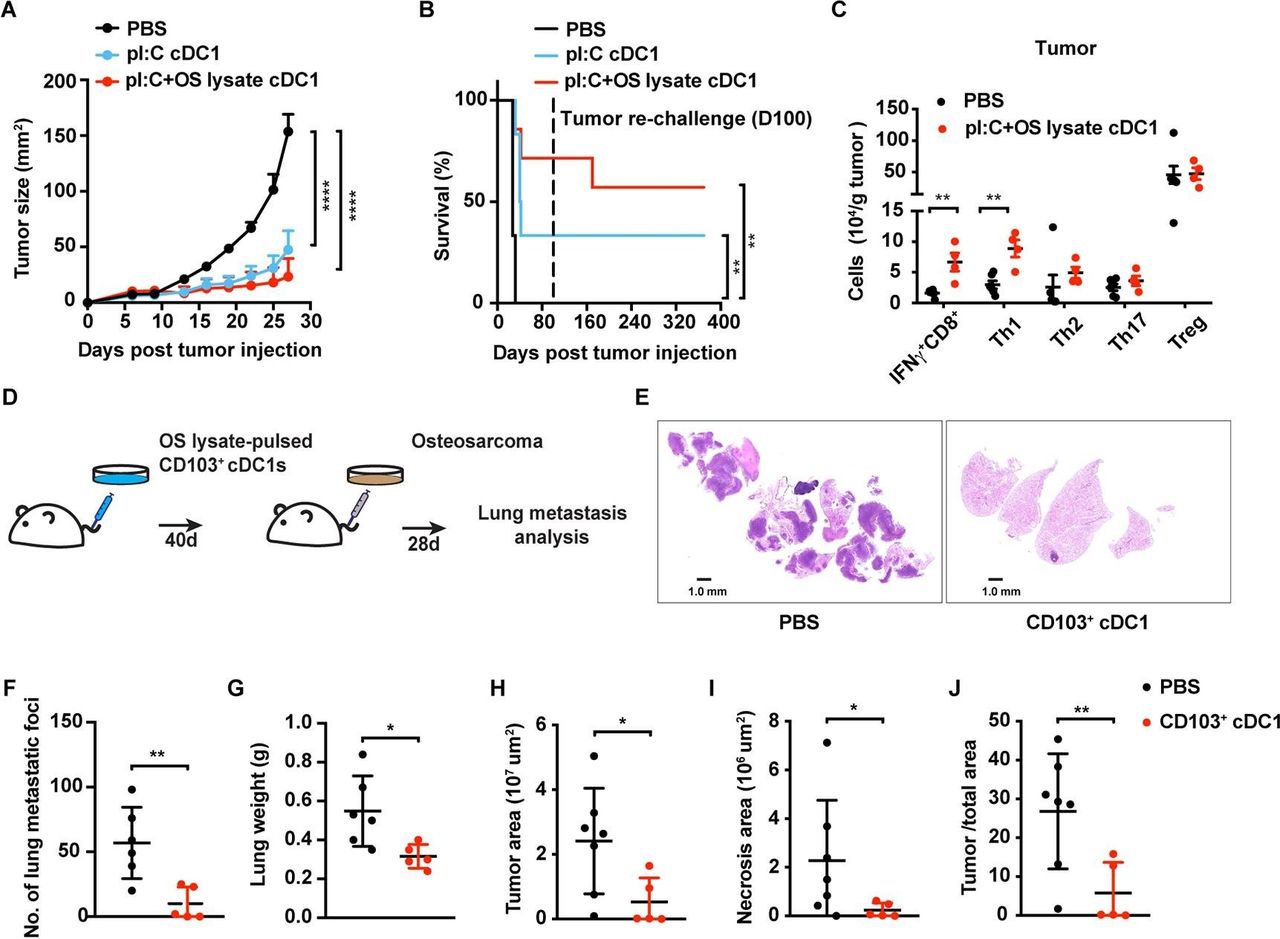
It is important to expand investigation of immunotherapeutic approaches in tumor types that are refractory to current treatments. We selected osteosarcoma for a second model since this tumor presents in pediatric populations and relapsed or metastatic disease has limited therapeutic options. K7M3 osteosarcoma-bearing mice (Balb/c) were vaccinated with Balb/c BM-derived CD103+ cDC1s following activation by poly I:C and incubation with K7M3 tumor lysate (tumor antigen). CD103+ cDC1s restrained osteosarcoma growth and prolonged mouse survival (figure 5A,B). Moreover, the majority of mice rechallenged with K7M3 cells did not develop tumors (figure 5B), suggesting CD103+ cDC1 vaccination elicits long-lasting protection against osteosarcoma. Notably, IFN-γ+ CD8+ T cell and Th1 infiltration into osteosarcoma tumors was increased on CD103+ cDC1 vaccination, as judged by analysis at maximum tumor burden, whereas tumor infiltration of other T cell subsets, B cells, myeloid subsets, or DC populations was not affected (figure 5C and online supplementary figure S5A). Moreover, TdLN T cell subsets were unchanged on CD103+ cDC1 vaccination (online