Article Text

Abstract
Background Despite clinical success with T cell engagers (TCEs) targeting hematological malignancies, achieving a safe and efficacious dose in patients with solid tumors remains challenging. Due to potency, low levels of target antigen expression on normal tissues may not be tolerated. To overcome this, we engineered a novel conditionally active TCE design called COBRA (Conditional Bispecific Redirected Activation). Administered as prodrugs, COBRAs bind to cell surface antigens on both normal and tumor tissues but are preferentially activated within the tumor microenvironment.
Methods A COBRA was engineered to target EGFR, TAK-186. The potency of precleaved TAK-186 relative to a non-cleavable control was assessed in vitro. Mice bearing established solid tumors expressing a range of EGFR levels were administered a single bolus of human T cells, and concurrently treated with TAK-186 and associated controls intravenously. We assessed the plasma and tumor exposure of intact and cleaved TAK-186.
Results TAK-186 shows potent redirected T cell killing of antigen expressing tumor cells. In vivo efficacy studies demonstrate regressions of established solid tumors, dependent on intratumoral COBRA cleavage. Pharmacokinetic studies reveal TAK-186 is stable in circulation, but once activated is rapidly cleared due to loss of its albumin-binding half-life extension domain.
Conclusions The studies shown support the advancement of TAK-186, and the pursuit of additional COBRA TCEs for the treatment of solid tumors.
- Therapies, Investigational
- Immunotherapy
- Antibodies, Neoplasm
- Tumor Microenvironment
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The potential of inherently active T cell engagers (TCEs) to treat patients with solid tumor indications has been limited by toxicities presented by the activity of these highly potent therapeutics toward critical healthy tissues expressing low levels of antigen, preventing a clinically active dose from being reached.
WHAT THIS STUDY ADDS
A solution to the limitation of inherently active TCEs is provided by the novel Conditional Bispecific Redirected Activation (COBRA) design, allowing activation and subsequent T cell engagement only in the tumor microenvironment due to the well-characterized dysregulation of protease activity by tumors.
TAK-186, the first therapeutic based on the COBRA design, regresses established solid tumors in mice at low dose as a result of tumor-specific cleavage, and further provides an additional safety mechanism via the loss of half-life extension in the active form.
The data presented supports that the dysregulation of protease activity by solid tumors can be used to enhance specificity of TCEs, that this activity is sufficient to regress established human tumors in mice, thus providing a mechanism with significant potential to achieve clinically active dose, and subsequent patient response.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
The reported activity of the COBRA TAK-186 supports further research on and advancement of protease-activated TCEs into clinical development, expanding the potential to treat solid tumors over a range of indications.
The results presented support further research of therapeutic prodrugs designed to be activated in
Introduction
The clinical success of blinatumomab in treating patients with certain B cell malignancies validated T cell engagers (TCEs) as a therapeutic modality and sparked an increase in the development of T-cell redirected therapies. While some early TCE programs targeted antigens expressed on solid tumor indications (catumaxomab, pasotuxizumab), most clinical studies initially focused on a range of hematological tumors.1 Of particular interest was the increased potency of TCEs relative to earlier therapeutic modalities, both monoclonal antibody therapeutics using enhanced antibody-dependent cellular cytotoxicity functionality as well as antibody-drug conjugates. Preclinical data have demonstrated the ability of TCEs to target tumor cells expressing antigen at fewer than 100 copies per cell.2 Notably, blinatumomab itself demonstrated around 100,000-fold increase in potency relative to rituximab when depleting B-cells in vitro.3 However, this increased potency toward tumor cells presents a double-edged sword, with a greater risk of damage to normal healthy cells where antigen may be expressed, although at low levels. This is particularly problematic as the potency of TCEs is often greater than the sensitivity of the immunohistochemistry (IHC) methods commonly used to screen normal tissues for potential risk.
