Article Text

Abstract
Checkpoint blockade immunotherapy established a new paradigm in cancer treatment: for certain patients curative treatment requires immune reinvigoration. Despite this monumental advance, only 20%–30% of patients achieve an objective response to standard of care immunotherapy, necessitating the consideration of alternative targets. Optimal strategies will not only stimulate CD8+ T cells, but concomitantly modulate immunosuppressive cells in the tumor microenvironment (TME), most notably regulatory T cells (Treg cells). In this context, the immunoregulatory receptor Neuropilin-1 (NRP1) is garnering renewed attention as it reinforces intratumoral Treg cell function amidst inflammation in the TME. Loss of NRP1 on Treg cells in mouse models restores antitumor immunity without sacrificing peripheral tolerance. Enrichment of NRP1+ Treg cells is observed in patients across multiple malignancies with cancer, both intratumorally and in peripheral sites. Thus, targeting NRP1 may safely undermine intratumoral Treg cell fitness, permitting enhanced inflammatory responses with existing immunotherapies. Furthermore, NRP1 has been recently found to modulate tumor-specific CD8+ T cell responses. Emerging data suggest that NRP1 restricts CD8+ T cell reinvigoration in response to checkpoint inhibitors, and more importantly, acts as a barrier to the long-term durability of CD8+ T cell-mediated tumor immunosurveillance. These novel and distinct regulatory mechanisms present an exciting therapeutic opportunity. This review will discuss the growing literature on NRP1-mediated immune modulation which provides a strong rationale for categorizing NRP1 as both a key checkpoint in the TME as well as an immunotherapeutic target with promise either alone or in combination with current standard of care therapeutic regimens.
- immunotherapy
- tumor microenvironment
- immunomodulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immuno-oncology opportunities beyond PD1 and CTLA4
The unprecedented success of checkpoint blockade immunotherapy has brought immune reactivation to the forefront of next-generation cancer therapeutic strategies, demonstrating that reinvigoration and/or prevention from immune exhaustion of CD8+ T cells is essential for a clinical response. In particular, the first checkpoint blockade immunotherapies offered as standard of care for patients with cancer, targeting either cytotoxic T-lymphocyte associated protein 4 (CTLA4) via ipilimumab1–5 or the programmed cell death protein 1 (PD1) pathway via nivolumab, pembrolizumab or atezolizumab,6–10 achieved overall response rates of up to 40%.
Despite these successes, the majority of patients fail to achieve objective clinical response.9 10 Numerous explanations for immunotherapy resistance have been posited, including low inhibitory receptor (IR) ligand expression,11 low prevalence of immunogenic neoepitopes,12–14 high tumor burden to inflammatory response ratio,15 and immune exclusion.16 Consequently, significant efforts in the field have centered on evaluating combinatorial strategies. In fact, combination of ipilimumab and nivolumab in advanced melanoma has demonstrated modest clinical benefit over nivolumab alone, extending median overall survival beyond 3 years.17 18 Continued evaluation of new targets and therapeutic combinations with standard of care immunotherapies is required to maximize the proportion of patients who mount a durable response.
Beyond CLTA4 and PD1, clinical investigation of additional IRs holds promise for bridging the gap in response rates. Major emerging targets include lymphocyte activation gene 3 (LAG3),19 20 T cell immunoreceptor with Ig and ITIM domains (TIGIT),21 22 and T-cell immunoglobulin and mucin-domain containing 3 (TIM3),23 among others. Although these receptors regulate antitumor immunity through different signaling pathways, the outcome on CD8+ T cell function is comparable and primarily mediated during the effector response.24 Thus, immunomodulatory receptors that impact unique T cell functions, subtypes, or differentiation stages may pose significant combinatorial therapeutic advantages.
The broad mechanism of action for checkpoint blockade therapy is reversal of intrinsic T cell inhibition via IRs. Such agents interfere with ligand binding to IRs expressed on CD8+ T cells in the tumor microenvironment (TME).25 Under normal physiology, IRs tune the immune response to pathogenic challenge by directly attenuating the T cell receptor (TCR) signaling cascade. As a consequence, IR expression limits off-target activation, minimizes host tissue damage, facilitates immune resolution, and enhances T cell memory formation.25 However in cancer, chronic antigen exposure sustains prolonged expression of multiple IRs, leading to extreme dysfunction marked by reduced effector function, that is, cytokine production and T cell proliferation in response to stimulation.26 Thus, IR blockade in patients with cancer leads to improved antitumor immunity.
Furthermore, the cell intrinsic impact of IR blockade on the function of immunosuppressive cells in the TME, most notably Treg cells, warrants additional consideration.27 Whereas CTLA428, TIGIT,29 30 and TIM331 32 mark more suppressive Treg cells, the roles of PD1 and LAG3 are equivocal and may be context-specific.33–36 In gastric cancer, a recent paper demonstrated that anti-PD1 therapy reinvigorates PD1+ intratumoral Treg cells in patients who have hyperprogression under treatment.34 Therefore, it is critical for the field to consider the balance between promoting CD8+ T cell activation and enforcing Treg cell suppression as a consequence of checkpoint blockade administration. Careful selection of therapeutic targets with this perspective in mind may elucidate superior clinical strategies for individual malignancies and perhaps even individual patients based on the prevalence of intratumoral Treg cells compared with CD8+ T cells.
Recently Neuropilin-1 (NRP1) has begun to garner significant interest in the immune-oncology field as a novel target for its potent dual function: augmentation of Treg cell suppression and restriction of durable CD8+ T cell responses. Though initially considered a Treg cell marker,37 new research indicates that NRP1 is not only required for intrinsic Treg cell stability in the TME,38 39 but it also substantially inhibits CD8+ T cell antitumor function.40 It is also highly expressed on myeloid subpopulations, but its function in this context is less well characterized. These preclinical observations justify further investigation of NRP1 antagonism in combination with established immunotherapies. This review will highlight the initial findings for immunomodulatory function via NRP1 and discuss emerging findings that NRP1 functions as a key immune modulator in the TME with attractive therapeutic opportunity.