supplementary figure S5B). These results are consistent with our findings in melanoma, suggesting CD103+ cDC1 vaccination induces IFN-γ+ CD8+ T cell and Th1 responses, as well as i.t. accumulation of IFN-γ+ CD8+ T cells, regardless of tumor type.
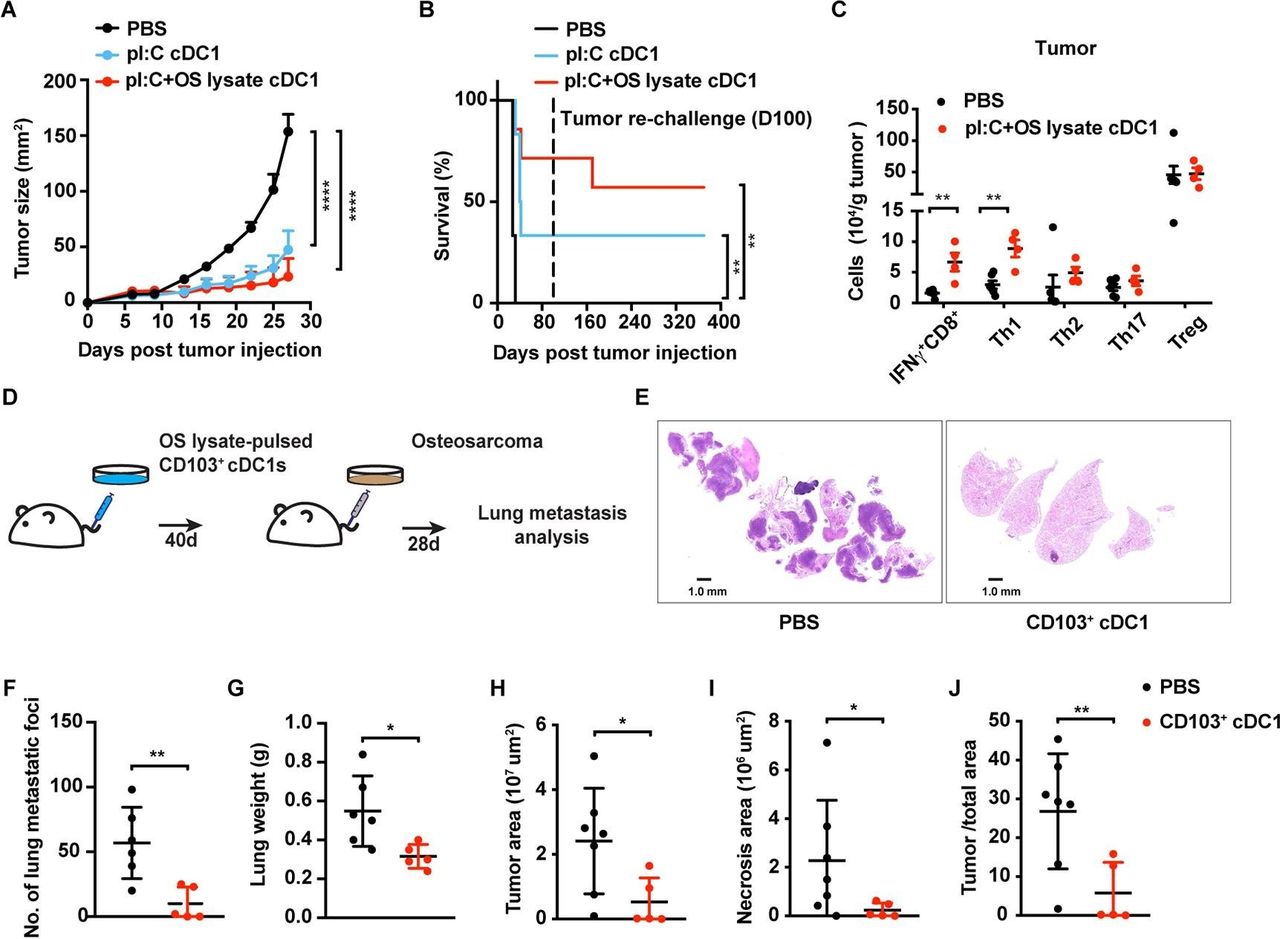
Response of K7M3 primary and metastatic osteosarcoma to the CD103+ cDC1 vaccine. (A, B) Tumor size (A) and survival rate (B) of Balb/c mice following vaccination at 4 and 7 days after tumor inoculation with CD103+ cDC1s or PBS (control). CD103+ cDC1s were pretreated with poly I:C (pI:C cDC1) or poly I:C+K7M3 osteosarcoma cell lysate (pI:C+OS lysate cDC1), as indicated; n=6 per group. Surviving mice were challenged with K7M3 cells on day 100 (dotted line), without receiving additional therapy (B). Cumulative survival curves shown. (C) T cell infiltration in osteosarcoma tumors in CD103+ cDC1 vaccine-treated mice (n=4) or PBS treated controls (n=6). Immune profiles were determined from mice euthanized when tumors reached maximum burden. (D) Schematic diagram of experimental metastasis approach. (E) Representative lung metastases as assessed by H&E staining. (F–J) Quantification of visible