Article Text

Abstract
Resistance to immune checkpoint inhibitors (ICI) and other anticancer therapies is often associated with the accumulation of myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) in the tumor microenvironment (TME). Therefore, targeting MDSC recruitment or function is of significant interest as a strategy to treat patients with ICI-resistant cancer. The migration and recruitment of MDSCs to the TME is mediated in part by the CD11b/CD18 integrin heterodimer (Mac-1; αMβ2), expressed on both MDSCs and TAMs. However, inhibition or blockade of CD11b/CD18 has had limited success in clinical trials to date, likely since saturation of CD11b requires doses that are not clinically tolerable with the agents tested so far. Interestingly, activation of CD11b with leukadherin-1 was found to reduce macrophage and neutrophil migration in animal models of inflammatory conditions. Preclinical studies with GB1275, a salt form of leukadherin-1, demonstrated that activation of CD11b improves the antitumor immune response and enhances the response to immunotherapy in mouse models of pancreatic adenocarcinoma, breast cancer and lung cancer. Based on the promising results from preclinical studies, a phase 1/2 clinical study (NCT04060342) of GB1275 in patients with advanced solid tumor types known to be resistant or less likely responsive to immuno-oncology therapies, including pancreatic, breast, prostate, and microsatellite-stable colorectal cancer, is ongoing. In this review, we examine targeting MDSCs as a therapeutic approach in cancer therapy, with a special focus on GB1275 preclinical studies laying the rationale for the phase 1/2 clinical study.
- myeloid-derived suppressor cells
- translational medical research
- tumor microenvironment
- drug evaluation
- preclinical
- therapies
- investigational
Data availability statement
Data are available upon request
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- myeloid-derived suppressor cells
- translational medical research
- tumor microenvironment
- drug evaluation
- preclinical
- therapies
- investigational
Background
Immune checkpoint inhibitors (ICIs) such as programmed cell death-1 (PD-1)-blocking antibodies have revolutionized cancer therapy; however, not all tumors are sensitive to such treatments, with many patients refractory or developing resistance during treatment.1 2 One of the factors contributing to resistance to ICIs and other anticancer therapies is the presence of immunosuppressive myeloid cells in the tumor microenvironment (TME).3 4 Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of immature myeloid cells, consisting of myeloid progenitors, monocytes, immature macrophages, and granulocytes that are primarily distinguished by their immunosuppressive functions.5 6 The two most common subsets of MDSCs are granulocytic MDSCs, also known as polymorphonuclear (PMN)-MDSCs, and monocytic MDSCs (mMDSCs).5 7
MDSCs accumulate in tumor and lymphoid tissues of patients with cancer, and can differentiate into tumor-associated macrophages (TAMs), which exhibit a wide spectrum of both tumor-inhibiting and tumor-promoting activities depending on the stimuli present.7 8 For simplicity, these phenotypes are often referred to as M1 (classically activated) or M2 (alternatively activated), with the majority of TAMs displaying an M2-like profile.9 Notably, diverse phenotypes across the polarization spectrum can be present within the same TME.9–11 TAM polarization is mediated by cytokines, with tumor-inhibiting (M1) phenotype driven by the Th1-derived cytokines interferon gamma (IFNγ), colony-stimulating factor 2, or toll-like receptor signaling. In contrast, polarization to the tumor-promoting (M2) phenotypes can be driven by the Th2-derived cytokines interleukin (IL)4, IL10, IL13, transforming growth factor-β (TGFβ), prostaglandin E2, colony-stimulating factor 1, or hypoxia.10 11 The tumor-inhibiting phenotypes are associated with an increased expression of MHC-II, CD80, and CD86, which promote T-cell priming, produce T-cell recruiting chemokine ligands CXCL9 and CXCL10, and secrete proinflammatory cytokines, such as tumor necrosis factor-α, IL1β, IL6, IL12, and IL23.11 In contrast, the tumor-promoting phenotypes often lose their antigen-presenting capabilities, express programmed death-ligand 1 (PD-L1), and secrete immunosuppressive factors such as IL10, TGFβ, and angiogenesis-inducing factors such as vascular epithelial growth factor and adrenomedullin.11
Although generally myeloid cells are clustered in niches within the tumor, in some rare cases of breast cancer, for example, myeloid cells can be the most abundant leukocyte population.8 12 MDSCs are potent suppressors of T-cell function and adaptive immune response and in accordance with this immunosuppressive effect, the presence of MDSCs and immunosuppressive TAMs in the TME is associated with poor prognosis and correlates with elevated neutrophil to lymphocyte ratio.5 7 13–16 Moreover, MDSCs are associated with elevated expression of several immunosuppressive mediators including PD-L1,17 inducible nitric oxide synthase,18 arginase-1,18 and reactive oxygen species.19 These mediators inhibit the activity of tumor-infiltrating lymphocytes and may also induce T-cell apoptosis.5
TAMs and MDSCs have been implicated both experimentally and in correlative clinical studies with resistance to both standard of care chemotherapies and immunotherapies. Elevated baseline levels of circulating MDSCs20–23 and tumor resident TAMs24 25 associate with advanced clinical stage, metastatic status, and worse treatment outcomes across various indications. Furthermore, treatment-induced modulations in baseline MDSC levels have been associated with response. For example, nivolumab treatment decreased circulating MDSCs in NSCLC responders and patients with stable disease, while values remained constant in the non-responder subset, suggesting MDSCs may be contributing to PD-1 blockade resistance.26 The precise mechanisms by which myeloid cells regulate resistance or susceptibility to treatment vary greatly by cancer type and microenvironment context and are reviewed in detail elsewhere.27–29
In this review, we examine the therapeutic approach of preventing the accumulation and immunosuppressive activities of MDSCs in the TME, with a special focus on GB1275, an allosteric modulator of CD11b, currently being developed for patients with solid tumors that are relapsed or refractory to standard of care (SOC) and ICI treatment.