Basic NRP1 biology
Neuropilins (NRPs) are single-pass transmembrane, non-tyrosine kinase surface glycoproteins found in all vertebrates and are highly conserved across species. Two homologous NRP isoforms are known to exist, namely NRP1 and NRP2, encoded by distinct neuropilin genes (Nrp1 and Nrp2) which arose due to a gene duplication event.41 Both NRPs were originally discovered as neuronal adhesion molecules participating in Semaphorin-mediated axonal guidance. They were later found to be fundamentally involved in vascular biology, with NRP1 required for normal embryonic vascular development42 43 and NRP2 involved in the formation of small lymphatic vessels and capillaries.44 Studies over the past decade have revealed NRPs are multifunctional proteins participating in a variety of biological processes beyond the nervous and vascular development, with NRP1 having a major role in immunity and tumorigenesis.45 46
Protein structure and binding ligands
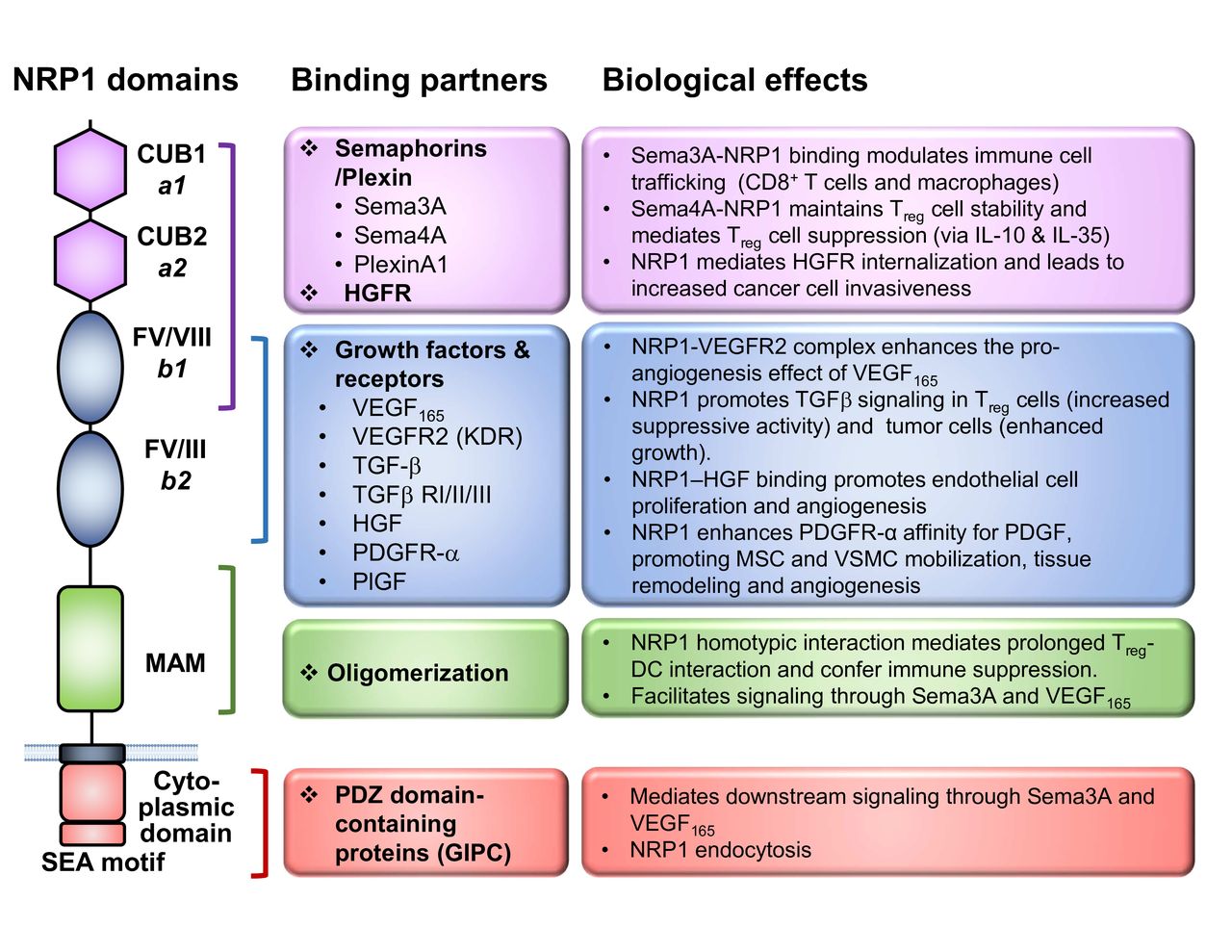
The NRP1 protein consists of a long N-terminal extracellular domain, followed by a transmembrane region and a very short cytosolic tail of 43–44 amino acids (figure 1). The large extracellular domain is comprised of three major segments, namely, two CUB (complement C1r/C1s, Uegf, bone morphogenetic protein 1) domains (denoted ‘a1/a2’) and two coagulation factor V/VIII domains (denoted FV/VIII or ‘b1/b2’), followed by a MAM (homologous to meprin protease, A5 antigen, receptor tyrosine phosphatase μ and К) domain (denoted ‘c’). The CUB domains are required for the binding of the Semaphorin group of ligands, while the FV/VIII domains are responsible for vascular endothelial growth factor (VEGF) binding as well as the docking site for different ligands (such as Semaphorins and VEGFs).47 48 The MAM domain is known to mediate homodimerization or heterodimerization of the receptors. Although it was initially thought that NRP1 was a non-signaling receptor due to its very short cytosolic tail, a conserved PDZ (PSD-95/Dlg/ZO-1 homology) domain-binding motif (SEA) at the c-terminus was later found to bind to the GAIP Interacting Protein C-terminus/synectin, which mediates intracellular signaling49 and receptor internalization.50
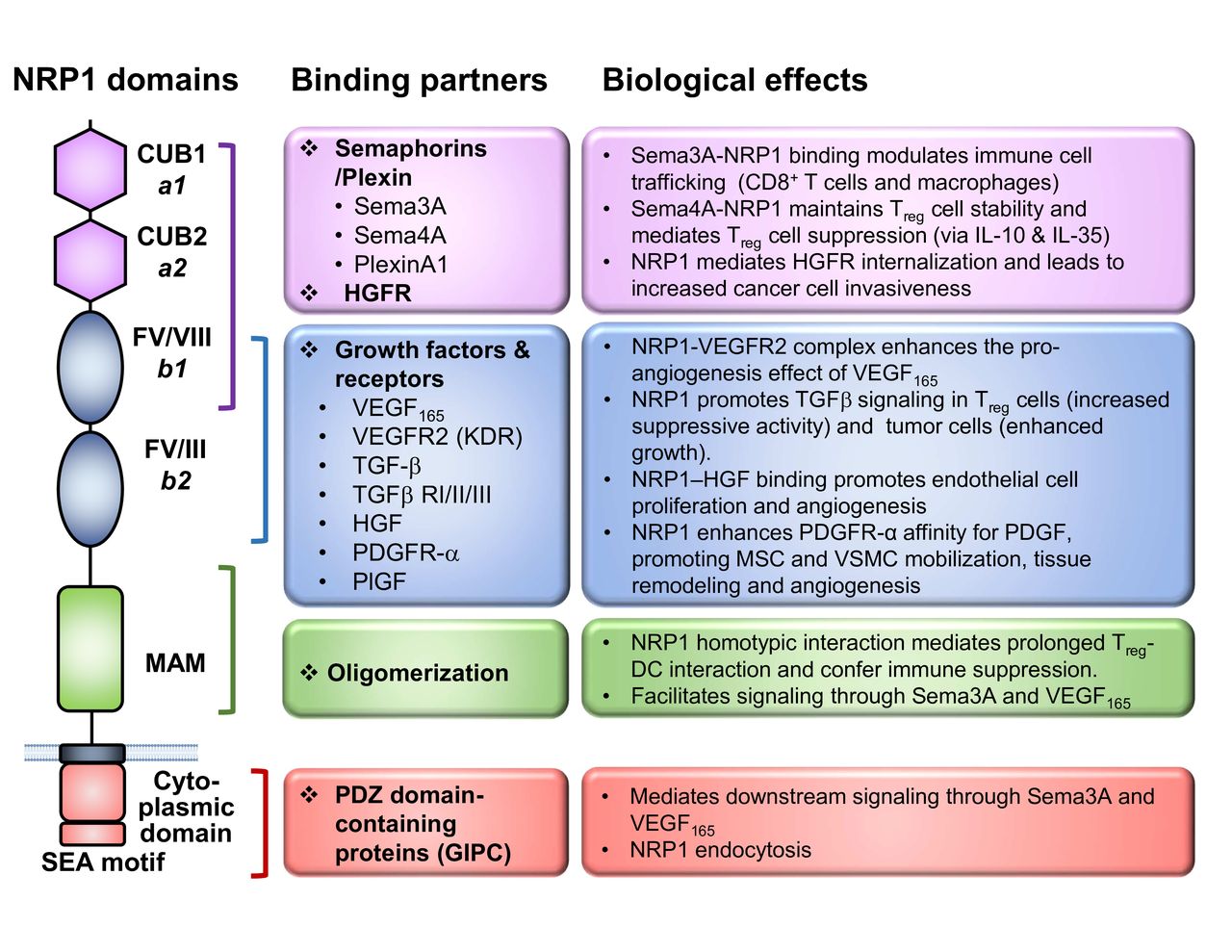