A notable distinction between target antigens expressed on hematological malignancies relative to solid tumors is the normal tissue types which also express the antigen. In the case of blinatumomab, patients are able to tolerate the transient depletion of normal B cells while receiving efficacious dose levels.4 Additionally, several TCEs targeting different antigens in multiple myeloma have demonstrated similar depletion of normal lineage cells and appear to be tolerated.5–7
For solid tumor indications, the antigens to which therapies are directed are frequently epithelial in origin and maintain a low level of expression on vital healthy tissues. Any toxicities induced by the TCE would likely not be tolerated, limiting the dose that can be administered, and subsequent patient response. Many TCEs covering a range of antigens targeting solid tumor indications have been discontinued, including EpCAM, CEA, P-cadherin, and gpA33.8–11 Notably, a TCE targeting epidermal growth factor receptor (EGFR) demonstrated kidney and liver toxicities at high dose in cynomolgus monkey studies, requiring early euthanasia.12 The tissue damage observed was consistent with low levels of EGFR expression in these tissues and supports the need for additional therapeutic designs to enhance tumor-specific activity.
In order to successfully move TCEs into the solid tumor space, a strategy to allow for increased tumor specificity and broadening of the therapeutic window is required. One such approach uses the unique properties of the tumor microenvironment (TME) to restrict activity of the TCE to the tumor, while sparing healthy tissues. The TME has been widely reported to demonstrate increased protease activity relative to normal tissues.13 14 Increased activity of matrix metalloproteinases (MMPs) specifically is associated with many solid tumor types and dysregulation of MMP activity is associated with increased invasion, metastases, and angiogenesis.15–17 MMP-2 and MMP-9 levels in particular have been explored as potential prognostic biomarkers in several tumor indications.18–20 Several groups have described conditionally active TCEs that are engineered to be administered as prodrugs, which become active on entering the TME.21–24 Typically, these designs incorporate a protease cleavage site within the TCE that unmasks antigen binding sites, allowing for coengagement of the tumor and the T cell, thereby inducing a cytolytic response against the tumor.
Here, we report the preclinical pharmacological characterization of TAK-186 (also known as MVC-101), a conditionally active Conditional Bispecific Redirected Activation (COBRA TCE) targeting EGFR, previously described.25 TAK-186 demonstrates highly potent activity against tumor cell lines expressing EGFR cocultured with T cells. Pharmacokinetic studies demonstrated stable and sustained exposure of intact TAK-186 over time in tumor-bearing mice, and rapid clearance of active species. Repeat administration in vivo resulted in regressions of established EGFR-expressing tumors in a dose-dependent and EGFR expression-dependent manner. Additionally, COBRA activity was dependent on a protease-mediated cleavage event within the TME.
Materials and methods
Protein design and expression
TAK-186 was designed by creating a constrained anti-CD3 scFv with a short (G3S)2 linker that functions to prevent the active anti-CD3 VH and VL domains from pairing into a functional, CD3 binding scFv. This constrained scFv is flanked on either side by anti-EGFR binding sdAbs linked by (G3S)2 linkers. TAK-186 utilized an MMP9-cleavable linker (SGGPGPAGMKGLPGS) to connect a second constrained scFv with a short (G3S)2 linker to prevent the inactivate anti-CD3 VL and VH domains from pairing into an scFv. This structure forces the COBRA into a single chain diabody.26 A his6-tagged anti-HSA sdAb was attached to the C-terminus to provide extended serum half-life. In NCL-186 a non-cleavable (G4S)3 linker replaces the MMP9 linker present in TAK-186. COBRA molecules were expressed via transient transfection using the Expi-293 system (ThermoFisher Scientific), purified by protein A using standard techniques, and analyzed as previously described.25
In vivo tumor efficacy and PK
LoVo, HT29 and SCC25 tumor cells were purchased from and cultured as recommended by the manufacturer. NSG mice (Jackson Laboratory) were implanted with tumor cell lines subcutaneously in the right flank. Human CD3 +T cells were isolated from leukopak via negative selection (StemCell Technologies, 17951). 20×106 T cells were expanded using G-Rex technology (Wilson Wolf G-Rex 100, 80 500S) in combination with T-cell expansion/activation beads (Miltenyi 130-091-441) in X-VIVO 15 media (Lonza) containing 5% human serum (Gemini), IL-2, NEAA, sodium pyruvate and hepes for 9–11 days. Tumor cells were implanted in 50% growth factor reduced matrigel (Corning) at densities of 10×106 LoVo, 5×106 HT29, or 5×106 SCC25 cells per mouse in the right flank. Once tumors reached 200–400 mm3, mice were randomized into groups of 6 based on tumor volume, expanded human T cells were implanted intravenously at 2.5×106 cells per mouse, and test articles administration was initiated. Test articles were dosed intravenously every 3 days for a total of seven doses. Tumor volume was assessed by caliper measurement (Mitutoyo CD-6” AX), and data were analyzed in Prism (GraphPad).
