Article Text

Abstract
Recent evidence indicates that ionizing radiation can enhance immune responses to tumors. Advances in radiation delivery techniques allow hypofractionated delivery of conformal radiotherapy. Hypofractionation or other modifications of standard fractionation may improve radiation’s ability to promote immune responses to tumors. Other novel delivery options may also affect immune responses, including T-cell activation and tumor-antigen presentation changes. However, there is limited understanding of the immunological impact of hypofractionated and unique multifractionated radiotherapy regimens, as these observations are relatively recent. Hence, these differences in radiotherapy fractionation result in distinct immune-modulatory effects. Radiation oncologists and immunologists convened a virtual consensus discussion to identify current deficiencies, challenges, pitfalls and critical gaps when combining radiotherapy with immunotherapy and making recommendations to the field and advise National Cancer Institute on new directions and initiatives that will help further development of these two fields.
This commentary aims to raise the awareness of this complexity so that the need to study radiation dose, fractionation, type and volume is understood and valued by the immuno-oncology research community. Divergence of approaches and findings between preclinical studies and clinical trials highlights the need for evaluating the design of future clinical studies with particular emphasis on radiation dose and fractionation, immune biomarkers and selecting appropriate end points for combination radiation/immune modulator trials, recognizing that direct effect on the tumor and potential abscopal effect may well be different. Similarly, preclinical studies should be designed as much as possible to model the intended clinical setting. This article describes a conceptual framework for testing different radiation therapy regimens as separate models of how radiation itself functions as an immunomodulatory ‘drug’ to provide alternatives to the widely adopted ‘one-size-fits-all’ strategy of frequently used 8 Gy×3 regimens immunomodulation.
- radiotherapy
- immunotherapy
- clinical trials as topic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Radiation therapy (RT) has significant major technological and biological advances in the last two decades, providing new opportunities in the era of ‘accurate, precision radiation medicine’.1 The ability to target and deliver radiation accurately in time and space, sparing organs at risk, may be particularly relevant to immuno-oncology given that: (a) the tumor microenvironment (TME) has a complex structure and cellular interactions, (b) the impact of radiation on the surrounding normal tissue including lymph nodes could alter the immune response, (c) a particular immunotherapy strategy might work very well with the proper priming and cytotoxic doses but not with an inappropriate cook-book schedule, (d) the tumor type and patient immune status will likely matter and (e) the biological adaptations by the patient’s immune system and tumor to radiation and other drugs will require adapting the immunotherapy in real-time to limit the risk of treatment resistance or relapse. The purpose of this commentary is to point out aspects of this complexity so that the need to study radiation dose, fractionation, type and volume is understood and valued.
While preclinical studies with combination immunotherapy and RT in murine transplantation tumor models have focused mainly on abscopal effects as surrogate end points of survival, the incidence of such abscopal effects in clinical experience has been relatively rare,2–4 thereby suggesting a need for re-evaluating the design of future clinical studies with particular emphasis on radiation dose and fractionation, immune biomarkers and selecting appropriate end points for combination RT plus immunotherapy. While early preclinical work suggested that a regimen of hypofractionated 8 Gy×3 is favored over a single fraction of 20 Gy3 in promoting abscopal effects of a combination of RT and anticytotoxic T-lymphocyte-associated protein 4 (anti-CTLA-4) immune checkpoint therapy, such studies compared a limited number of fractionation schemes. This article provides a framework for considering different RT regimens as distinct immunomodulatory ‘drugs’ to provide alternatives to a ‘one-size-fits-all’ strategy with the frequently used 8 Gy×3 regimens for immunomodulation. In the earliest years of immuno-oncology clinical trials, efforts were made to standardize the RT so that this fractionation was selected. However, with more immuno-radiotherapy and immunotherapy experience, this is the opportune time to examine a broader range of hypothesis-driven options.
‘Classical’ radiation tumor and cellular biology built on the four Rs of repopulation, repair, reoxygenation and redistribution (cell cycle) have had a variety of Rs added, including radioresistance and immune response, among others. These are not irrelevant in the same manner that ‘classical’ pharmacology is not irrelevant such that dose, timing, schedule, and concentration at the critical target can determine success or failure, even of a very effective drug. The development of new radiation biology focuses on tumor vasculature damage, cancer stem cell response, immunomodulation, metabolic changes, tissue plasticity and radiation-induced molecular adaptation and, indeed, leads to the paradigm of using ‘radiation as a drug’.5 6
RT is gaining importance in immunotherapy, including both the direct tumor effect and the sought after abscopal effect, so now is a critical juncture to delve deeper into the mechanistic and biological questions that need to be addressed so that the appropriate doses and schedules can be investigated in preclinical studies that will inform the clinical regimen. To miss this opportunity in immuno-oncology would be unfortunate. There is the intersection of great potential and enthusiasm, and a clear need for improvement for non-limited or limited responders to immunotherapies. RT can cause significant immunomodulation by increasing antigen presentation (including human leukocyte antigen), expression of CD80 together with increased DNA damage leading to the type I interferon (IFN-I) response, pro-inflammatory effects and T-cell-mediated immunogenic killing.7 In the decade preceding the Food and Drug Administration approval of the first immune checkpoint inhibitor, ipilimumab, for metastatic melanoma, RT was shown to generate an in situ tumor vaccine and enhance the effect of immunotherapy in preclinical studies.8 9 With the introduction of multiple immune checkpoint inhibitors as the standard of care for an increasing number of tumor types in the next decade,10 abscopal effects were reported in some patients who received radiotherapy during immunotherapy.11 12 Multiple clinical trials of RT with immunotherapy were started to determine the ability of RT to enhance responses to immunotherapy reproducibly. Such increased use of RT in immunotherapy trials, mainly to improve immune checkpoint blockade (ICB) therapy, led to three critical scientific challenges, namely determining the: (1) influence of radiation characteristics on immune modulation of the TME and tumor cells; (2) effective combinations of radiation and immunotherapy and (3) biomarkers of effective immunogenicity after radiation-immunotherapy combinations. Before discussing radiation dose and fractionation, we summarize these three questions for which in-depth analysis and research are needed.
Influence of radiation characteristics on immune modulation of the TME and tumor cells
RT is a double-edged sword that can augment immune activation and cause immune suppression. Hence, a shift in favor of the immune stimulatory versus the immunosuppressive effects of radiotherapy is critical to achieving the immunogenic modulation of TME.13 RT is emerging as a valuable partner of cancer immunotherapy. As this science of immunobiology of radiation is in development, many critical questions remain unanswered, including those in box 1.
Tumor microenvironment response to radiation: influence of radiation-induced immunomodulation in regulating response
Influence of radiation-induced immunomodulation on tumor microenvironment
Radiation: Does the quality of radiation (high vs low linear energy transfer), dose, size, fractionation (low-dose fractionation vs high-dose fractionation) and dose rate (high-dose rate vs low-dose rate) and schedule (hypofractionation vs multifractionation) influence effective modulation of tumor immune microenvironment?
Tumor cell susceptibility: Is the tumor cell susceptibility per se to immunotherapy impacted by the above radiation schedules?