NRP1 structure and ligands. Graphical depiction of NRP1 structure to scale with key functional domains annotated. The associated ligands for each domain are listed and grouped into classes, whereas the A1 and A2 domains primarily facilitate Sema interactions, the B1 and B2 domains contribute to NRP1 binding a number of growth factors. The MAM domain uniquely functions to oligomerize NRP1 in trans. The cytoplasmic tail contains a C-terminal SEA motif which serves as a docking site for PDZ domain-containing proteins. Lastly, the key biological functions of the interactions referenced are listed, with specific emphasis on known immune-related and cancer cell biology functions. DC, dendritic cell; NRP-1, Neuropilin-1; Sema, Semaphorin; Sema-3A, Semaphorin-3A; TGFβ, transforming growth factor beta; VEGF, vascular endothelial growth factor; PDGFR, platelet-derived growth factor receptor; PlGF, placenta growth factor; VSMC, vascular smooth muscle cell; MSC, mesenchymal stem cell; HGF, hepatocyte growth factor; GIPC, GAIP Interacting Protein C-terminus.
NRP1 is capable of binding to a broad repertoire of ligands, accounting for its diverse biological functions. First identified as the coreceptor for the secreted class III Semaphorins, such as Semaphorin 3A (Sema3A),51 NRP1 was later shown to bind to a variety of growth factors, most notably VEGF165, as well as others including transforming growth factor beta,52 53 platelet-derived growth factors C and D,54 55 and c-Met.56 Interestingly, recent studies have revealed that NRP1 also binds to extracellular microRNA/AGO2 complexes and facilitates their internalization,57 adding another mechanism by which NRP1 modulates cellular functions.
Physiological consequence of genetic deletion
Nrp1-null mice are embryonic lethal due to severe developmental defects,43 58 mainly manifested in the neuronal and cardiovascular systems. While the signals conveyed by Semaphorin and VEGF through NRP1 are responsible for the neuronal and vascular defects, respectively, both signals are required for normal heart development.59 Studies utilizing advanced genetic tools including a Nrp1 variant knock-in mouse strain (Nrp1-sema) in which the Semaphorin binding was disrupted without affecting the VEGF binding, as well as an endothelial cell conditional Nrp1 knockout (Tie2 Cre Nrp1 L/L) strain,59 indicate that NRP1 acts as a receptor ‘hub’ for sorting signals from diverse ligands. This canonical mode of action is key to determining the context and cell-type specific function of NRP1.