Targeting MDSCs and TAMs
Considering the role of MDSCs in conferring resistance to ICIs and other anticancer therapies, reduction or prevention of the migration of MDSCs into the TME and/or their differentiation into TAMs is of significant interest as a therapeutic strategy in patients with cancer; however, results of studies investigating this approach have been mixed to date. For example, chemotaxis inhibition, via blockade of the chemokine receptors type 1 (CXCR1), type 2 (CXCR2), or C-C chemokine receptor type 2 (CCR2), was shown to reduce the migration of MDSCs into the TME, and thereby enhance the efficacy of PD-1 targeting therapy in animal models.30–32 However, in patients with pancreatic ductal adenocarcinoma (PDAC), inhibition of CCR2 with small molecule antagonists had limited clinical efficacy to date.33 Preclinical models suggested that resistance may be mediated by influx or expansion of alternate CCR2-negative MDSC subsets, compensating for the loss of the targeted myeloid population.31 32 To counteract these compensatory mechanisms, CCR2/CCR5 dual inhibitors that simultaneously target multiple chemotaxis pathways on different myeloid subsets are currently being evaluated in the clinic.34–36 Nonetheless, the potential for compensatory action by an untargeted chemokine axis or myeloid subset may limit this approach.31 32
Colony-stimulating factor 1 receptor (CSF-1R),37 phosphoinositide 3-kinase-gamma (PI3Kγ),38 and integrin39 signaling pathways also play an important role in MDSC migration and/or TAM polarization in the TME. Inhibition of CSF-1R reduced TAM infiltration and survival in the TME in preclinical and clinical studies.40–43 Although signs of efficacy have been seen with inhibition of CSF-1R in phase 1 (NCT01346358, NCT02265536),40 44 45 phase 1b/2 (NCT02713529),46 phase 2 (NCT01349036, NCT01217229, NCT03336216),41 47–49 and phase 3 (NCT02371369)43 clinical trials, activity was limited to very select indications. These outcomes suggested that targeting only TAM polarization may be insufficient across broader solid tumor indications due to other compensatory immunosuppressive mechanisms in the TME (eg, PMN-MDSCs).41 42 As such, exploration of additional combination strategies for CSF-1R inhibitors is currently underway.41 In addition, the inhibition of PI3Kγ was shown to reduce MDSC migration and accumulation in the TME,50 reverse TAM polarization, and was synergistic with PD-1 blockade in preclinical studies.38 50–53 These preclinical results have led to the investigation of PI3Kγ inhibitors in several phase 2 clinical studies (NCT03980041, NCT03795610, NCT03961698), and the results are forthcoming.
The CD11b/CD18 integrin heterodimer (Mac-1; αMβ2), which is expressed on MDSCs, TAMs, and subsets of dendritic cells (DCs), mediates myeloid cell adhesion to the vasculature, transendothelial migration, tissue recruitment under inflammatory conditions, phagocytosis, and survival.54 55 Blocking of CD11b/CD18 and its ligands, or deletion of the CD11b or CD18 genes, was shown to decrease the severity of inflammatory response in animal models of vascular injury.56–58 However, blocking of CD11b/CD18 with monoclonal antibodies such as rovelizumab,59 erlizumab,60 or efalizumab,61 has had limited success in clinical trials to date, likely since saturation (>90% occupancy) of CD11b is not achievable at clinically tolerable doses.59 62 63 Interestingly, stabilizing integrins α4β1, αLβ2, and α4β7 in an activated state has been shown to increase cell adhesion and decrease eosinophil migration,64–67 suggesting that stabilizing CD11b/CD18 in an activated state may have a similar effect on cell adhesion and migration. Indeed, leukadherin-1, a small molecule allosteric modulator of CD11b, was shown to increase cell adhesion and reduce migration in vitro and effectively reduce macrophage and neutrophil migration in several animal models of inflammatory conditions.63 68–71 These findings warranted further investigation of CD11b modulation on MDSC migration and function in the TME.