Treatment volume: Is irradiation of the complete tumor volume necessary? Or is irradiation of partial volume adequate for effective modulation of tumor immune microenvironment and minimize normal tissue injury? Should one treat gross tumor volume alone or including involved lymph nodes for effective immune modulation?
Abscopal effect: Is distant effect (abscopal effect) a marker of effective modulation of tumor immune microenvironment?
Tumor cell immune characteristics: Does human leukocyte antigen class I loss or low T-cell repertoire diversity or checkpoint expression influence radiation effects of immunomodulation? Does tumor immunogenicity predict radiation-induced tumor immune modulation?
Cytokines: What dictates the balance between radio-induction of immunesuppressive cytokines and radio-induction of immune-activating cytokines?
Effective combinations of radiation and immunotherapy
Several components dictate the effectiveness of RT plus immunotherapy. These include tumor burden (tumor-induced immune suppression and low immunogenicity), immune tolerance (regulatory T-cells (Tregs), myeloid-derived suppressor cells) and modulatory effect of standard therapies that can inhibit/enhance tumor immunity due to dependence on drug, dose and schedule with RT.14 Preclinical models are essential to understanding the above components and inform clinical trial design, which can help accelerate clinical testing. Current reported literature raises several critical questions regarding combining radiation with immunotherapy (box 2).
Radiation therapy (RT) and immunotherapy combinations
Effective combinations of radiation and immunotherapy
RT plus immunotherapy: Can radiation-induced immunogenic death be augmented with checkpoint blockade/biological immunotherapy? What is the optimal sequence and radiation dose per fraction when combining with checkpoint blockade/biological therapy?
Tumor models: What mouse models are relevant to translate radio-immunotherapy into clinical trials and to understand the results of clinical trials (reverse translation)? Which animal models are best to test such checkpoint blockade/biological therapy with radiotherapy? Is orthotopic in syngeneic background better than genetically engineered mouse models or humanized patient-derived xenografts models?
Spontaneous tumors: Can spontaneous canine cancer models be used for generating preclinical data to support clinical concepts?
When are checkpoint blockades most effective: Does checkpoint blockade cure diseases rendered into complete response by standard-of-care RT, but destined to relapse, that is, antiprogrammed cell death protein 1 (anti-PD-1) as adjuvant therapy postcurative intent radiation? Does radiation render anti-PD-1 failures (not previously irradiated) responsive to anti-PD-1, that is, in patients progressing on anti-PD-1 will hypofractionated radiation to all or as much as possible known disease render disease responsive?
Normal tissues: How does the radiation effect on normal tissues impact the efficacy of radiation+checkpoint blockade therapy? Is there a risk of triggering autoimmunity in irradiated normal tissues receiving checkpoint blockade therapy?
Clinical trials: Are traditional end points of safety and efficacy used for drug trials suitable to study radiation immunomodulation? How are the different schedules tested and validated in vivo before large clinical trials?
Proof of principle: given the complexity of interaction, will there be one seminal trial combination that proves radiation as potent enhancer of immunotherapy, or will it require logical sequencing so that careful stepwise advances will be the way ahead.
Biomarkers of effective immunization after radiation-immunotherapy combinations
Comprehensive immune monitoring strategies are critical to understanding the underpinnings of how different RT fractionation approaches mediate other immune-modulatory effects. This understanding is necessary to guide and inform future RT and immunotherapy designs. Obtaining systemic and irradiated tumor samples preradiotherapy and postradiotherapy for this type of correlative analysis is required to improve our understanding. Multiplexing and sample-sparing techniques allow in-depth interrogation of such samples to address the complexity of RT-induced immune responses and suppression. Dose and delivery of RT, alone or in combination, affect the mechanisms of action. Novel technologies will allow for an era of biomarker discovery for companion diagnostics and patient preselection.15 Specific questions relevant to the above points are listed in box 3.
Radiation and immunotherapy biomarkers and biological processes
Biomarkers and biological processes after radiation-immunotherapy combinations
Cytokines: Are biomarkers of cytokines, chemokines and chemokine receptors relevant to radiation-induced immunogenic death? Does immunogenetics (immune polymorphism, genes behind human leukocyte antigen and cytokine network) play a role in radiation response?
Antigen presentation: Does major histocompatibility complex class I expression in tumor and metastatic lesions reflect abscopal response? Can T-cell repertoire diversity modeling predict radiation-enhanced antigen presentation?
Mechanisms of tumor cell death: Are there crosstalk biomarkers of immunogenic death with apoptosis, autophagy and necroptosis? Do radiation-induced oxidative stress biomarkers play a role in immunosensescence?
Immunity and radiation response: Are there pan-prognostic biomarkers of innate and adaptive immunity in radiation and combined modality settings? Can tumor-infiltrating lymphocytes topography form biomarkers to predict the immunogenic response to radiation?
Radiation dose and fractionation: considerations for imminent and future clinical trials
Based on robust supporting preclinical data in the immunobiology of 8 Gy×3 dose-fraction,9 the National Cancer Institute (NCI)’s Clinical Trials and Evaluation Program approved several clinical letters of intent involving combinations of checkpoint agents with 8 Gy×3 times radiation dose-fractions. Later, this dose-fractionation schedule was considered as the standard fractionation scheme for immunotherapy combinations (personal email communication from Jeffrey Buchsbaum, NCI on July 24, 2020).
The field has expanded substantially in complexity and clinical experience, so it is critical to explore how the dose, fractionation and schedule be selected as a significant critical decision requiring an in-depth understanding. Published literature indicates that hypofractionated RT is best suited for some forms of immunomodulation.16 However, controversies do exist in terms of using ablative or subablative dose and fractionation.17 18 It has been argued with robust preclinical data that subablative, immune-modulatory fractionation of 8 Gy×3 is better than a single ablative fraction of 20–30 Gy.9,19 However, a single fraction but not fractionated radiation was found equally effective with immunotherapy.20 In the clinical arena of stereotactic ablative radiotherapy (SABR)/stereotactic radiosurgery/stereotactic body radiation therapy (SBRT), there is the use of ablative and subablative terms for defining the local control (online supplemental table 1). The ablative dose can be defined as a one to five fraction(s) that can achieve a 90% or greater local control rate.21 22 Any dose to be delivered with twoto five fractions to achieve the same local control rate is considered a ablative total dose or each fraction dose is considered subablative.23 Both ablative and subablative dose-fractionations have been used in the clinic, often with 8 Gy×3 fractions (online supplemental table 2). However, the clinical efficacy of such doses and fractionation in several clinical trials has not been demonstrated.24–26 In particular, Theelan et al24 showed doubled responses and survival following RT and pembrolizumab treatment in patients with lung cancer, the prespecified end point (overall response rate) was not met. Hence, RT may have had a beneficial effect. Still, it was of lower magnitude than expected, and subgroup analysis suggests this effect may have been influenced by the degree of programmed death-ligand 1 (PD-L1) expression in the tumor. Although the reasons for the overall negative results in these three studies are unclear, it is possible that a different dose and fractionation of radiation would have been more effective. Thus, more preclinical research is necessary to identify the underlying biology of different RT doses and delivery schedules to determine if there is an optimal window of RT dose-fractionation (both ablative and subablative that is defined with dose-fractions in online supplemental table 2 that can be appropriate for combination with immunotherapies and formally tested in prospective clinical trials.