NRP1 in the immune system
Myeloid cells
Whereas NRP1 is ubiquitously expressed across multiple tissue and cell types within the central nervous and vasculature systems in both human and mouse, NRP1 expression in the immune system is more restricted and regulated. NRP1 was identified as a marker for human dendritic cells (DCs), known as the blood DC antigen 4 (BDCA4, or CD304), which is expressed in all plasmacytoid DCs (pDCs).60 Known as the ‘professional’ type I interferon (IFN) producing cell type, human pDCs showed reduced IFN-α release on virus infection when treated with anti-NRP1 antibody, suggesting an immunoregulatory role of NRP1 in the context of antivirus immunity.61 Other antigen-presenting cells (APCs) that express NRP1 include monocytes and macrophages, in particular several types of tissue-resident macrophages, such as microglia and adipose tissue macrophages (ATMs).62–65 Monocyte/macrophage NRP1 expression is generally considered proangiogenic and anti-inflammatory, thereby contributing to tissue remodeling and wound healing.66 In a recent report, the NRP1+ ATMs were found to confer protection against obesity and metabolic syndrome by maintaining glucose homeostasis.63
T cells
Within the adaptive immune compartment, NRP1 has been known as a marker for murine thymically derived Treg (tTreg) cells, although this does not extend to human Treg cells (to be discussed later). Conversely, NRP1 is expressed at very low levels on resting CD4+ helper T cells and CD8+ T cells. In a mouse model resistant to the pathogenesis of experimental autoimmune encephalitis (EAE), NRP1 is upregulated in the EAE-tolerant CD4+ T cells (both the Foxp3+ and Foxp3– compartments) and functionally contributes to their suppressive phenotype.67 Other T cell subsets where NRP1 expression is reported include the T follicular helper cells68 and IL-17 expressing invariant natural killer T cells cells.69 Lastly, NRP1 is constitutively expressed on human thymic epithelial cells (TECs) and upregulated on immature thymocytes during TEC–thymocyte contact, which is subsequently blocked by Sema3A to direct thymocyte migration.70
Myeloid–T cell interactions
NRP1 is also expressed by conventional DCs isolated from human peripheral blood, where it may promote early T cell priming by mediating the formation of immunological synapse between DCs and T cells via homotypic interactions.71 As Treg cells preferentially express NRP1, Treg cells gain advantage over conventional CD4+ T cells by engaging DCs longer at immune synapses through NRP1–NRP1 interactions in the absence of inflammation or foreign antigen exposure, a mechanism that maintains immune tolerance at homeostatic state.72 Moreover, in the context of transplantation, intercellular membrane protein transfer from APCs to T cells, called trogocytosis, sensitizes CD4+ T cells to inhibitory signals through several known NRP1 ligands, such as Sema3A and VEGF.73 Furthermore, VEGF, known as an immunosuppressive cytokine, can inhibit lipopolysaccharide-induced DC maturation in a NRP1-dependent manner.74
To summarize, it is evident that NRP1 mediates important immunoregulatory functions including self-tolerance and immune homeostasis, resembling the activity of some known IRs. However, NRP1 is distinguished from classical IRs by exerting such impacts on a variety of cell types, in particular immunosuppressive cells. These features underscore why examining the function of NRP1 in the context of cancer may be a key next step for improving immunotherapy.
NRP1 expression and function in the mouse and human TME
The established association between NRP1 and cancer is supported by three key observations: (1) elevated NRP1 expression was reported in malignant cells of multiple human cancer types;45 (2) NRP1 is abundantly expressed within the TME, including both the stromal and immune compartments75 ; and (3) NRP1 expression is generally associated with poor clinical prognosis.76 From the perspective of tumor cells, NRP1 supports tumor cell growth via multiple axes, including cell survival, neoangiogenesis, and metastasis.45 76 77