GB1275, a potential first-in-class CD11b modulator
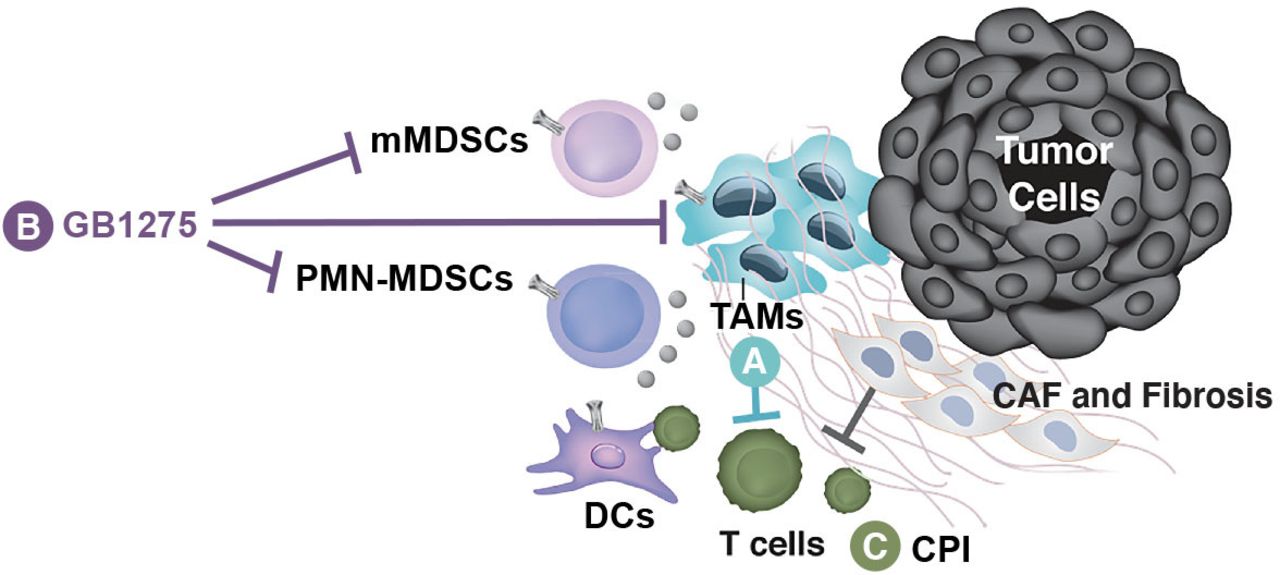
GB1275 (formerly ADH-503) is the salt form of leukadherin-1 designed to stabilize CD11b in an active state.63 68 72 73 Unlike inhibition, activation of CD11b does not require saturation of the integrin and, therefore, it is hoped that use of GB1275 will be able to overcome the dosing challenges of earlier CD11b-targeted strategies that focused on inhibition. GB1275 binds to the allosteric pocket in the αA domain of CD11b and stabilizes CD11b in the active conformation required for its ligand binding63 68 74 (figure 1). By altering CD11b conformation, GB1275 enhances CD11b-mediated adhesion to intercellular adhesion molecule 1 (ICAM-1, CD54) on vascular endothelium and thereby impairs extravasation and transendothelial myeloid cell migration into inflamed tissues.63 68 71 In addition, by modulating CD11b signaling in TAMs, GB1275 increases expression levels of interferon-related factors (IFNα1, IFNβ, CXCL9 and CXCL10) and antigen presentation/co-stimulation complexes (MHC-I, MHC-II, CD80, and CD86), while reducing the expression of immunosuppressive genes (IL10, TGFβ, and arginase-1).72 Thus, GB1275 allosteric modulation of CD11b, leads to a shift in TAM polarization towards pro-inflammatory and antigen-presenting phenotypes, potentiating an interferon response and facilitating T-cell recruitment and activation at the tumor site (figure 2).72 74 75
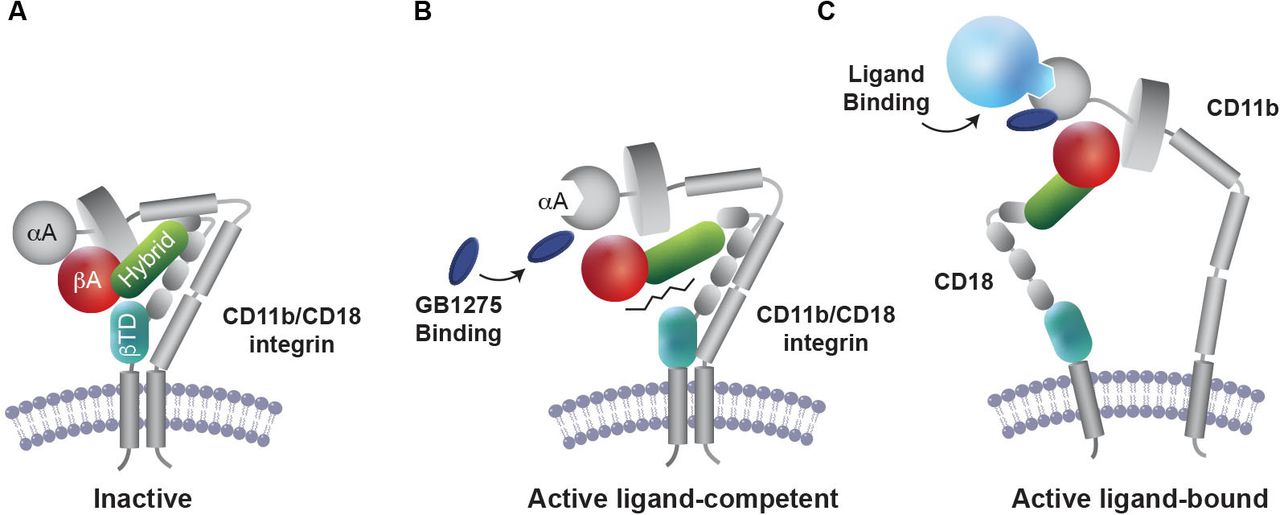