To assess pharmacokinetics, test articles were administered intravenously to NOD-SCID mice (Jackson Laboratory) and blood was collected and processed to plasma. Plasma concentration was determined by MSD (MesoScale Discovery) using anti-idiotype antibody 13H4 as capture, and anti-HIS detection as previously described.25 Data were analyzed using Prism (GraphPad Software). To assess tumor concentrations of TAK-186, tumor growth was established, and test articles were dosed intravenously Mice were perfused with PBS, tumor were collected, and flash frozen prior to analysis.
T-cell-dependent cellular cytotoxicity and cytokine release assays
LoVo, HT29 and SCC25 cells were purchased from ATCC, engineered to express luciferase via lentiviral transduction (Biosettia), and cultured per manufacturer’s recommendation. Human PBMC were isolated from leukopak utilizing EasySep technology (Stem Cell Technologies). Cells were plated at an E:T ratio of 5:1, combined with NCL-186 and cTAK-186 and incubated for 48 hours at 37° in AIM-V media (Thermo Fisher). To assess T-cell-dependent cellular cytotoxicity (TDCC), SteadyGlo (Promega) was added to quantitate the amount of luciferase labeled target cells remaining after the duration of the assay. RAJI cells engineered to express luciferase as well as human EGFR were also tested in this manner. Cytokine release was assessed by combining tumor cells, PBMC and MVC-NCL and pcTAK-186 as above; supernatants were collected after 24 hours. Respective cytokine concentrations were determined using MSD Proinflammatory panel 1 (human) kit for detection (MesoScale Discovery). Data were analyzed using SoftMax Pro (Molecular Devices) and Prism (GraphPad).
EGFR quantitation and TAK-186 binding
Cells were trypsinized from flasks, resuspended in culture media, then washed in FBS Stain Buffer (BD Biosciences). Tumor cells were combined with TAK-186 and incubated on ice for 30 min, then washed prior to detection via anti-HIS-FITC (Genscript). EGFR surface levels were assessed using custom fluorescence labeled anti-EGFR (AY13) using Quantibrite Beads PE Fluorescence Quantitation Kit (BD Biosciences), and data collection on CytoFLEX LX. Data was analyzed using FlowJo (BD Biosciences) and Prism (GraphPad).
Detection of tumor-mediated TAK-186 cleavage
Homogenization of tissues
Tissues were placed in T-PER (Thermo Fisher, 78510) with 1X protease inhibitor (ThermoFisher, 87785) and 5 mM EDTA. Tissues were homogenized with an Omni BeadRuptor using 5 mm ceramic beads; samples were centrifuged at 10 000 rpm for 10 min at 4C. Supernatant was collected and analyzed for protein concentration.
Immunoprecipitation
Anti-idiotype to TAK-186 (7A8) was biotinylated and bound to magnetic beads (Dynabeads Streptavidin M270). Bound biotinylated antibody/beads were washed with 1X PBS twice. Homogenates (supernatant) were normalized to a fixed protein concentration and incubated with the biotinylated antibody bound Dynabeads. Mixture was washed then eluted in 0.1X SDS buffer. KingFisher was utilized to perform all steps (Thermofisher).