metastatic foci (F), lung weight (G), tumor area (H), lung necrotic area (I), and the ratio of tumor area to total area of the lung (J), 28 days after delivery of K7M3 cells i.v. (A, C, F–J). Data shown as mean±SEM. (A–J) Results from two independent experiments. Results were analysed by two-way ANOVA (A), log-rank (Mantel-Cox) test (B), and unpaired Student’s t-test (C, F–J). *P<0.05, **P<0.01, ****P<0.0001. ANOVA, analysis of variance; cDC1, type 1 conventional dendritic cells.
To examine whether CD103+ cDC1 vaccination protected against osteosarcoma pulmonary metastases, we delivered poly I:C-activated and K7M3 tumor lysate-pulsed CD103+ cDC1s i.v. and challenged mice 40 days later with K7M3 tumor cells i.v. (figure 5D). After 4 weeks, pulmonary metastatic foci were visible in controls, yet less frequent in CD103+ cDC1-vaccinated mice (figures 5E,F; online supplementary figure S6). Lung weights, as well as total tumor and necrotic areas in lung, were lower in mice vaccinated with CD103+ cDC1s versus controls (figure 5G–J, online supplementary figure S6). These data indicate CD103+ cDC1 vaccination effectively protects animals from experimental osteosarcoma lung metastases.