Proposed mechanism of action for GB1275. (A) Inactive CD11b/CD18 integrin heterodimer expressed in circulating leukocytes. (B) GB1275 binding to the allosteric pocket in the α-A-domain of CD11b stabilizes CD11b in the active conformation that is required for ligand binding. (C) On exposure to ligand, GB1275-bound CD11b displays a lower threshold for activation, thereby promoting CD11b-mediated cell adhesion and reduced myeloid cell migration.

Proposed impact of GB1275 on the tumor microenvironment (TME). (A) Tumor-associated macrophages (TAMs) and monocytic myeloid-derived suppressor cells (mMDSCs) suppress T cells in the TME. (B) GB1275 binds CD11b on TAMs and MDSCs, leading to reduced expression of immunosuppressive phenotypes and an influx and activation of tumor-attacking T cells. (C) Exhausted T cells in the TME may then be reinvigorated by checkpoint inhibitor (CPI) therapy. GB1275 combined with a CPI is expected to support effective antitumor immunity. CAF, cancer-associated fibroblast; DC, dendritic cell; PMN-MDSC, polymorphonuclear MDSC.
Preclinical studies with GB1275
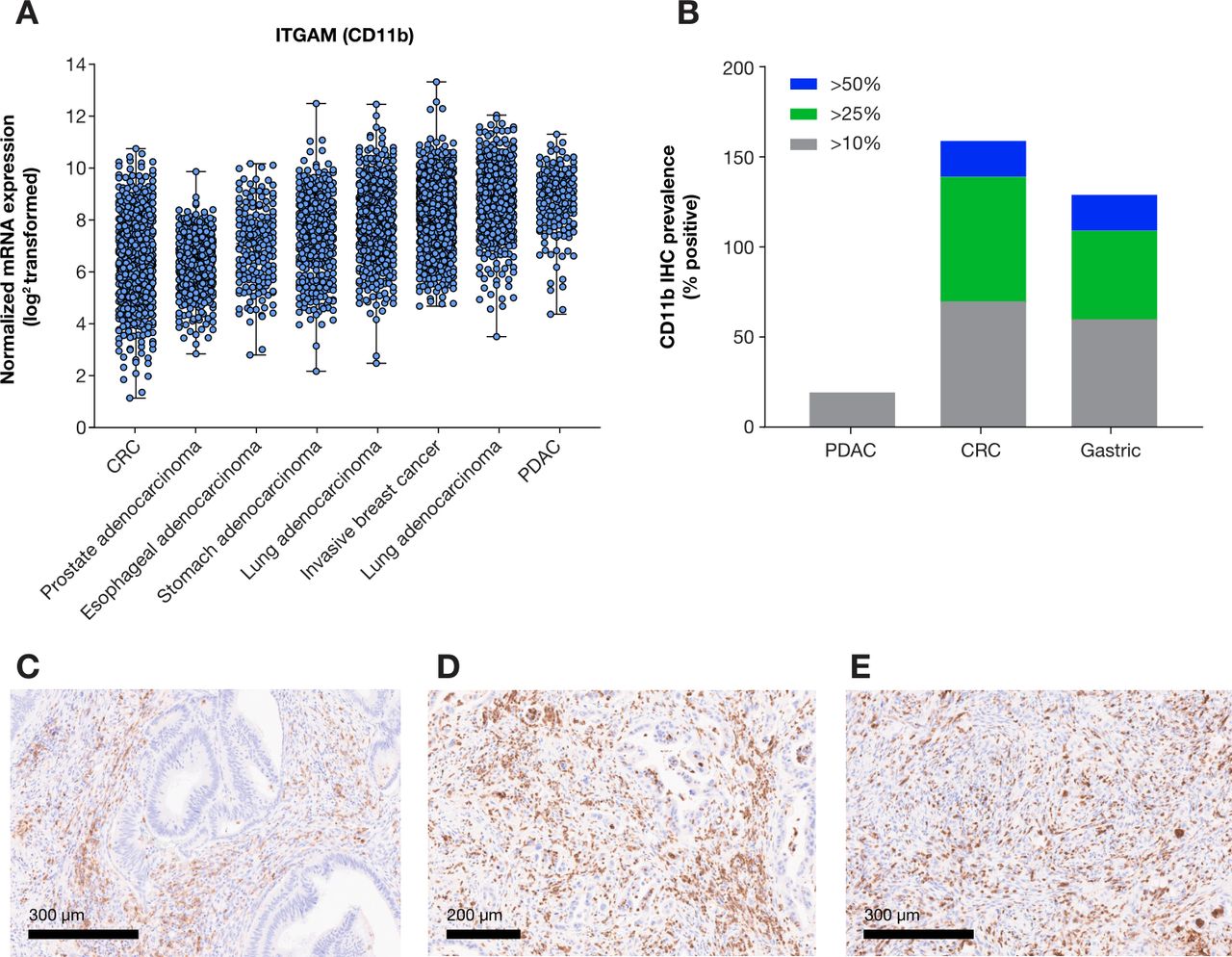
Based on these initial mechanistic findings, tumor types that are characterized by abundant infiltration by CD11b+ MDSCs and display dependence on MDSCs in checkpoint inhibitor resistance may potentially benefit from GB1275, including PDAC,72 lung cancer,74 breast cancer,76 prostate cancer,13 77 colorectal cancer (CRC),78 79 gastric cancer,80 and esophageal cancer.81 82 