At the tumor–immune interface, NRP1 promotes immune evasion by orchestrating multiple inhibitory processes within the immune compartments of the TME (figure 2). On the one hand, the physiological roles of NRP1 in DCs and macrophages, which contribute to an anti-inflammatory phenotype of these cells, are ‘hijacked’ by tumors to promote tumor angiogenesis and tumor-associated immunosuppression.46 On the other hand, NRP1 has a direct impact on adaptive antitumor immunity, which is achieved by impinging on both cell-extrinsic and cell-intrinsic inhibitory pathways regulating intratumoral Treg cells and CD8+ T cells. This review will focus on the latter topic, in particular observations substantiated by data derived from human patients and preclinical mouse models in order to better contextualize the importance of NRP1 on these two lymphocytic populations that are heavily regulated by immune-based therapies.
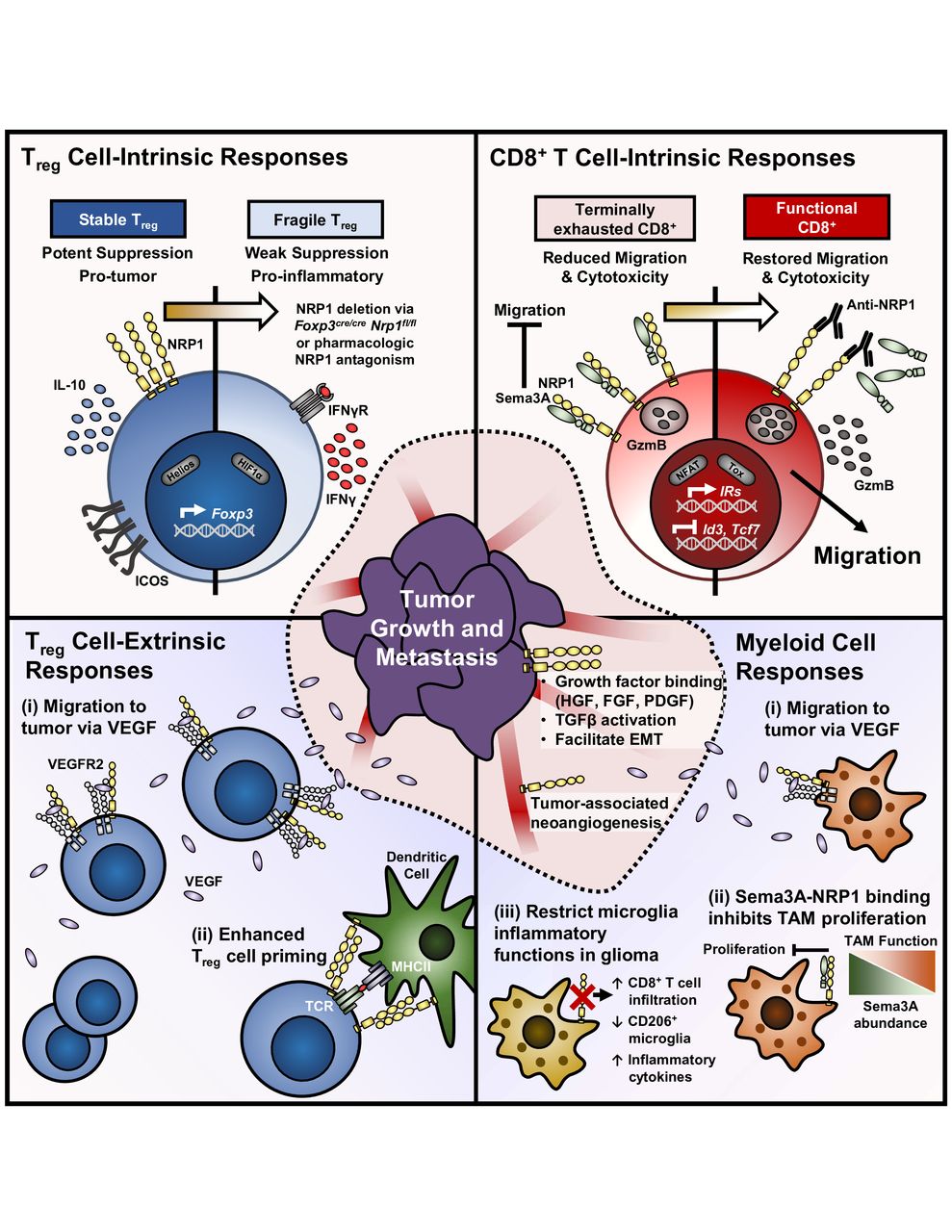
NRP1 contributes to central immune inhibitory mechanisms on T cells in the TME. (Top left) NRP1 marks stable intratumoral Treg cells. Genetic deletion of NRP1 in mouse Treg cells restore antitumor immunity by cell-intrinsically restraining Treg cell function. NRP1-deficient Treg cells are reprogrammed to a proinflammatory phonotype marked by interferon-γ (IFNγ) production. (Top right) Terminally exhausted CD8+ T cells express high levels of NRP1 with a module of additional inhibitory receptors. Sema3A binding to NRP1 on CD8+ T cells inhibits migration to tumors and cytotoxic function, both of which are restored with NRP1 blockade. (Bottom left) Beyond Treg cell fragility (i) NRP1 enhances Treg cell migration and retention in tumors secreting vascular endothelial growth factor (VEGF) by acting as a coreceptor with VEGFR2. (II) Furthermore, homotypic interactions between NRP1 on Treg cells and DCs facilitate enhanced Treg cell priming in response to low antigen availability and maintained peripheral tolerance. (Bottom right) NRP1 expression by tumor-associated monocytes, macrophages, and microglia (collectively TAM) tunes their inflammatory response. (i) NRP1 expression enhances chemotaxis to tumor-derived VEGF. (II) Sema3A complexes with NRP1 on TAM to inhibit their proliferation—even favoring antitumor M1 macrophage function. (III) NRP1 deletion on microglia restore antitumor immunity, marked by increased CD8+ T cell infiltration, decreased CD206 expression, and increased proinflammatory cytokine production. (Center) NRP1 has many known functions in promoting tumor growth and invasiveness, both when expressed on transformed cells or tumor-associated blood vessels. DCs, dendritic cells; NRP-1, Neuropilin-1; Sema-3A, Semaphorin-3A; TME, tumor microenvironment; Treg cells, regulatory T cells; VEGF, vascular endothelial growth factor.
NRP1 in Treg cells: the guardian of intratumoral Treg cell stability
NRP1 expression in human tissues and immune cell subsets mirrors that found in mouse models. The most notable exception is Treg cells. NRP1 was initially considered a marker for tTreg (or natural) cells in mice,37 in part due to its apparent coexpression with Helios.78–80 Indeed, the murine Nrp1 gene is a direct target of Foxp3-mediated transcriptional regulation, demonstrated by ectopic expression and chromatin immunoprecipitation experiments.81–83 However, subsequent investigation revealed that NRP1 is not expressed by human peripheral Treg cells in blood or lymph nodes.84 Instead, healthy donor Treg cells upregulate NRP1 on in vitro activation,84 indicating that immune processes may regulate NRP1 expression in vivo. Though NRP1 regulation may have species-specific determinants, results discussed below suggest that its impact on Treg cell phenotype and function remains conserved.
In the context of cancer, Treg cell expression of NRP1 potentiates immune suppression through at least two parallel pathways: Treg cell recruitment to the tumor by acting as a coreceptor for VEGF,85 and maintaining tumor-specific Treg cell stability via Semaphorin-4A (Sema4a) ligation.38 39 Initial analysis of the effects of T cell-restricted Nrp1 deletion in tumors utilized Cd4 Cre Nrp1 L/L mice. Though bulk Nrp1-deficient Treg cells retained equal in vitro suppressive function, the proportion and function of intratumoral CD8+ T cells was dramatically increased. This led to enhanced tumor-free survival and reduced tumor growth kinetics in both implantable and spontaneous tumor models. As NRP1 is known to act as a VEGFR2 coreceptor in the context of angiogenesis,86 87 it was demonstrated that NRP1+ Treg cells migrated along a VEGF gradient in vitro. Additionally, in vivo reduction in tumor growth in Cd4 Cre Nrp1 L/L mice could be recapitulated in wildtype mice by administering Vegf –/– fibrosarcoma tumors subcutaneously in contrast to VEGF replete tumors. These observations highlight the importance of Treg cell chemotaxis in response to VEGF as a critical component of Treg cell-established immune suppression in tumor models.