Supplemental material
The preclinical findings that hypofractionated RT (6 Gy×5 and 8 Gy×3) was superior to a single RT dose (20 Gy) in promoting antitumor immune responses were obtained in studies aimed at eliciting systemic antitumor responses (abscopal responses) in combination with CTLA-4 blockade.9 In experimental settings with two mouse tumors treated when they were well established (2 weeks postinoculation), the anti-CTLA-4 antibody had no effect. However, in the same setting, when anti-CTLA-4 was given with RT to one tumor, there was an improved response of the irradiated tumors and regression of non-irradiated synchronous ones. Of note, these effects have been reproduced in some case reports and clinical trials using similar RT dose and fractionation (9.5 Gy×3 and 6 Gy×5) combined with the antihuman CTLA-4 antibody ipilimumab.11 27 28 Thus, it is worth considering if the RT dose that optimally enhances responses may also vary depending on the type of immune checkpoint inhibitor (ie, anti-CTLA-4 vs antiprogrammed cell death protein 1 (anti-PD-1)), as discussed below.
Preclinical studies investigating the mechanisms of radiation-induced antitumor T-cell responses19 led to identification of IFN-I induction by RT as a critical factor.19 29–31 In many viral infections, IFN-I is induced by cytosolic DNA that stimulates cGAS to produce cGAMP leading to STING activation.32 IFN-I activates conventional dendritic cells type 1 (cDC1) that cross-present viral antigens to CD8 T-cells,33 required to eliminate the infected cells.
A similar mechanism was demonstrated in irradiated tumors. In multiple syngeneic murine models, recruitment of cDC1 was key to the synergy of RT and anti-CTLA-4 or anti-PD-1.19 These preclinical findings are consistent with the growing evidence that cDC1’s presence in tumors is required for the development of spontaneous T-cell responses to immunogenic cancers and response to ICB.34 35 The degree of cDC1 infiltration in human tumors has also been associated with patient survival.35 These data provide an example of the complexity of the interaction between the host immune system and the cancer cells: optimal dose and fractionation of immunogenic radiotherapy depend on each given tumor’s unique immune features and its TME.36
One important consideration when considering radiotherapy regimens is also the total treatment duration. Re-oxygenation, which occurs as a tumor responds to standard fractionated radiotherapy, increases radiosensitivity and response. It has been suggested that this effect is also essential when considering ablative or hypofractionated regimens.37 The importance of re-oxygenation may be even more pronounced when combining radiotherapy with immunotherapy as hypoxia compromises radiation sensitivity and can limit the anticancer immune response.
Another critical variable is the type of immunotherapy used in combination with RT. For instance, among ICBs, anti-CTLA-4 and PD-1/PD-L1 have distinct effects.38 Whereas both have been shown to have synergistic or additive effects with RT in some preclinical tumor models, the underlying mechanisms are likely to differ.
Finally, the issue of targeting multiple tumor sites may dictate the likelihood of successful immunization. Given the importance of tumor burden for the response to PD-1 blockade,39 the best synergy between RT and PD-1 may be seen with doses that more efficiently ‘debulk’ the tumor, at least in some cases. Besides, when RT is used to induce an in situ vaccine, sequencing may be critical. A recent study showed that PD-1 blockade before vaccination was detrimental, while it was beneficial when given after T-cell priming.40 Elegant studies in a mouse model of colorectal cancer have shown that PD-1 blockade concomitant with RT, given at 2 Gy×5, was more effective than a delayed administration of this ICB,41 and the work of Verma et al40 41 suggest that response may depend on the status of CD8+ T-cell priming at the time of initiation of anti-PD-1 therapy.
Overall, emerging data indicate that multiple factors influence the RT’s immunogenicity and that dose and fractionation may need to be tailored to the characteristics of the tumor and the immunotherapy used.
Other important questions that require attention are how RT is delivered and optimized to benefit from the immune modulation effect. These include lymphocyte sparing to avoid lymphopenia that can negate RT-induced pro-immunogenic effect42 43; identifying the right metastatic site for optimal immune system activation44; the impact of underdosing the tumor and partial tumor radiation on immunosuppression45 and finally the role of RT dose heterogeneity (such as spatial fractionation) in immune priming.46
Critical to the application of novel treatments in the clinic is comparison to the appropriate standard-of-care. Chemoradiation remains standard-of-care management for many solid tumors. Hence, optimization of systemic therapy with immunotherapy is necessary, whereby the sequencing and timing of immunotherapy and immunomodulation with chemoradiotherapy should be tested in preclinical models.
The above challenges provide directions to existing questions in need of further evaluation. The following are the key points that require urgent attention and are central points to boxes 1–3:
Timing of immunotherapies with RT may depend on the type of immunotherapy (eg, vaccines vs ICB). In-depth studies are warranted to determine the optimal RT regimen for combination approaches (ablative and subablative dose and fractionation). Although there may be a few candidates, there is a need to identify reliable biomarkers for response and mechanisms of action. Novel pathways can be targeted in combination with approaches to improve synergy between RT and immunotherapy (eg, autophagy,47 epigenetic, tyrosine kinase and DNA-repair).
Need for more focused collaboration between industry, academia and government on clinical studies. Need for shared access to data that has already been captured concerning RT and immunotherapy.48 Development of an immunotherapy registry to collect data on RT use during immunotherapy. Need for an efficient and novel clinical trial design that still ensures patient safety. Need for access to resources to help bank patient samples for future studies.
In addition to identifying patient populations, biomarker research can provide insight into the mechanisms of action for immunotherapy plus RT and identify new therapeutic targets. Genetic immune signatures or measures of immunogenicity can be used to individualize RT treatment regimens. There is a need for tissue banking to support exploratory studies to aid in biomarker research.