Elevated levels of Itgam (gene coding for the CD11b subunit of Mac-1 complex) messenger RNA were confirmed across a variety of tumors types in The Cancer Genome Project PanCancer profiling data set, where PDAC and lung adenocarcinoma displayed especially increased relative expression. Assessment of CD11b+ infiltrates by immunohistochemistry across PDAC, CRC and gastric cancer demonstrated heterogenous infiltration of CD11b+MDSCs across representative tumor sections, in line with prior published data.72 Importantly, subsets of CRC and gastric tumors displayed elevated presence of CD11b+ infiltrate, in some cases representing greater than 25%–50% of the cell population (figure 3).
{kind=link}
{kind=link}
{kind=link}
CD11b expression profile in solid tumors. (A) Relative Itgam (CD11b) mRNA expression across solid tumors from select indications in TCGA PanCancer Atlas (www.cbioportal.org). (B) Quantification of CD11b positive cell prevalence at three different cut-offs (>10%, >25%, or >50% positive) in representative sections from PDAC, CRC, and gastric cancer (n=10/indication). Representative images of CD11b positive infiltrates in PDAC (C), CRC (D) and gastric cancer (E). CRC, colorectal cancer; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma; TCGA, The Cancer Genome Atlas.
Antitumor activity of GB1275 as a single agent, and in combination with chemotherapy or ICI therapies, was evaluated in several syngeneic tumor models, including PDAC72 and lung cancer74 75 and is reviewed below.