A complimentary finding from our group revealed that beyond cell trafficking, NRP1 was essential for maintaining intratumoral Treg cell function and phenotype.38 Disruption of the NRP1 pathway, either by antibody blockade or by Treg cell-specific genetic deletion (via Foxp3 Cre Nrp1 L/L), impeded intratumoral Treg cell suppressive function, thereby restoring antitumor immunity, without permitting off-target peripheral autoimmunity. Compared with their Nrp1 +/+ wild-type counterparts, Nrp1 –/– intratumoral Treg cells had decreased Bcl2 and increased active caspase-3, indicating dysregulation of survival pathways. Furthermore, Nrp1 –/– intratumoral Treg cells downregulated activation markers (including Helios, IL-10, ICOS, CD73) and adopted characteristic T helper lineage markers (such as Tbet, CXCR3, IRF-4, and RORγt). Our group showed that NRP1 localized to the immunologic synapse during T cell activation to restrain Akt (aka protein kinase B or PKB) activity through phosphatase and tensin homolog (PTEN), thereby relieving Akt-mediated antagonism on Foxo1/3 to stabilize Treg cell function. Follow-up work demonstrated that although intratumoral Nrp1-deficient Treg cells retain Foxp3 expression, their loss of suppressive function is potentiated by adoption of a pro-inflammatory phenotype, marked by increased production of IFNγ.39 Of further interest, IFNγ production by Nrp1 –/– Treg cells initiated dysfunction of neighboring NRP1+ Treg cells, in a process termed infectious fragility. The clinical significance of this finding was demonstrated by the requirement for Treg cell sensitivity to IFNγ in order to mediate tumor clearance on anti-PD1 immunotherapy in the MC38 tumor model. Indeed, mice harboring Treg cells that lack the IFNγ receptor were insensitive to anti-PD1. These findings provide substantial clinical rationale for further investigation of NRP1 antagonism as a therapeutic agent.88 89 In fact a recent report detailed how NRP1 therapeutic blockade mirrors the effects of Treg cell-restricted NRP1 genetic deletion in murine models, both in vitro and in vivo.90
Although Treg cell expression of NRP1 differs significantly between mice and humans, numerous studies have reported increased NRP1+ Treg cells in patients with cancer.39 90–95 This Treg cell phenotype was anecdotally reported in peripheral blood of pancreatic adenocarcinoma and liver metastases from colorectal cancer.91 In chronic lymphocytic leukemia, elevated NRP1 on B cells and Treg cells was observed in patient blood that is decreased following thalidomide treatment, linking antiangiogenic therapies to alleviation of antitumor suppression.92 It has also been reported that there is an enrichment of NRP1+ Treg cells in tumor-draining lymph nodes (TDLN) of patients with cervical cancer, particularly TDLN where tumor metastases are established.93 94 Furthermore, human NRP1+ Treg cells are more functionally suppressive and both FOXP3 and glucocorticoid-induced TNFR-related protein (GITR) expression are increased in NRP1+ Treg cells. This observation matches recent findings from our group. Interestingly, NRP1 antagonism at the VEGF-binding domain reduced human intratumoral Treg cell suppression by approximately 20% by inducing NRP1 protein internalization.90 For human Treg cells, NRP1 expression may also reflect exposure to ongoing inflammation as enrichment of NRP1+ Treg cells has also been reported in the synovial fluid of patients with rheumatoid arthritis.96 It is also worth noting that the frequency of NRP1+ Treg cells decreases following therapeutic intervention, both pharmacologic and surgical.91–93 Lastly, our group reported elevated NRP1+ Treg cells in the peripheral blood and tumors of treatment-naive melanoma and patients with head and neck squamous cell carcinoma.39 The prevalence of intratumoral NRP1+ Treg cells negatively correlated with disease-free survival in both cohorts. Together, these findings support the notion that NRP1+ Treg cells are functionally enhanced in cancer. Furthermore, increased NRP1+ Treg cells in peripheral blood was observed in patients with cancer.39 92 This finding is unique among IRs, which tend to have minimal expression in blood samples and may suggest that NRP1 expression on circulating Treg cells could serve as a pretreatment or on-treatment prognostic biomarker for clinical outcomes.97
NRP1 on CD8+ T cells: the link between T cell dysfunction and aberrant tumor-specific CD8+ T cell memory
In contrast to the constitutive expression of NRP1 on thymically derived murine Treg cells, its expression on naive CD8+ T cells is undetectable (both mouse and human) and is only induced on T cell activation. Transcriptional upregulation of NRP1 by CD8+ T cells was first documented in an early molecular and functional profiling of CD8+ T cell differentiation using LCMV-specific (P14) TCR transgenic T cells,98 where Nrp1 transcription peaked at the effector CD8+ T cells and the effector-to-memory transition phases. Nrp1 upregulation coincided with a group of genes encoding proteins involved in T cell migration and adhesion, such as CCR5, CD44, and p-selectin glycoprotein ligand 1 (PSGL-1). This raises the question of whether NRP1 also modulates CD8+ T cell migration, as it does in neuronal or endothelial cells. Consistent with this finding, our group observed upregulation of NRP1 expression (both gene transcription and protein level) on polyclonal intratumoral effector CD8+ T cells as well as activated tumor-antigen specific CD8+ T cells. Therefore, TCR engagement seems to be necessary to drive NRP1 expression in CD8+ T cells, a feature shared by most known T cell coreceptors. However, despite the observed upregulation, the functional role for NRP1 during the early priming of CD8+ T cells is unknown.