Simple western
Samples were diluted in 0.1X SDS buffer and then mixed with fluorescent master mix (prepared as non-reduced, DTT substituted with ddH2O) at a ratio of 4:1. The prepared samples were heated at 95C for 5 min and placed on ice. The primary antibody, anti-VHH (GenScript, A01860) was prepared at a working concentration of 25 ug/mL in diluent 2. Secondary antibody, anti-rabbit HRP were used per manufacturer recommendation. The samples, primary and secondary antibodies and chemiluminescent substrate were dispensed into their designated wells (12-well, 230 kDa separation plate). Simple Western assay buffers, capillaries and assay plates were placed in the instrument per manufacturer’s instructions. Peak of interests (AUC) from samples were analyzed and back-calculated to a standard curve using Compass (Protein Simple).
Statistics
All data are represented as mean±SD, or ±SEM as noted. Statistics applied are described in the figure legends.
Results
TAK-186 design
TAK-186 is a conditionally active TCE that is engineered to bind EGFR expressing cells on administration. Its design, illustrated in figure 1A, consists of two anti-EGFR single domain antibodies (sdAbs), the first located N-terminal and the second in the middle of the linear peptide. A third C-terminal sdAb binds human serum albumin (HSA), which functionally extends the in vivo half-life of TAK-186 in prodrug form. The anti-EGFR sdAbs flank anti-CD3 variable heavy (VH) and variable light (VL) domains. The short 8 amino acid (a.a.) linker separating these VH and VL domains does not allow for proper folding into a single-chain fragment variable (scFv), therefore, preventing formation of an anti-CD3 antigen binding site in its intact prodrug state. A 15 a.a. protease cleavable linker separates the second anti-EGFR sdAb from another pair of VH and VL domains, which include mutations in their complementary determining regions (CDRs), rendering them inactive. Like the previous set of VH and VL domains, the inclusion of a short 8 a.a. linker does not allow for folding into an scFv. We believe this forces the linear peptide to fold in such a way that the anti-CD3 VH and VL pair with the corresponding inactive VL and VH domains, thereby forming a stable structure that can bind to both EGFR and HSA, but not to CD3, as depicted in figure 1B. Once the central 15 a.a. linker is cleaved by proteases in the TME, the fragment containing the (2) anti-EGFR sdAbs and the anti-CD3 VH and VL domains can separate from and release the HSA-binding half-life extension sdAb from the fragment which is bound to EGFR on the cell surface. Due to the short linker constraining the anti-CD3 VH and VL domains, this single fragment alone cannot form an active, CD3-binding, TCE. Only after dimerizing with a second cleaved fragment on the tumor cell surface can the COBRA coengage EGFR and CD3 (figure 1B). Per design, if the active dimer were to escape the tumor it will be rapidly cleared due to the loss of the anti-HSA sdAb, reducing potential exposure and subsequent toxicity to any normal EGFR-expressing tissue.