Pancreatic cancer
The effect of GB1275 in PDAC was studied in syngeneic orthotopic murine PDAC cell lines (KP2 or KI) or in the genetically engineered KPC (p48-CRE/LSL-KrasG12D/p53flox/flox) mouse models that closely resembles the pathology and genomic features of human PDAC.72 83 In all of these PDAC models, GB1275 significantly improved survival compared with a vehicle control, demonstrating efficacy as a single agent.72 In the same mouse models, GB1275 reduced tumor infiltration of CD11b+ MDSCs and increased influx of activated CD103+ DCs and CD8+ T cells. GB1275 was found to upregulate the expression of PD-1/PD-L1 checkpoint molecules on CD8+ T-cells, TAMs, and PDAC cells, suggesting that GB1275 may act concurrently with PD-1-blocking antibodies or other ICI therapies.72 Indeed, in syngeneic orthotopic PDAC mouse models a combination treatment of GB1275 with a PD-1-blocking antibody improved survival to >120 days in all animals, while survival in the vehicle control was 0% after 40 days.72 Notably, single-agent PD-1 blocking antibody failed to improve survival outcomes relative to the vehicle control, which aligns with clinical reports of limited efficacy of PD-1 pathway targeted therapies in patients with PDAC,84 providing further evidence for the relevance of the PDAC mouse model. Similar results were obtained in the KPC model for PDAC.72 Furthermore, a combination of GB1275 with SOC chemotherapy (gemcitabine plus paclitaxel) displayed improved survival when compared with SOC alone or with single-agent GB1275 treatment.72 These data suggest that GB1275 may be effective for the treatment of patients with PDAC as a single agent, or in combination with ICIs or SOC, and warrant further investigation in clinical studies.
Lung cancer
Similar to findings in the PDAC model, GB1275 was shown to reduce TAM infiltration to the TME, increase the proportion of pro-inflammatory M1 TAMs, and inhibit tumor growth in a CD8 T-cell dependent manner in the Lewis lung carcinoma syngeneic lung cancer model. These effects were demonstrated to be dependent on CD11b modulation, as neither leukadherin-1 nor GB1275 were effective against LLC tumors propagated in CD11b knockout mice (Itgam –/–).74 75 Combinations with ICI were not evaluated in these studies; however, observed impact of GB1275 monotherapy on macrophage polarization and induction of T-cell mediated immunity, suggests that further studies may be warranted. Although treatment with ICIs significantly improves survival in patients with lung cancer, these agents are not effective in all patients.85–87 These preclinical studies suggest CD11b modulation with GB1275 may be a potential strategy for the treatment of lung cancer subsets, where TAM immune suppression may be contributing to ICI resistance.
Breast cancer
The effect of leukadherin-1 on breast cancer was evaluated in two breast cancer models: a syngeneic orthotopic breast cancer model (CL-66) and a human triple negative breast cancer (TNBC) xenograft model (MDA-MB-231) in mice that lack T-cells. In the immune-competent model, leukadherin-1 displayed single agent antitumor activity and demonstrated improved outcomes in combination with either irradiation or paclitaxel.75 Furthermore, leukadherin-1 treatment led to a reduction in tumor CD11b+ MDSCs, and a repolarization of macrophages from the CD206+ to the MHCII+ subtype, which was further potentiated in combination with SOC chemotherapy (paclitaxel).75 Leukadherin-1 also reduced tumor growth in the human xenograft model, suggesting that modulation of the innate immunity alone was sufficient to delay tumor growth and that further studies to understand effects of GB1275 in breast cancer are warranted.75
Prostate, colorectal, gastric, and esophageal cancers
Although GB1275 has not been assessed to date in animal models of prostate, colorectal, gastric, or esophageal cancers, there is a strong rationale for its investigation in these tumor types. Patients with prostate cancer often acquire resistance to androgen deprivation therapy and move to a castration-resistant prostate cancer (CRPC) phenotype.3 13 Resistance to androgen deprivation therapy is attributed to the activation of the androgen receptor pathway by IL23, which is secreted by PMN-MDSCs.3 Elevated levels of CD11b+ MDSCs or CD163+ TAMs in the TME are associated with poor prognosis in patients with prostate cancer.16 77 88 In addition, patients with elevated pretreatment levels of CD11b+CD33+HLA-DR–CD14+ mMDSC were less responsive to combination treatment.16
In patients with CRC, the accumulation of CD11b+ PMN-MDSCs in the TME is associated with resistance to ICIs and poor outcomes.21 79 89 The involvement of CD11b+ MDSC in the development of CRC was studied in the ApcMin/+ mouse model for the development of CRC. The mice were crossed with CD11b–/– mice to generate ApcMin/+ CD11b knockout mice (ApcMin/+/CD11b–/–).54 In the ApcMin/+/CD11b–/– mice, the lack of CD11b+ cells resulted in reduced PMN-MDSC infiltration into the TME, peripheral blood, and spleen compared with the ApcMin/+ mice.54 Furthermore, the levels of IFNγ and CXCL9 were upregulated in the ApcMin/+/CD11b–/– mice tumor tissues, compared with tumor tissues in ApcMin/+ mice.54 Therefore, targeting the infiltration of PMN-MDSCs may render the tumors more susceptible to SOC therapy and given poorer outcomes associated with disease progression and the dependence on cytotoxic chemotherapy, it would seem prudent for investigation of GB1275 in CRPC and CRC.