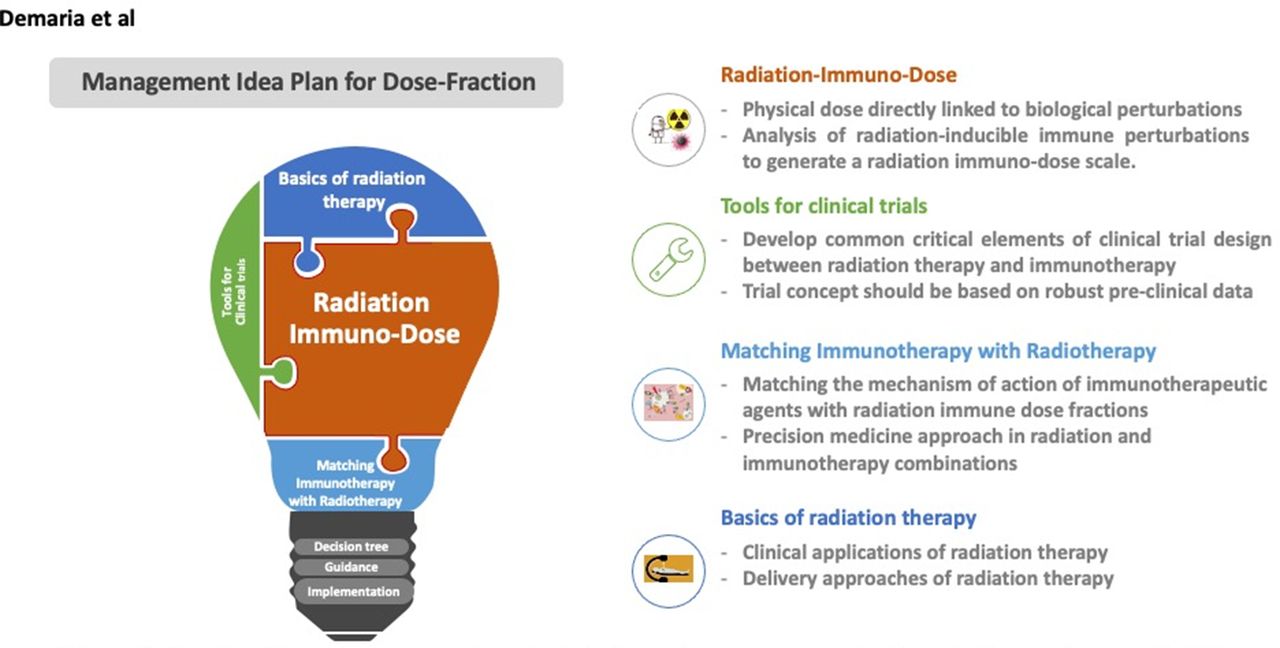
Four key intersecting categories of bridge ideas to manage dose-fractions in immunotherapy. Radiation immuno-dose is a major focus that can use clinical trails tools to match immunotherapy with radiation based on clinical delievery approaches and applications of radiotherapy, together will form optimal management for dose-fraction.
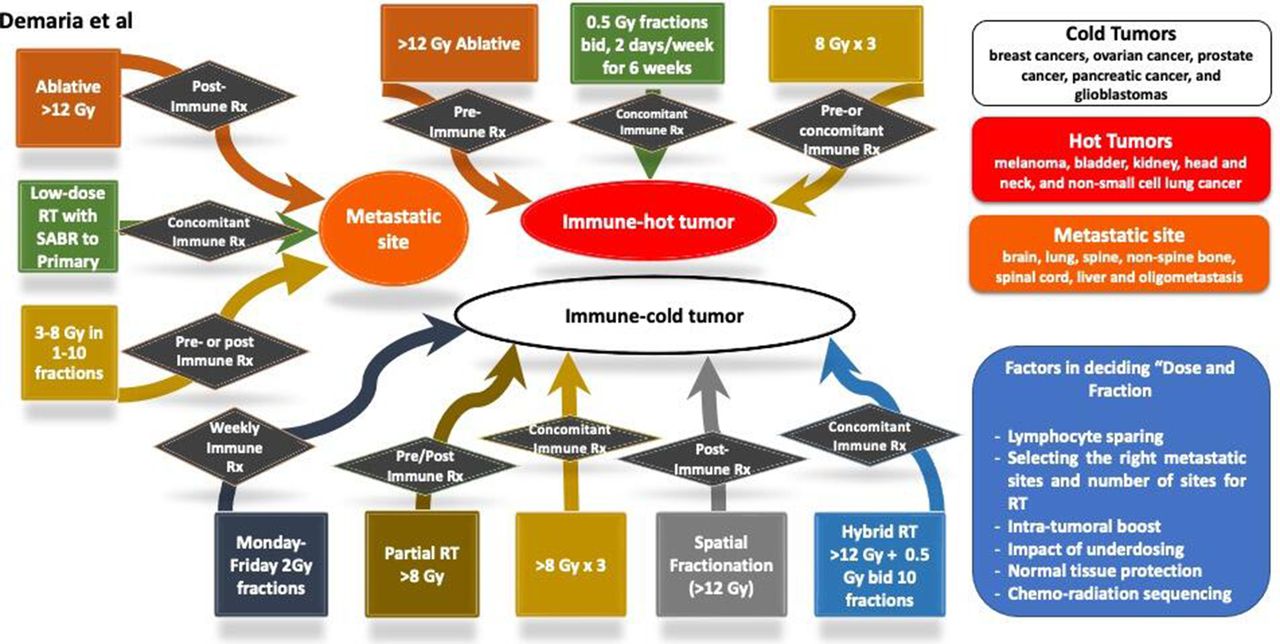
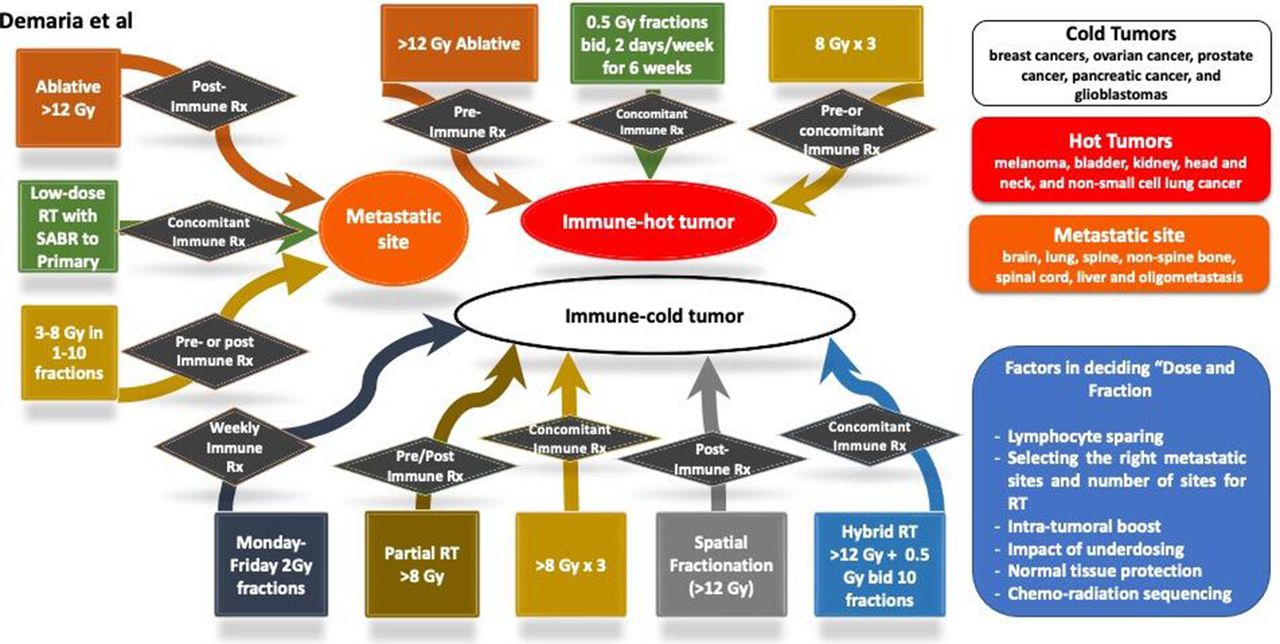
Decision tree chart aid for radiation oncologists in deciding ‘dose and fraction’ for combining with immunotherapy. There are three general scenarios, immune hot tumor, immune cold tumor and metastatic site. For each of these settings, a number of options are considered. Some of the dose-fraction suggestions are based on the published preclinical and clinical data. RT, radiation therapy; SABR, stereotactic ablative radiotherapy.