Combination treatment of osteosarcoma-bearing mice with CD103+ cDC1 vaccination and checkpoint blockade
CTLA-4 and PD-1 checkpoint blockade have modest impact in osteosarcoma.27 28 35 To examine whether combination treatment with CD103+ cDC1 vaccination improves their efficacy, mice were treated with the CD103+ cDC1 vaccine alone or in combination with CTLA-4 or PD-1 antibody (figure 6A). CTLA-4 or PD-1 blockade alone restrained osteosarcoma growth in some but not all mice (figure 6B). Similar results were found for animals vaccinated with CD103+ cDC1s alone or CD103+ cDC1 vaccination and PD-1 blockade (figure 6B). By contrast, CD103+ cDC1 vaccination and CTLA-4 blockade significantly impaired osteosarcoma growth and led to complete tumor regression in 100% of mice approximately 2 weeks following initiation of therapy (figure 6B,C) These results suggest combination therapy with CD103+ cDC1s and CTLA-4 checkpoint blockade effectively restrains osteosarcoma.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combination therapy with CD103+ cDC1 vaccine and immune checkpoint blockade in osteosarcoma. (A) Schematic diagram of experimental approach. (B–D) Tumor growth in individual mice (B), median tumor size (C), and survival rate (D) following CD103+ cDC1 vaccine and immune checkpoint inhibitors, as indicated. n=6–7 for each group. Data shown as mean±SEM, from two independent experiments. Results were analysed by two-way ANOVA (C) and log-rank (Mantel-Cox) test (D). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANOVA, analysis of variance.
To examine whether individual or combination immunotherapies induced long-lasting antitumor immunity, we rechallenged the surviving mice from each therapeutic group with K7M3 tumors. Tumor-free survival was observed in the majority of mice up to 370 days following tumor rechallenge (figure 6D). These data indicate single or combination immune therapy is capable of establishing immunological memory responses against murine K7M3 osteosarcoma, with the most potent response mediated by combining CD103+ cDC1 vaccination and CTLA-4 blockade.
Discussion
The cDC1 subset mediates antitumor immune responses in mice and is associated with improved outcomes in human cancer.6 12–14 Nonlymphoid organ cDC1s (ie, CD103+ cDC1s in mice or CD141+ DCs in humans) infiltrate solid tumors and transport tumor antigens to LNs to induce tumor immunity.4 15–18 Thus, we employed a previously described culture system29 to produce large numbers of CD103+ cDC1s for use as a cancer vaccine. Our data indicate in vitro-generated CD103+ cDC1s produce T cell activating cytokines and chemokines, cross-present exogenous antigen to naïve CD8+ T cells to induce their proliferation, and migrate to TdLNs in vivo. Significantly, in vitro-generated CD103+ cDC1s control primary and metastatic melanoma and osteosarcoma tumors on vaccination. The CD103+ cDC1 vaccine also shows improved activity over a MoDC vaccine, and enhances the response of osteosarcoma to checkpoint blockade. These results support the idea that functional CD103+ cDC1s can be generated in abundance in cultures to provide an effective form of immunotherapy.
MoDC-based vaccines have been employed for years, as these cells can be expanded efficiently in culture. Furthermore, MoDC vaccines are relatively safe and can induce antitumor immunity; however, their clinical efficacy is limited.36 Hence, recent efforts have focused on evaluating distinct DC types in cancer immunotherapy.37 38 Vaccines based on pDC or cDC2 subsets have shown some promise.10 11 Moreover, purified murine lymphoid organ cDC1s demonstrated effective tumor control.20 The number of naturally occurring DCs that can be harvested for clinical use is limited, however, posing challenges for effective translation.39 40 In mice, this hurdle was overcome by generating abundant amounts of CD103+ cDC1s in culture29; BM cells from one mouse can generate sufficient quantities of CD103+ cDC1s for vaccination of 30–50 tumor-bearing animals. Importantly, murine and human DC populations share developmental and functional traits, including similar transcriptional networks, cytokine responses, and T cell-activating abilities.31 41 These data suggest culture systems that effectively expand human cDC1s may be feasible; for instance, optimizing use of Flt3L in cultures with blood-derived DC progenitors to generate human CD141+ DCs, and testing the ability of this population in tumor vaccine strategies.