Some early observations have suggested NRP1 may be an IR-like molecule in CD8+ T cells. It was first found highly induced on a subset of immunosuppressive intestinal CD8+ T cells (the Foxp3+ CD8+ Treg cells), along with molecules known to be associated with CD4+ Treg cells such as PD1 and CD103. These CD8+ Treg cells may contribute to maintaining intestinal homeostasis in vivo by down-modulating effector functions of T cells.99 Consistently, in a later report using Gag-specific (TCRGag) CD8+ T cells to understand cell intrinsic mechanisms regulating CD8+ T cell tolerance versus immunity,100 it was determined that NRP1 was preferentially expressed on tolerant, self-reactive CD8+ T cells, mirroring PD1, LAG3 and CTLA4, although NRP1 was dispensable for tolerance. Additional evidence suggested that NRP1 may have a role in T cell dysfunction, a term used to describe T cells that are anergized or exhausted as a result of lacking costimulation or persistent antigen exposure. T cell dysfunction is phenotypically characterized by high IR coexpression and reduced effector marker expression,101 and it was found that NRP1 belongs to a core transcriptional signature of 174 genes shared by all aforementioned T cell dysfunctional states.102
Indeed, a recent report indicated that CD8+ T cell NRP1 expression in mice and humans is exclusive to a subset of intratumoral CD8+ T cells marked by high expression of PD1, whereas NRP1 is minimally detected on the PD1neg intratumoral CD8+ T cells.40 Compared with the NRP1–PD1– and NRP1–PD1+ counterparts, the NRP1+PD1hi cells exhibited higher expression of classical IRs (eg, LAG3, TIM3, TIGIT, 2B4), as well as markers related to cell proliferation (eg, Ki67) and cytotoxicity (eg, Granzyme B). They also express higher levels of exhaustion-associated transcription factors, such as NFATc1, TOX, Blimp1 and IRF4, but decreased levels of genes associated with cell survival (Bcl2) and memory/exhaustion precursor cells (TCF1). This is highly reminiscent of ‘terminally exhausted’ CD8+ T cells that have been defined in both chronic viral infection and tumor models.103 Importantly, NRP1 was functionally involved in the terminal exhaustion of intratumoral CD8+ T cells, rather than a mere consequence of this dysfunctional state. Specifically, the CD8+ T cells recovered from B16F10 tumors treated with a neutralizing anti-NRP1 antibody, expressed higher levels of Perforin and Granzyme B, the key molecules mediating the cytotoxic activity of CD8+ T cells, and exhibited enhanced cell killing towards autologous tumor cells ex vivo.40 In the same study, in vivo NRP1 blockade led to reduced tumor growth, which further synergized with PD1 blockade, although such synergy was not observed in terms of enhanced cytotoxic activity ex vivo. The differences between in vivo and ex vivo settings may be due to enhanced tumor recruitment of recently activated CD8+ T cells with anti-NRP1 treatment through blockade of Sema3A–NRP1 axis. Importantly, there might also be the contribution from blocking NRP1 on cell types (eg, Treg cells) other than CD8+ T cells under an in vivo anti-NRP1 regimen. Indeed, the latter seems to be supported by our study utilizing a CD8+ T cell-specific Nrp1-deficient mouse strain (E8I Cre Nrp1 L/L) in which primary tumor growth was similar to wild-type counterparts. Nevertheless, with the E8I Cre Nrp1 L/L mice we also observed clear synergy between genetic ablation of Nrp1 and PD1 blockade in the MC38 colon adenocarcinoma model, further supporting the notion that NRP1 contributes to, although indirectly, the defective CD8+ T cell-mediated primary tumor control.104
In fact, the most striking phenotype of the E8I Cre Nrp1 L/L mice is their improved protection against secondary tumors in a mouse model of postsurgical tumor immunity.104 This observation suggested a primary role for NRP1 in CD8+ T cells may be it contributes to the defective CD8+ T cell-mediated immunological memory against tumors. Of clinical significance, this model resembles control of disease relapse in patients with cancer. Further investigation revealed that Nrp1 –/– intratumoral CD8+ T cells were more capable of sustaining a stem cell-like, memory/exhaustion T cell progenitor phenotype, as opposed to the irreversible differentiation of a terminally exhausted T cell phenotype. Consequently, a larger pool of tumor-specific memory CD8+ T cells are formed from these Nrp1 –/– memory precursors, a scenario which is not reported when blocking any of the other IRs, either by genetic deficiency or by antibody blockade.
In contrast to effector CD8+ T cells, the reported expression of NRP1 on memory CD8+ T cells varies substantially between studies. During acute viral infection, Nrp1 gene transcription was downregulated (although higher than naive cells) on long-lived memory cells compared with T effectors.98 Such downregulation of NRP1 expression was even more striking on tumor-specific memory CD8+ T cells, wherein Nrp1 transcription decreased to baseline level in naïve cells (unpublished data). Contrary to these observations, one report described NRP1 as a surface marker for liver-primed memory CD8+ T cells generated through liver sinusoidal endothelial cells cross-presenting antigen under non-inflammatory conditions, while it was functionally dispensable for these memory CD8+ T cells.105 The discrepancies between these studies may be explained in part by different in vivo conditions (eg, inflammatory milieu and the type of antigen) under which functional memory is generated. Thus, the expression of NRP1 on memory CD8+ T cells (likely the same for other IRs) is dictated by the environmental milieu during memory formation.
In summary, the evolving biology of NRP1 in CD8+ T cells contributes to a hypothesis that independent sets of IRs, or immune ‘checkpoints’, seem to exist, which respectively control the effector versus memory functions of intratumoral CD8+ T cells. Further mechanistic elucidation of this hypothesis is critical, in the hope of improving the durability of T cell-targeted immunotherapeutics, including immune checkpoint blockade and adoptive T cell transfers.
NRP1-targeting in cancer therapy: a next generation checkpoint molecule
Clinical investigation of NRP1 has predominantly focused on its contributions to tumor angiogenesis by acting as a coreceptor with VEGFR2, rather than its immune regulatory function through binding semaphorins or other ligands. Initial pharmacokinetic studies of an anti-NRP1 monoclonal antibody (MNRP1685A) demonstrated potent inhibition of the human VEGF pathway with additive efficacy in combination with anti-VEGFA, bevacizumab.106 However, subsequent safety analysis in Phase I studies revealed high levels of adverse events, most notably grade 2 or 3 proteinuria (protein accumulation in the urine) in over 50% of subjects in one study, that ultimately terminated therapeutic investigation of this agent.107–109 A recent investigation of an anti-VEGFR2 agent, ramucirumab, found marked impacts on the Treg cell compartment, leaving room for speculation about whether NRP1 function and expression contributed to the observations.110
To date, only one anti-NRP1 monoclonal antibody is being clinically assessed for its inhibitory effect of Treg cell function. ASP1948 (human IgG4, Astellas Pharma Inc) in combination with Nivolumab is under Phase Ib evaluation (NCT03565445) for patients with advanced solid tumors. Results from this trial are expected in 2022 and will be critical to further assess the clinical significance and potential of targeting NRP1. Immune monitoring of Treg cell and CD8+ T cell alterations, even in the patient periphery, could be informative for therapeutic response and mechanism of action.