Decision tree combining radiation therapy (RT) with immunotherapy in metastastic disease. SABR, stereotactic ablative radiotherapy.

Examples of clinical trials (basket trial of radiation dose). PD-1, programmed cell death protein 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Radiation as an immunomodulatory drug. CTL, cytotoxic T lymphocyte; DC, dendritic cell; IART, immune ablative radiation therapy; ICD, immunogenic cell death; IM, immunomodulatory; ImRT, immunogenic radiation therapy; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; RT, radiation therapy; SABR, stereotactic ablative radiotherapy; SBRT, stereotactic body radiation therapy; SRS, stereotactic radiosurgery; TMEM, tumor microenvironment modulation.
In practical terms, these above key points can be addressed in four intersecting categories as summarized in table 1 and illustrated in figure 1. Applying these categories in real-time can help generate a decision tree that will help guide and construct new clinical trial concepts when combining radiation with immunotherapy (figure 2) discussed below in four sections.
Key guiding factors to decide dose-fraction with immunotherapy
Basics of radiation therapy
Designing combination trials of RT and immunotherapy requires careful considerations of radiation dose and fractionation. If conventional fractionation of 1.8–2.0 Gy fractions is delivered daily for 6–8 weeks, immunotherapy can be administered 3–6 weeks after the completion of RT, as was offered in the PACIFIC trial.49 50 The release of tumor antigens after a conventional RT course induces the activation of tumor-specific T-cells that can be further amplified by ICB. Concurrent administration of immunotherapy with conventional fractionation may not be desirable because it was shown that conventional fractionation could reduce tumor-infiltrating CD8+ cytotoxic T lymphocytes (CTLs) during treatment, possibly because of radiation-induced cell death of tumor-infiltrating, immune-effector cells.51 In patients with metastatic disease, ICB has been combined with hypofractionated RT, often delivered in one to five fraction sizes of SABR or SBRT. Although the dose-fractionation of SBRT is usually guided by the normal tissue tolerance of RT, recent studies have proposed ‘hybrid’ regimens designed for immunomodulation.52 We have grouped SBRT regimens into three categories based on their postulated dominant effects on the crosstalk between the tumor and the immune system: (a) immunogenic ablative, (b) immunomodulatory subablative and (c) TME modulatory (TMEM) low-dose fractions. Immunogenic ablative SABR regimens deliver a single fraction of 25–34 Gy or three fractions of 18–20 Gy, or five fractions of 10 Gy for the treatment of pulmonary metastases. At times, because of consideration of normal tissue tolerance, large ablative fractionation is not considered. Instead, hypofractionated SBRT regimens of 5–8 Gy×5 or 8 Gy×3 are delivered to accommodate radiation tolerance of organs-at-risk. At times, such a potentially subablative regimen is deliberately prescribed for immunomodulation because it is immunomodulatory and induced abscopal effects when combined with ICB therapy in preclinical studies.9 19 Clinical trials have been designed to irradiate one index lesion to achieve abscopal regression in unirradiated tumors. On the other hand, ‘multisite’ irradiation is designed to achieve total metastatic ablation of tumors and cause tumor debulking and achieve the efficacy of immunotherapy in improving the overall survival of metastatic patients.3 53 Recent preclinical studies have shown that tumor-resident T cells can be relatively radioresistant and can be amplified to control irradiated tumors.54 Further studies have shown that low-dose ‘scatter’ irradiation to a non-index lesion from high-dose irradiation (such as SBRT) to an index lesion improved the systemic response rates for combination therapy.55 A ‘hybrid’ regimen was recently investigated preclinically, where postablative modulation with low-dose TMEM regimen of 0.5 Gy×4 fractions improved the local tumor control rates of single high-dose ablative RT.52 Furthermore, Savage et al52 demonstrated that a low-dose TMEM radiation regimen could be delivered to the whole organ that is at risk for metastatic spread of tumors, such as, lungs to increase the survival after immunogenic ablative fractionation of the primary tumor by reducing the infiltration of Tregs, while increasing the infiltration of CD8+ CTLs in micrometastatic lesions. Figures 2–5 show potential applications of RT dose-fractionation regimens in combination trials of immunotherapy and RT.
Immune mechanisms
Radiation-inducible adaptations are dependent on the dose and fractionation that encompass both non-immune mechanisms such as DNA repair response (which might be immunomodulatory), signal transduction pathways (eg, prosurvival) and metabolic reprogramming, and immune mechanisms (such as changing of cell surface proteins and receptors). For example, it has been shown that although both single (SD) and multiple fractions (MF) of radiation-induced changes in gene expression and microRNA profiles, a higher number of alterations occurred following MF of radiation compared with SD.56 Importantly, these alterations induced by radiation change dynamically and depend on the type of fractionation. While MF induced more early changes in integrin expression, SD induced delayed alterations in cells that survived irradiation,56 57 some of which persisted for many months (and possibly longer, to be studied). An important aspect of understanding these dynamic inducible cellular adaptations is that these changes can be targeted at the right time to improve the therapeutic outcomes. An in vitro study in DU145 cells showed upregulation of phospho-AKT and mammalian target of rapamycin at 2 hours after MF RT (3×2 Gy), but not after an SD of 6 Gy.56 Accordingly, the AKT inhibitor’s efficacy was significantly increased when given 2 hours after MF RT but not when given before or during the MF RT. Similar inducible metabolic changes have been observed following MF and SD RT in several cancer cell lines, especially in fatty acid metabolism.56
In addition to the inducible non-immune mechanisms, the immune-modulating potential of RT has been recognized. The soluble and cellular mediators modulate the antitumor immune response, involving response from the tumor, antigen-presenting (APC) and T cells. These responses are shown to induce both local and systemic abscopal effects.58–60 Several mechanisms, including the release of tumor antigens/neoantigens or damage-associated molecular patterns (DAMPs) like calreticulin, high mobility group box protein 1, ATP and enhancement of APCs function, have been associated with radiation-induced effects on the immune system.61–63 Furthermore, activation of the innate and adaptive immune system, improved infiltration of the immune-competent cells into the tumor, enhancement of T-cell effector activity and so on are responsible for RT-mediated modulation of antitumor immune responses.7 58 59 64 65 Focal irradiation of the tumor has also been shown to reduce the immunosuppressive potential of Tregs while increasing effector cytokines.66 Interestingly, irradiation of human macrophages reprogrammed them towards a pro-inflammatory phenotype, although the irradiated macrophages enhanced cancer cell invasion and cancer cell-induced angiogenesis.67 Furthermore, it has been suggested that radiation enhances tumor immunogenicity and act in synergism with immunotherapeutics when tumor cells have an existing immune responsive phenotype.58 Together, these studies raise several questions such as whether different doses and dose-fractionation of radiation may induce an effective immune response independent of the underlying immune responsive phenotype of the tumor cells or avoid the detrimental immune effects of radiation such as irradiated macrophages promoting cancer cell invasion. Essentially, can we select a radiation dose or fractionation that can convert an immunosuppressed tumor to a highly immunogenic tumor? This would then act synergistically with immunotherapeutics when used in the right schedule, improving the therapeutic outcomes. However (and not surprising given the range of potential mechanisms related to dose and timing of radiation and different target cells), a consensus optimal radiation dose or fractionation to stimulate the immune system has not been achieved. A single schema may not be possible, although several preclinical studies have been conducted68 to inform future preclinical and clinical research. These studies have shown that conventional 2 Gy dose fractions, a single fraction of a large dose or high-dose hypofractionated radiation all are effective in tumor control (reviewed in the study by Carvalho and Villar68). Therefore, systematic preclinical and clinical studies are needed to measure the inducible immune events by varying radiation dose and fractionation regimen dynamically.