Similarly, elevated levels of CD14–CD11b+ PMN-MDSCs in the peripheral blood of patients with gastric and esophageal cancers may be associated with reduced overall survival.80 81 In patients with esophageal squamous cell carcinoma (ESCC), infiltrating CD68+CD163+ TAMs were associated with resistance to chemotherapy and radiation.82 The role of these infiltrating TAMs was validated in a murine model for ESCC, where incidence and number of tumors were reduced with genetic approaches targeting TAM recruitment or polarization.90
Taken together, these preclinical studies demonstrated that CD11b modulation may render tumors with elevated MDSC infiltration more sensitive to ICIs and other SOC therapies. In general, most of the tumor types investigated in animal models to date have a poor response to anti-PD-1 therapy, and other effective treatment options are limited; consequently, outcomes are poor and there is a large unmet need for new strategies in these cancers.
First-in-human phase 1/2 clinical study with GB1275
Based on the promising preclinical evidence described above, GB1275 is currently being tested in a first-in-human, open-label, phase 1/2 clinical study (NCT04060342) as monotherapy, and in combination with pembrolizumab (Keytruda) in patients with specified advanced solid tumor types that are known to be resistant or less likely responsive to immuno-oncology therapies. The rationale for the selection of the tumor types in the dose escalation phase (phase 1) included the following: tumor types with high unmet need for effective treatment, or with moderate or poor response to ICIs therapy; tumor types with preclinical evidence of GB1275 antitumor efficacy; tumor types in which myeloid suppressor biology is known to be involved based on literature and The Cancer Genome Atlas analyses; and tumor types in which preclinical data suggested mechanistic rationale for combination with chemotherapy.
Patient eligibility and study design
Adults (age ≥18 years) with Eastern Cooperative Oncology Group performance status 0 or 1 and prespecified relapsed or refractory tumors are eligible to enroll in the study. Patients with history of another malignancy within 2 years prior to enrollment, known history of testing positive for HIV, positive for hepatitis B or hepatitis C infection, unable to take oral medication, and women who are pregnant or nursing are excluded from the study. The study is divided into two phases: phase 1 (dose escalation and expansion) and a phase 2 basket expansion. The dose escalation phase, based on a standard 3+3 design, enrolled patients with microsatellite-stable colorectal cancer (MSS-CRC), gastric cancer, metastatic PDAC (mPDAC), TNBC, metastatic CRPC, or esophageal cancer. Oral GB1275 is given at escalation dose levels alone (regimen A) or with intravenous pembrolizumab 200 mg every 3 weeks (regimen B). Once a recommended phase 2 dose (RP2D) of regimen B is determined, up to 40 patients will be enrolled into the phase 1 expansion to be treated at the RP2D in combination with pembrolizumab to further assess the safety, tolerability, clinical and biological activities at the selected RP2D. Phase 2 basket expansion will use a Simon’s two-stage optimal design to evaluate GB1275 in combination with pembrolizumab.
The objective of the dose escalation phase is to evaluate safety and tolerability to determine the maximum tolerated dose or RP2D and to examine the pharmacokinetic and pharmacodynamic profile of GB1275 alone and in combination. The objective of the expansion phase is to determine the safety of the selected dose and schedule of GB1275 with intravenous pembrolizumab (regimen B) in patients with previously treated specified advanced solid tumors. The objective of phase 2 basket expansion is to evaluate the efficacy and safety/tolerability of GB1275 in combination with pembrolizumab.
Exploratory objectives for the study are to evaluate changes in biomarkers to assess target engagement, including mMDSCs and PMN-MDSCs in blood and tumor, immune cell distribution by flow cytometry, CD11b+ cells at the tumor site, and CD8+ cells at the tumor site.
To date, enrollment has commenced in six sites in the USA and one site in the UK, and additional sites are being recruited in the USA. The study is ongoing and is estimated to complete in March 2023.
Discussion
Many patients develop resistance to ICIs and other anticancer therapies, which is often associated with the accumulation of MDSCs and TAMs in the TME.3 4 Several therapeutic approaches with an aim to target MDSCs infiltration and/or TAM polarization have been investigated to date. However, attempts to impact these targets have been largely unsuccessful so far, possibly due to the targeting of individual subsets of myeloid cells in isolation.