In addition to this, insight into the potential outcomes of NRP1-directed trials may be gleaned from examining how the semaphorin family, which is the primary group of NRP1 ligands implicated in its immune regulation, is being therapeutically targeted. To this end, although the primary ligand for NRP1 on T cells, Sema4A, has yet to be targeted in the clinic, preclinical evaluation of agents against Sema3A (a known NRP1 ligand) have yielded promising results. Sema3A is considered a vasculature normalizing factor through its interactions with NRPs complexed with plexins. While multiple studies have shown that Sema3A function inhibits tumor cell growth and migration,111 112 one recent study in glioblastoma suggests that neutralization of Sema3A impedes tumor growth in patient-derived xenograft (PDX) models.113 The discrepancies between these findings remains to be reconciled; however, it is possible that differences in tumor vasculature requirements and immune infiltration across tumor types could determine whether Sema3A has pro-tumor or antitumor function. In fact, Sema3A intrinsically regulates T cell activation114 115 and thus therapeutic blockade of this interaction may mediate enhanced antitumor immunity.
Whereas prior small molecule or peptide antagonists of NRP1 primarily targeted the b1 domain interaction with VEGF-A to reduce tumor cell migration and angiogenesis,77 116 117 novel candidates also appear to intrinsically regulate Treg cell function in vitro.118 This combinatorial action may permit lower dosing regimens to mitigate potential side effects; however, the therapeutic efficacy and safety profile of the lead candidate (EG01377) has yet to be evaluated with preclinical in vivo models. Although human Treg cells selectively express NRP1 in the context of cancer, making it an attractive tumor-specific immune target, NRP1 is constitutively expressed by subsets of both hematopoietic and non-hematopoietic cells including pDCs and endothelial cells. The impact of EG01377 on the function of these cell types in steady state and disease will further shape its clinical applicability.
Conclusions and future directions
A growing body of literature demonstrates that NRP1 is a unique immune modulator in cancer immunotherapy (figure 3). In the TME, NRP1 intrinsically regulates both Treg cell and CD8+ T cell function to collectively impede antitumor immunity and is expressed concurrently with multiple IRs. NRP1 benefits the function of intratumoral Treg cells by both facilitating their recruitment to the tumor bed and enforcing their functional stability amidst ongoing inflammation. Patients with cancer have a higher abundance of NRP1+ Treg cells across malignancies and therapeutic intervention is associated with decreased NRP1 expression in peripheral Treg cells. Though of relatively minor consequence to CD8+ T cell effector function, the impact of NRP1 on memory formation and durable response is distinct compared with the function of other IRs. These novel characteristics may translate into non-overlapping clinical efficacy with existing standard of care checkpoint inhibitors, thereby providing informed rationale for therapeutic combinations.

{kind=link}
{kind=link}
{kind=link}
NRP1 is a unique checkpoint target with diverse impacts on tumor immunity. Comparison between NRP1 and benchmark IRs, PD1 and CTLA4, as a checkpoint target in the setting of cancer immunotherapy. 1 In murine studies, NRP1 ligation was found to recruit PTEN to the immunological synapse to antagonize Akt activation. This has not yet been confirmed in CD8+ T cells. 2 Whereas mouse tTreg cells constitutively express NRP1, resting human Treg cells do not express NRP1, though it is upregulated with T cell stimulation. CTLA4, cytotoxic T-lymphocyte associated protein 4; IRs, inhibitory receptors; NRP-1, Neuropilin-1; PD1, programmed cell death protein 1; tTregcells, thymically-derived regulatory T cells.
As the opportunity rises to improve NRP1-targeted cancer therapy by focusing on its immunoregulatory roles, key questions remain regarding fundamental NRP1 biology and translational application.
Can we improve our understanding of the molecular basis underlying how NRP1 signals in subsets of T cells (ie, Treg cells, CD4+ and CD8+ T cells), particularly in humans? The delineation of these mechanisms is crucial for developing specific intervention strategies, facilitating optimal design for combinatorial immunotherapies, as well as informing ways to reduce the therapy-induced immune-related adverse events.
Among the numerous ligands for NRP1, which are the most relevant within tumors, particularly for intratumoral Treg cells and CD8+ T cells? The answer to this question will help inform blocking strategies that specifically target the relevant tumor-associated NRP1–ligand interactions while sparing physiological NRP1 functions.
Will NRP1-targeted approaches provide new strategies to improve the durability of adoptive T cell therapy, such as the chimeric antigen receptor (CAR) T cell therapy? NRP1 may be a viable genetic target in this setting to overcome adopted CD8+ T cell dysfunction and enhance in vivo duration of response.
Does NRP1 antagonism intrinsically impact the function of other cells in the TME including APCs and stromal cells? If so, is the effect to potentiate antitumor immunity?
Will combining NRP1-targeted therapy with standard of care checkpoint inhibitors, such as anti-PD1/PD-L1, achieve better clinical efficacy? What will be the optimal dosing regimen?
Can peripheral T cell NRP1 expression (either Treg cells or CD8+ T cells) be a diagnostic or prognostic biomarker for human patients with cancer? Can it be used to identify optimal candidates for immunotherapy or evaluate on-treatment response?
In conclusion, the translation of NRP1 biology into viable therapeutic interventions for patients with cancer holds substantial future promise, particularly in combination with immunotherapies that do not directly target Treg cell function or CD8+ T cell memory formation. However, a more holistic view of NRP1 in the complete TME, namely tumor, stroma and immune cells, is necessary to design the most optimal clinical strategies surrounding this promising next generation immune modulator.
Acknowledgments
The authors would like to thank Anthony Cillo, Hiroshi Yano and Lawrence Andrews for their discussion regarding the contents of this manuscript.
References
Footnotes
CAC and CL contributed equally.
Contributors CAC and CL performed the literature search and produced the initial drafts of the manuscript. TB and CJW revised the first draft. DAV, TB, and CJW revised subsequent drafts. All authors read and approved the final manuscript. CAC drafted the figures which CL revised. Additional figure edits were made by TB, CJW, and DAV.
Funding This work was supported by the National Institutes of Health (R01 CA203689 and P01 AI108545 to DAAV), and an NCI Predoctoral Fellowship Award (F31 CA243168 to CAC).
Competing interests DAV has submitted patents covering NRP1 that are licensed or pending and is entitled to a share in net income generated from licensing of these patent rights for commercial development.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.