The activity of current DC vaccines may be affected by DC survival rates, persistence in tumors, and migratory activity to TdLNs. We found CD103+ cDC1s remain longer in melanoma tumors, and accumulate in TdLNs more efficiently than MoDCs, as judged by their relative abundance 40 hours after vaccination. Moreover, CD103+ cDC1s showed prolonged survival following antigen stimulation, TLR agonist treatment, and exposure to the melanoma environment versus MoDCs. The delivery of tumor antigens to TdLNs is critical for effective induction of antitumor immunity by DCs.42 Taken together, these results suggest functional traits exhibited by CD103+ cDC1s in the tumor environment contribute to their superior efficacy as a tumor vaccine.
Vaccination with CD103+ cDC1s was also more efficient than MoDC vaccination in stimulating IFN-γ-producing CD8+ and CD4+ (Th1) T cell responses in vivo. While Tregs were increased on CD103+ cDC1 vaccination, Treg frequency within the total tumor-infiltrating CD4+ T cell population decreased, consistent with the vaccine efficacy. CD103+ cDC1 vaccination also preferentially elicited tumor antigen-specific CD8+ T cells, and was associated with increased amounts of tumor-infiltrating CD8+ T cells; vaccine-origin CD103+ cDC1s also preferentially stimulated OT-I CD8+ T cell proliferation in vitro. Moreover, IFN-γ-producing CD8+ T cells persisted in CD103+ cDC1 vaccinated mice until maximum tumor burden was reached in control groups. CD103+ cDC1s recruit T cells to tumors by production of T cell chemoattractants such as CXCL10, in addition to their roles in stimulating tumor antigen-specific T cell responses in TdLN.6 12 Collectively, these data suggest the CD103+ cDC1 vaccine stimulates an effective antitumor immune response that encompasses tumor antigen presentation and transportation to TdLN, activation of tumor antigen-specific CD8+ T cells, and increased CD8+ T cell recruitment to tumors.
The MoDC vaccine preferentially induced IL-4-producing CD4+ (Th2) T cells in TdLN, and MoDCs isolated from TdLNs were significantly more effective in stimulating OT-II CD4+ T cell proliferation in vitro versus CD103+ cDC1s. Moreover, MoDC vaccination was generally associated with increases in T cell amounts in TdLNs relative to CD103+ cDC1 vaccination. At first glance, the latter data seem inconsistent with the superior efficacy of the CD103+ cDC1 vaccine. Importantly, however, MoDC vaccination did not significantly expand tumor-infiltrating T cell populations, and MoDCs isolated from tumors were inferior to CD103+ cDC1s in eliciting CD8+ T cell proliferation in vitro. MoDC vaccination also failed to induce tumor antigen-specific T cell responses to an appreciable amount in vivo. Collectively, these results indicate the CD103+ cDC1 vaccine is significantly more potent at stimulating antitumor immune responses relative to MoDCs. Furthermore, as Th2 cells produce cytokines such as IL-10, IL-4, and IL-5, which limit cytotoxic T cell proliferation and drive macrophages to an M2 phenotype,43 our data suggest MoDCs have potential to promote an immune suppressive environment. Additional work to elucidate underlying immune mechanisms activated by each vaccine, such as determination of tumor macrophage phenotypes and cytokine profiles, is necessary to fully understand and improve DC-mediated vaccine responses.