In contrast to prior approaches, GB1275, a first-in-class allosteric modulator of CD11b, is directed toward a target that is expressed on multiple subsets of immunosuppressive myeloid cells and may simultaneously impact mMDSCs, PMN-MDSCs, and TAMs. Preclinical studies with GB1275 demonstrated that GB1275 reduced MDSC infiltrates and reprogramed TAMs within the TME,63 68 71 72 74 75 suggesting modulation of CD11b as a promising approach for targeting these immunosuppressive mechanisms. In animal models of cancer that closely resemble human disease, including PDAC and lung cancer, GB1275 demonstrated efficacy both as a single agent and in combination with a PD-1-blocking antibody or chemotherapy.72 74 75 Preclinically, GB1275 was well tolerated as monotherapy and in combination with either a PD-1-blocking antibody or SOC. Furthermore, GB1275 effects were shown to be localized to the tumor microenvironment, with no changes in circulating myeloid cells or their progenitors observed in blood, bone marrow or spleens of treated mice.72 No dose-limiting toxicities (DLTs) were identified in the investigational new drug-enabling good laboratory practice toxicology studies (unpublished data). These encouraging results have led to the investigation of GB1275 in an ongoing phase 1/2 study of patients with tumors that are either resistant to ICI or less likely responsive to ICI. As reported recently, GB1275 demonstrated an acceptable safety and tolerability profile as monotherapy and in combination with pembrolizumab (no DLTs observed in preliminary analysis). In addition, downregulation of peripheral MDSCs was seen with both regimens.91 Recruitment for the phase 1 expansion is currently ongoing.
Exploratory readouts assessing modulation of MDSC frequencies and functional signatures in the periphery and tumor biopsies, pretreatment and post-treatment with GB1275, will be critical to understand in which settings modulation of CD11b will have the most benefit. CD11b is a promiscuous receptor with greater than 50 reported ligands and a variety of functional roles influenced by the microenvironment.92 CD11b engagement enhances ICAM-1-mediated73 and fibrinogen-mediated93 myeloid cell adhesion and impacts IFN signaling modulation.72 74 75 Furthermore, CD11b plays a role in phagocytosis and antigen presentation, supports CD14, Fc gamma receptor, and TLR signaling, and influences inflammatory response to damage-associated and pathogen-associated molecular pattern signals.92 Gene expression and multiplex immunohistochemistry tumor biopsy profiling approaches should provide insights into which TME parameters will influence the GB1275 response signature. It will be interesting to understand the impact of the relative expression density of CD11b, MDSC density, and localization throughout the tumor site, along with the expression levels and profile of the different ligands associated with CD11b signaling on antitumor response.
The selection of tumor types (MSS-CRC, mPDAC, TNBC, metastatic CRPC, gastric, and esophageal cancer) in the phase 1/2 clinical study was based on high unmet need, particularly in terms of poor response to ICIs, poor prognosis, and lack of other current effective treatment. As the study includes tumors with high expression levels of CD11b and those with reported involvement of TAMs and MDSCs in tumor progression, both qualities considered to be good candidates for GB1275 therapy, it is well placed to provide further information on the utility of CD11b modulation in cancer therapy.
Supplemental material
Data availability statement
Data are available upon request
Ethics statements
Patient consent for publication
Acknowledgments
Medical writing support was provided by Miriam Cohen, PhD, and editorial support was provided by Elke Sims, MLangTrans, all of Paragon (Knutsford, UK), supported by Gossamer Bio according to Good Publication Practice guidelines (http://annals.org/aim/article/2424869/good-publication-practice-communicating-company-sponsored-medical-research-gpp3). The ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DGD and AG contributed equally.
Correction notice This article has been corrected since it was first published. The Y axis of Figure 3B has been updated to ‘CD11b IHC prevalence (% positive)’.
Contributors All authors conceptualised the content, reviewed, and contributed to each draft, and approved the publication.
Funding The study was funded by GB006, Inc., a wholly owned subsidiary of Gossamer Bio, Inc, in collaboration with Merck & Co., Inc., Kenilworth, New Jersey, USA.
Competing interests AG: Employee of Gossamer Bio. JD: Formerly employee of Gossamer Bio. LZ: Consultant of Gossamer Bio. JB: Did not receive personal research or consulting funds. Received food/beverage/travel funds from Gilead, Genentech/Roche, Bristol Myers Squibb, Lilly, Merck, MedImmune, Celgene, Taiho Pharmaceutical, Novartis, OncoMed, Boehringer Ingelheim, ARMO BioSciences, Ipsen, Oncogenex, and FORMA Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.