Irrespective of the lack of a consensus on an optimal radiation dose and fractionation, RT has been combined with several immune checkpoint inhibitors, CAR-T cells and other immunotherapeutics in different schedules (reviewed in the study by Buchwald et al69). In general, based on these studies, it has been suggested that for high-dose RT (single or hypofractionated), immunotherapy should begin with RT to use the release of neoantigens at that time; however, for conventional radiation schedule, the timing of immunotherapy is not very crucial due to continuous release of tumor antigens.69 Of note, several other factors need to be considered to identify an optimum schedule for a combination of RT with immunotherapy, such as the tumor subtype, the immune responsive status of the tumors and patients’ underlying immune status, to name a few.
Immune modulators
Standard conventional doses (1–3 Gy fractions) increase vascular access and upregulate local production of cytokines and tumor cell surface expression of stress markers.70 Hypofractionated moderate doses (eg, 8 Gy×3) elicit many of the effects of standard fractionation while producing a greater IFN-I response.19 29 Finally, ablative doses (eg, 20 Gy/fx) lead to profound cell death while depleting radioresistant suppressive immune cells in the TME, but may also induce increased levels of fibrosis and chronic inflammatory/immune suppressive pathways (figure 5).
One challenge to identifying an ‘optimal’ schedule for immuno-radiotherapy is the difficulty of resolving seemingly discordant findings that sometimes emerge from preclinical studies using different tumor models and treatment regimens. For example, prior studies have demonstrated strong induction of antitumor T-cell response with radiation alone when using a single ablative 20–30 Gy dose and a cooperative therapeutic interaction between this dose and the anti-CTLA-4 checkpoint inhibitor resulting in abscopal tumor responses in a B16 melanomal model.29 51 71 In contrast, preclinical studies of radiation and the anti-CTLA-4 immune checkpoint inhibitor examining dose-fractionation in syngeneic murine breast and colon cancer models showed that multiple moderate-dose fractions (eg, 8 Gy×3) were superior to a single 20 Gy dose, with the latter failing to generate an abscopal response.9 While these observations may reflect unknown biological differences between these tumor models, such preclinical studies of fractionation are also confounded by covariant effects of fractionation on the timing of radiotherapy delivery relative to immunotherapy. Such effects may be directly impacted by differences in the rates of tumor growth and response for a given model.
On the other hand, seemingly discordant observations related to the ‘optimal’ radiation dose and fractionation may also reflect the unique sensitivity of tumors and/or immunotherapies to the activation of distinct dose-dependent immune mechanism. Low-dose radiation at 2 Gy stimulates nitric oxide synthase by tumor-associated macrophages and creates an immunogenic environment.72 Such low-dose radiotherapy may also trigger transient local depletion of lymphocyte lineages in the TME73–76 as well as local release of inflammatory cytokines77 78 that results in repopulation of radiated tumors by a distinct milieu of tumor infiltrating immune cells. Dose escalation and/or fractionation may alter the magnitude, quality or dynamics of these effects and this could bear critical implications for the optimal timing of immunotherapy delivery. High doses of radiation may activate tumor-promoting macrophages79 and cause severe vascular damage, decreasing recruitment of immune cells to the tumor.80 Effects of radiation on vasculature include activation of endothelial cells that recruits circulating cells and infiltration of immune-inflammatory cells.81 In particular, low-dose irradiation remodels the vasculature, reprograms the macrophages and increases lymphocytes penetration.72 On the other spectrum, hypofractionated radiation causes vascular remodeling, affected diffusion and influences hypoxia.82
Dose escalation of radiotherapy may also optimize the induction of immunogenic tumor cell death83 84 and increase expression of pro-immune markers such as major histocompatibility complex class I.62 85 Recent mechanistic studies also shed light on negative feedback pathways that may antagonize the immunogenic effects of radiation at high doses (20 Gy) but not at moderate fractional doses (8–12 Gy).19 29 Moderate radiation dose induces DNA damage more effectively than lower doses, leading to greater formation of micronuclei and cytoplasmic leakage of DNA.19 29 This is detected by cGAS/STING and activates primordial viral response pathways leading to production of IFN-I in the radiated tumor cell. At higher doses, however, further stimulation of DNA damage may lead to negative feedback expression of 3-prime repair endonuclease 1 (TREX1), which digests cytosolic DNA and reduces radiation-induced activation of a IFN-I response.19 Radiation doses above 12–18 Gy strongly induce TREX1, as determined by the size of the SD rather than total dose. Therefore, fractionation and dosage can alter the immune response to radiotherapy but by being able to control the radiation delivery, enhance immunotherapy to address the specific tumor immune-phenotype.
Because the immunological effects of radiotherapy are not unidimensional and because these diverse effects exhibit distinct dose-response profiles, one dose-fraction scheme for radiation cannot provide optimum immunomodulation in all situations. When designing combination strategies of immunotherapy and radiotherapy, extensive consideration must be given to the immunotherapy agent(s) employed and the mechanism of action in the context of data from prior clinical trials and preclinical studies. The delivery of radiotherapy should be specifically tailored to the desired outcome. For example, where T-cell priming is the objective, the in situ vaccine effect of radiation may be optimized by moderate-dose hypofractionation (eg, 8 Gy×3) that activates a IFN-I response and upregulates expression of MHC class I.62 85 In situations where an in situ vaccine effect of radiation is inadequate or where hypofractionated moderate-dose radiation is not feasible to deliver, combination of moderate-dose radiation with toll-like receptor (TLR) agonists,86 tumor-specific antibodies or supportive cytokines such as granulocyte-macrophage colony-stimulating factor, interleukin (IL)-2, IL-12, or IL-15 could also be considered.87 In tumors deficient in IFN response or lacking MHC class I, it is possible that dose escalation could be advantageous for in situ vaccination by stimulating greater immunogenic cell death, although additional immunotherapies such CD73 inhibitors may be critical to overcome detrimental effects of high-dose radiation on the TME at these doses.36
In contrast, where natural killer cell activation is desired, the dose-dependent increase in expression of MHC class I on tumor cells could be counterproductive. In this setting, single fraction moderate or even low-dose radiation may be optimal to activate a IFN-I response without marked increase in MHC class I expression. Such lower dose regimens do not optimally activate IFN-I but may augment the susceptibility of TMEs to the propagation of an existing adaptive or innate antitumor immune response or to exogenous cell therapies. Where this is the goal of radiotherapy, in situ vaccine-activating doses may not be needed or practical. In contrast to in situ vaccine approaches in which targeting a single tumor site may be adequate for priming a systemic antitumor immune response, approaches that aim to augment tumor cell or TME susceptibility to existing or exogenous antitumor immunity may require delivery of immune-modulatory radiation to as many tumor sites/cells as possible while avoiding systemic lympho-depletion. This may necessitate advanced radiation targeting approaches or targeted radionuclide therapies and may also still benefit from combination with an in situ vaccine approach targeting a single or few tumor sites with higher dose radiation. Ultimately, it may be that no single immunological mechanism or radiation dose is optimal for the activation of a robust and diversified antitumor immune response. Rather, dose heterogeneity within single or multiple tumors may be preferable. Advanced techniques including brachytherapy and heterogeneous dosing of conformal external beam radiotherapy may be capable of optimally engaging multiple distinct dose-sensitive mechanisms within a single tumor site.88 Further preclinical and clinical investigations of these approaches is warranted in the context of tumor immunomodulation.