Our work and studies by others suggest that multiple routes of cDC1 vaccine delivery are effective. A prior study used intradermal delivery of lymphoid organ CD8α+ cDC1s loaded with dead tumor cell-derived antigens,20 while we employed i.t. as well as i.v. delivery mechanisms. The i.t. delivery route was expected to enable CD103+ cDC1 migration to TdLNs and subsequent activation of antitumor immune responses; however, it was unclear a priori whether administration of CD103+ cDC1s i.v. would lead to effective antitumor immunity since cDC1s terminally differentiate in lymphoid organs or tissues.40 41 As delivery of poly I:C-stimulated and tumor antigen-loaded CD103+ cDC1s i.v. suppressed experimental lung metastases, the results suggest this route supports CD103+ cDC1 survival, antigen presentation, and elicitation of durable T cell responses. Thus, i.v. delivery mechanisms may be effective for DC vaccines that use tissue-resident cDC1s, therefore facilitating DC-based vaccine approaches for disseminated or metastatic tumors.
Although checkpoint blockade is a current treatment for many cancers including melanoma, osteosarcoma has been largely refractory to immunotherapy with the exception of mifamurtide.22 44 Mifamurtide is also effective in melanoma,45 suggesting melanoma and osteosarcoma share features of therapeutic responsiveness. We examined the effect of CD103+ cDC1 vaccination on primary and metastatic osteosarcoma tumor growth, as well as the efficacy of combination treatment with checkpoint blockade. Significantly, combination of CD103+ cDC1 vaccination and CTLA-4 blockade led to osteosarcoma tumor regression in all mice. Moreover, this combination treatment appeared to induce long-term immune memory, as animals rechallenged with osteosarcoma did not develop tumors. Combination treatment with CD103+ cDC1s and PD-1 blockade was less effective, by contrast. CTLA-4 and PD-1 have distinct roles in limiting T cell priming or effector function, respectively.46 47 Hence, we expect CD103+ cDC1 vaccination operates in part by priming naïve T cells, particularly CD8+ T cells, and this function may be enhanced by combination treatment with CTLA-4 blockade. Further studies are required to delineate the immunological mechanisms by which CD103+ cDC1 vaccination and CTLA-4 blockade elicit anti-tumor immunity; however, these results suggest the exciting potential for combination immunotherapy in the treatment of osteosarcoma.
Conclusions
Our findings reveal potent systemic and anti-metastatic efficacy of an in vitro-generated CD103+ cDC1 vaccine. CD103+ cDC1 vaccination elicits IFN-γ-producing CD8+ T cell and Th1 responses, as well as long-lasting tumor antigen-specific T cell activity. CD103+ cDC1 vaccination was superior to MoDC vaccination, and enhanced response to checkpoint blockade therapy in osteosarcoma, a tumor type that is refractory to the majority of current immunotherapies. Our data suggest the potential for novel cellular immunotherapies based on use of cDC1s alone or in combination with checkpoint blockade.
Acknowledgments
We thank Kathryn Newton and Dr. Yuanzheng Yang for technical assistance, Dr. Bhakti Patel for discussion and review of the manuscript, Drs. Gabriela Raso and Ximing Tang for evaluating osteosarcoma pulmonary metastasis, and the MD Anderson South Campus Flow Core for experimental advice and assistance.
References
Footnotes
Contributors YZ conceptualized the studies, designed and performed experiments, collected and analyzed data, and wrote the manuscript. NS and ESK conceptualized studies and designed experiments. NS, TTC, OK, RLB, YBM and HSL developed methodology, performed experiments and collected and analyzed data. SSW conceptualized the studies, designed experiments, wrote the manuscript, contributed to funding acquisition, administered the project, and supervised the study. All authors read, edited and approved the final manuscript.
Funding This work was supported by grants from the NIH NIAID (R01AI109294 and 3R01AI109294-04S1 to SSW), the MD Anderson Center for Inflammation and Cancer (to SSW and HSL), a Research Training Award from the Cancer Prevention and Research Institute of Texas (CPRIT RP170067 to TTC, OK and RLB), and the MD Anderson NIH NCI P30CA016672 grant (supporting the MD Anderson South Campus Flow Core).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplementary information. Additional information regarding data may be obtained from the authors on reasonable request.