While the immunotherapeutic approach will help dictate the optimal radiotherapy approach chosen for a combined modality treatment regimen, it is also important to consider that the effects of a given dose/fractionation of radiotherapy might differ across tumor types and between patients. This is also true for all cancer therapies that aim to be part of immunotherapy. Additionally, while preclinical models are helpful for identifying complementary mechanisms of action, and developing first-pass combination approaches, caution must be taken when extrapolating dose/fractionation schemas from murine models since the inherent radio-sensitivity and the immune system of mice are different than humans. It is clear that additional data, particularly clinical data, examining the immune-modulatory effects of different radiotherapy dose/fractionation schemas across various tumor types is needed to help inform clinical trial design. The importance of biomarkers is apparent.
A persistent challenge to the clinical translation of studies evaluating dose-dependent effects of radiotherapy on antitumor immunity is the limited funding available to support clinical and preclinical testing of questions about radiation dose and delivery. Exploring questions of radiotherapy dose, fractionation, volume, heterogeneity and several lesions to target can be problematic in clinical trials since insurance companies might deem that the radiotherapy approach is experimental, requiring the study to bear the cost of the radiotherapy. Standard-of-care is evolving from conventionally fractionated radiation 1.8 Gy–2.2 Gy to a more hypofractionated radiation as seen in SBRT/SABR. However, in clinical practice, there is tremendous variability in the dose, fractionation, volume, heterogeneity and number of lesions targeted in the standard-of-care palliative radiotherapy and thus means should be sought to compare these clinically relevant standard approaches in combination with immunotherapy. At the same time, insurers, funding agencies and regulatory reviewers should encourage such approaches as a cost-effective means to advancing clinical practice within a well-defined hypothesis testing trial. An additional barrier to maximizing the knowledge gained from clinical trials is focusing exclusively on determining clinical activity without including the detailed biospecimen banking and correlative studies necessary to understand the immune effects of different combinations, and identify signatures of response and mechanisms of cancer immune escape.
Tools for clinical trials
Hypothesis-driven, thoughtfully designed clinical trials are needed to optimize combinations of radiation with immunotherapy. These require input and collaboration between basic and translational scientists, clinical practitioners including both medical and radiation oncologists and patient groups. Consideration of radiation treatment parameters such as dose and fractionation are critical to the success of these trials. Therefore, the scientific rationale, appropriate clinical setting, study end points and study design must be considered, at least within the hypothesis of a study, in conjunction with the radiation parameters most appropriate for the scientific question being explored.
The scientific rationale supporting radiation immuno-oncology trials originates from many sources mentioned in more detail above, including in vitro studies that demonstrate immunogenic cell death following radiation83; in vivo studies that demonstrate enhanced local and systemic immune-mediated tumor regression induced by radiation and immunotherapy combinations41 71 89–91; case reports, retrospective studies and early prospective trials.11 12 27 28 Whereas murine models provide unique tools to address mechanisms of action, they do not recapitulate the complexity and heterogeneity of human tumors. This gap can be bridged, at least in part, by performing studies in large animals. For example, canine trials are being performed to test some immunotherapy agents,92 and radiotherapy could be easily incorporated.
Scientific rationale can help guide the clinical trial setting. To maximize patient beneficence, and minimize patient risk, novel immunotherapy radiation combinations are tested in patients with more advanced disease that have failed standard-of-care treatments. The prior lines of therapy received by these patients and a more extensive disease burden could impair antitumor immunity and impact radiation dosing,39 93 94 but is not typically accounted for in preclinical models. In addition, the bar is very high in advanced stages since the measure of success often used is systemic tumor control after irradiation of one metastasis, in face of substantial tumor heterogeneity and immunosuppression. Radiotherapy employed with the goal of stimulating antigen cross-presentation to enhance systemic antitumor immunity may also be less effective in a tumor type with a low mutational burden and fewer potential tumor antigens. A radiation immunotherapy combination shown to enhance local effects in preclinical models would be more effectively tested in settings where local control with radiation is often suboptimal, for example, locally recurrent head and neck cancer and unresectable sarcoma.
When the risk of added toxicity is not a significant concern, it is reasonable to test new radiation and immunotherapy combinations as part of neoadjuvant or window of opportunity trials. The advantage of this setting is the availability of tissue obtained at surgery to interrogate the effects of radiation and explore candidate biomarkers of response. However, the potential for curative standard treatments in this group of patients makes it more challenging to administer radiation dosing that is not standard along with immunotherapies that are unproven.
Finally, in the definitive and adjuvant settings conventionally fractionated radiation delivered over multiple weeks is most standard, but is likely to be relatively more immunesuppressive compared with shorter regimens,42 and thus it is not the optimal choice to combine with immunotherapy. Trials conducted in the definitive and adjuvant settings generally require sufficient patients treated to achieve adequate power, and are also more expensive; therefore, these trials are often led by industry and/or cooperative trial groups who may be more at adverse risk and less inclined to evaluate novel dose/fractionation regimens. But, there is risk in using a suboptimal regimen if others could be tested and potentially improve the trial outcome.
New end points have been developed to encompass the varied patterns of response to immunotherapy that are associated with clinical benefit, such as immune response criteria, immune Response Evaluation Criteria in Solid Tumours and immune response assessment in neuro-oncology.95–97 When radiotherapy is added to immunotherapy, end points designed for systemic and immune agents remain appropriate, but the irradiated lesion(s) should be measured separately to determine local response and tease out systemic and abscopal effects. Toxicity end points are also important and include both immune-related toxicity and monitoring for enhanced local toxicities such as radionecrosis and stricture that have been observed in previous studies.98 99 Importantly, toxicities following both immunotherapy and radiation can be delayed over months to years.
For correlative end points, in addition to interrogating samples of tumor, normal tissue and blood for treatment effects and for evidence of antitumor immune responses, collecting stool and/or saliva to investigate the microbiome, and extracting data from radiological images will provide comprehensive information to help identify mechanisms of response and resistance. Obtained data can also be hypothesis-generating and lead to reverse translation from the bedside to the bench, an important path to further our understanding of the interactions between radiation and antitumor immunity in patients. The optimal design and interpretation of these complex data in the context of clinical response and other patient characteristics requires a multidisciplinary team approach, including mathematicians who can model this complexity to identify patterns and key factors influencing outcome.
Well-designed studies that include thoughtful correlative measures do not need to be positive to be informative, a negative or null result is equally informative. As with other agents given in combination with ICB, it is challenging to identify additive or synergistic benefit with single-arm radiation-immunotherapy studies given immune checkpoint inhibitors have variable monotherapy response rates that depend on the tumor type, patient characteristics as well as increasingly appreciated germline polymorphisms. Randomized phase II or ‘pick the winner’100 studies can help address these limitations and are feasible when asking radiation dose/fractionation questions, but these types of trials require larger number of patients. Additionally, interpretation of randomized or multiarm trials can remain challenging given significant heterogeneity in tumor burden at baseline, the specific sites irradiated and other patient and treatment factors. As the number of immunotherapy combination trials continues to expand, there is increasing urgency to maximize the amount learned from each study. Hence, repository of data from such combination trials will be useful.
Decision tree for potential rationale approach in deciding RT dose and fractionation with immunotherapy
Taking into consideration of the above four categories (as succinctly summarized in table 1), radiation immuno-oncologists should consider the following aspects while deciding the dose per fraction, the number of fractions and total dose when combing with immunotherapy: (a) the entire time duration of RT (such as Monday to Friday or 6 weeks in synchrony with immunotherapy schedule); (b) tumor characteristics (such as IFN resistance, ‘hot and cold’ tumors and how that is defined); (c) type of immunotherapy can dictate synergy with specific radiation characteristics; (d) intrinsic tumor resistance; (e) oligometastases dose (and is it based on number of metastases); (f) type of radiation; (g) location of metastases (bone/liver/lung/brain) for radiation and (h) relative biological effectiveness for photon, charged particles and dose per fractionation and total dose.
The above aspects are captured in figure 2, which provides a chart highlighting a number of considerations about the underlying rationale for selecting appropriate dose-fraction for trials and management of solid tumors with radiation and immunotherapy. Immunologically, tumors are classified as hot and cold tumors,101 and this is the first factor to consider.
Characteristics of hot immune tumors include increased infiltration of T-cells and CTL (high immunoscore) with checkpoint activation proteins. On the contrary, cold tumor characteristics include the absence of T-cells and CTLs within tumor/tumor edges (low immunoscore, based on the percentage of immune tumor infiltrates) with the low mutational burden and poor antigen presentation. Other factors that can interfere with immunoscore include oncogene activated pathways, epigenetic regulation, reprogramming of the TME (including mutational burden) with aberrant tumor vasculature and/or stroma and hypoxia,101 and key regulators of macrophages reprogramming.102
Next part of the decision tree is the ‘radiation dose’ that can be divided into three dose ranges: low-dose RT (0.1–1 Gy), high-dose RT including ablative and subablative (8 Gy and above) and the more standard clinical dose (1.8–2.2 Gy) to define immune modulation effects and how these effects can be exploited with cancer immunotherapy.103 At low doses of radiation from 0.1 to 1 Gy, immune activation is achieved by an increased T-helper 1 response that attracts naïve T-cells and promotes its differentiation and activation.104 Since the D10 (radiation dose required to reduce clonogenic survival by 90%) for CD4+/CD8+ lymphocytes range from 3.32 to 3.84 Gy,105 low-dose chronic and acute irradiation stimulates enhanced immune function leading to in situ vaccination, homing of activated T-cells, trafficking, infiltration and tumor cell-killing. Low-dose radiation at 0.5 Gy is associated with the highest number of infiltrating T-cells with a decline at >1 Gy, and this was accompanied by redirecting macrophage differentiation from a ‘tumor-promoting/immunosuppressive state’ to one that enables cytotoxic T-lymphocytes to infiltrate tumors and kill cancer cells. All these immune modulation conditions can be harnessed with drugs that augment DC maturation, such as TLR or CD40 agonist or IFN-β, to intensify antitumor immunity effects further.
In addition, both subablative doses (5–10 Gy) and ablative doses (>12 Gy) (online supplemental table 1) with fractionations can play important role in immune modulation events. With radiation doses of 8 Gy and above in SD or MF, cancer cells undergo an immunogenic cell death associated with release of DAMPs, including cytosolic DNA that induces to release IFNs to help DC mature and promote T-cell activation. The resultant antitumor immune response has the potential to act distally to the irradiated tumor and significantly increase the incidence of distal (so-called abscopal) effects. These events lead to T-cell priming, trafficking, infiltration, in situ vaccination and immunogenic killing. The immune modulation events by high-dose RT can be exploited to enhance immunotherapeutic efficacy by activating T-cells using antibodies targeted against co-inhibitory T-cell receptors/ligands such as PD-1/PD-L1 and T-cell immunoglobulin and mucin-domain containing-3 and transforming growth factor-β and lymphocyte activation gene 3 protein blockers.
Doses at 1.8–2 Gy in fractionated settings are standard-of-care for several solid tumors. Such fractionation extends several weeks to minimize toxicity to normal tissue. At the same time, lymphocytes are rapidly cleared from the irradiated field, diminishing tumor antigen-specific T-cell populations through persistent site-specific cytotoxicity. Such tolerogenic immunosuppressive events of radiation can be exploited by repletion of T-cells in a T-cell-deficient environment that can lead to proliferative expansion of T-cells with activated phenotype and thus increase cytolytic activity to self and tumor antigens. Other immunotherapy combinations with 2 Gy fractions that can potentially partner for synergy include TLR and CD40 agonist, IFN-β and cancer vaccines.103
The final part of the decision tree is to decide the dose-fraction and type of immunotherapy that will fit for the hot or cold or metastatic site, as shown in figure 2. Certain limitations such as lymphocyte sparing, sequencing of chemoradiation, the impact of underdosing and normal tissue damage can affect decisions on the selection of dose-fraction scheme with immunotherapy. Such clinical experimentations can be adapted for metastatic cancers (where the immunobiology is different) as there is far more latitude for testing (as highlighted in figures 3–5), but that has not been exploited.
In summary, careful design of combination trials of RT and immunotherapy should consider alternative dose-fractionation schemes testing immunomodulation and immunogenic ablation as RT as drugs with appropriate immunological end points as a surrogate for overall survival as well as clinical end points. Along with dose-fractionation for SBRT, randomized phase II trials should also consider the number and sites of metastases, and avoidance or delaying elective nodal irradiation for effective immunomodulation (figure 3). Such approaches will lead to effective partnership, whereby RT’s direct action on reducing tumor burden including enhanced antigen presentation (figure 5) synergizes with immunotherapy’s sustained immune activation to achieve both systemic and local control.
Acknowledgments
The authors would like to thank Drs Jeffrey Buchsbaum and Charles Kunos at NCI for highlighting the use of 8 Gy×3 as standard dose-fraction in NCI-CTEP clinical trials letters of intent. The authors would like to thank Pataje Prasanna at NCI for the critical reading of the manuscript. The co-organizers and writing committee gratefully acknowledge the timely input of the many participants. The ideas, discussion and preparation of this document would not have been possible without the aggregate expertise.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MA, NC and BV initially conceived the idea of addressing these important questions. Rest of the coauthors are the writing team from the participants at an NCI that organized the “Radiation Dose and Fraction in Immunotherapy: A Consensus Discussion Meeting” April 28, 2020 to discuss the issue of radiation dose and fractionation being used in immunotherapy clinical trials. Appendix A lists the participants in the discussion including participants at the NCI who contributed to the organization of the meeting and the publication.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.