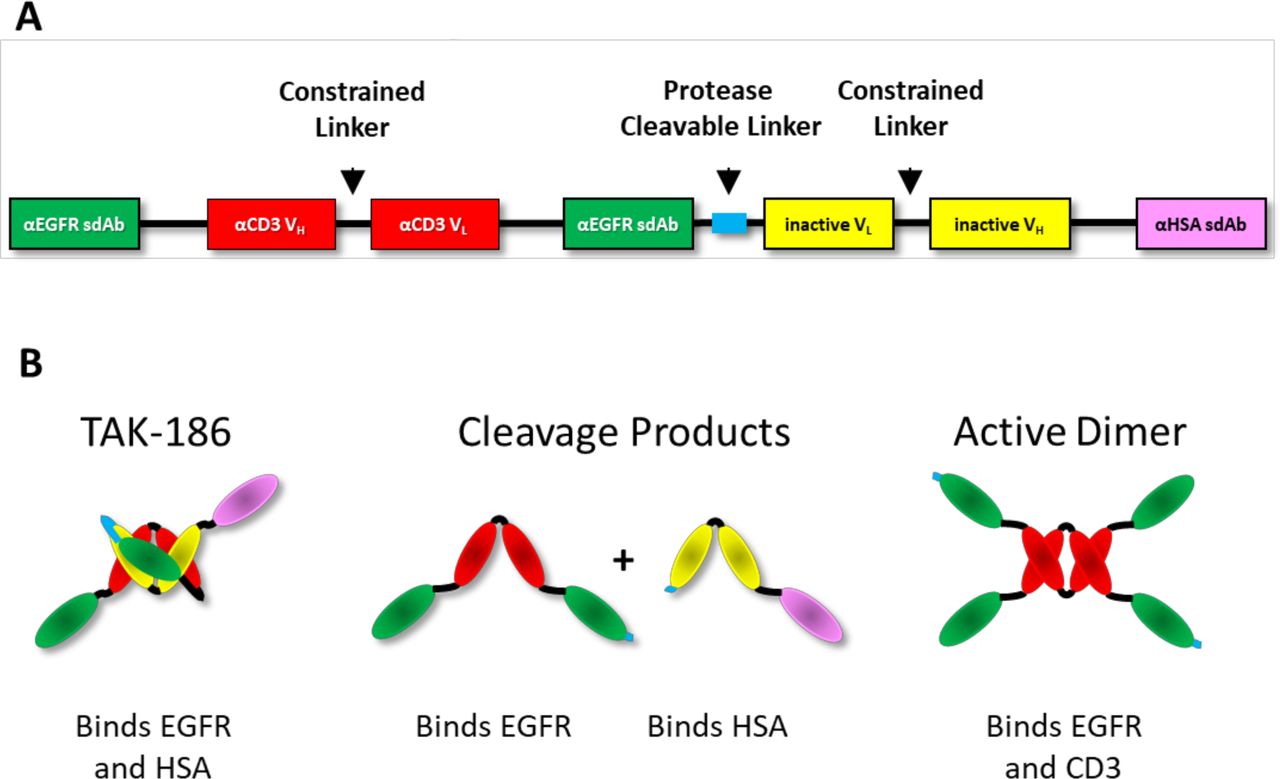
Design of EGFR-targeted conditionally activated COBRA TAK-186. (A) The linear construct design of TAK-186 is shown here, with the N-terminal (2) EGFR sdAbs separated by the αCD3 VH and VL, constrained and separated from the C-terminal Inactive VH and VL and HLE αHSA sdAb by a single protease cleavable linker. Both VH and VL are prevented from forming an active scFv by a constrained 8 a.a. linker. (B) In cartoon form is the predicted structure of the intact and cleavage products of TAK-186. The inactive VH and VL are associated with the αCD3 VH and VL. in TAK-186 prodrug form. Following cleavage of TAK-186, the (2) EGFR binding sdAbs and αCD3 VH and VL are separated from the Inactive VH and VL, which remain fused to the αHSA sdAb, providing HLE. The active dimer of TAK-186 forms when a single αCD3 VH and VL pairs with another αCD3 VH and VL on the surface of EGFR-expressing tumor cells. The Active Dimer contains in total (4) EGFR-binding sdAbs and (2) αCD3 scFvs, forming an active TCE.
TAK-186 mediates in vitro activation and potent T cell-mediated killing of EGFR expressing cell lines
We measured the binding of TAK-186 to CD3 on T cells by flow cytometry (figure 2A). Human T cells were incubated with (1) TAK-186, (2) a control version of TAK-186 which has replaced the protease cleavable linker with a non-cleavable linker (NCL-186), (3) TAK-186 that has been precleaved (pcTAK-186) in solution with MMP-9 prior to incubation, and (4) with the recombinantly-expressed active dimer of TAK-186 (adTAK-186). The highest level of binding was measured with adTAK-186, 1.4-fold higher compared with pcTAK-186, as active dimer formation and binding to CD3 with pcTAK-186 is expected to be less efficient in solution compared with the recombinantly expressed adTAK-186, as the anti-CD3 VH and VL domains in this molecule have already paired into an active TCE.
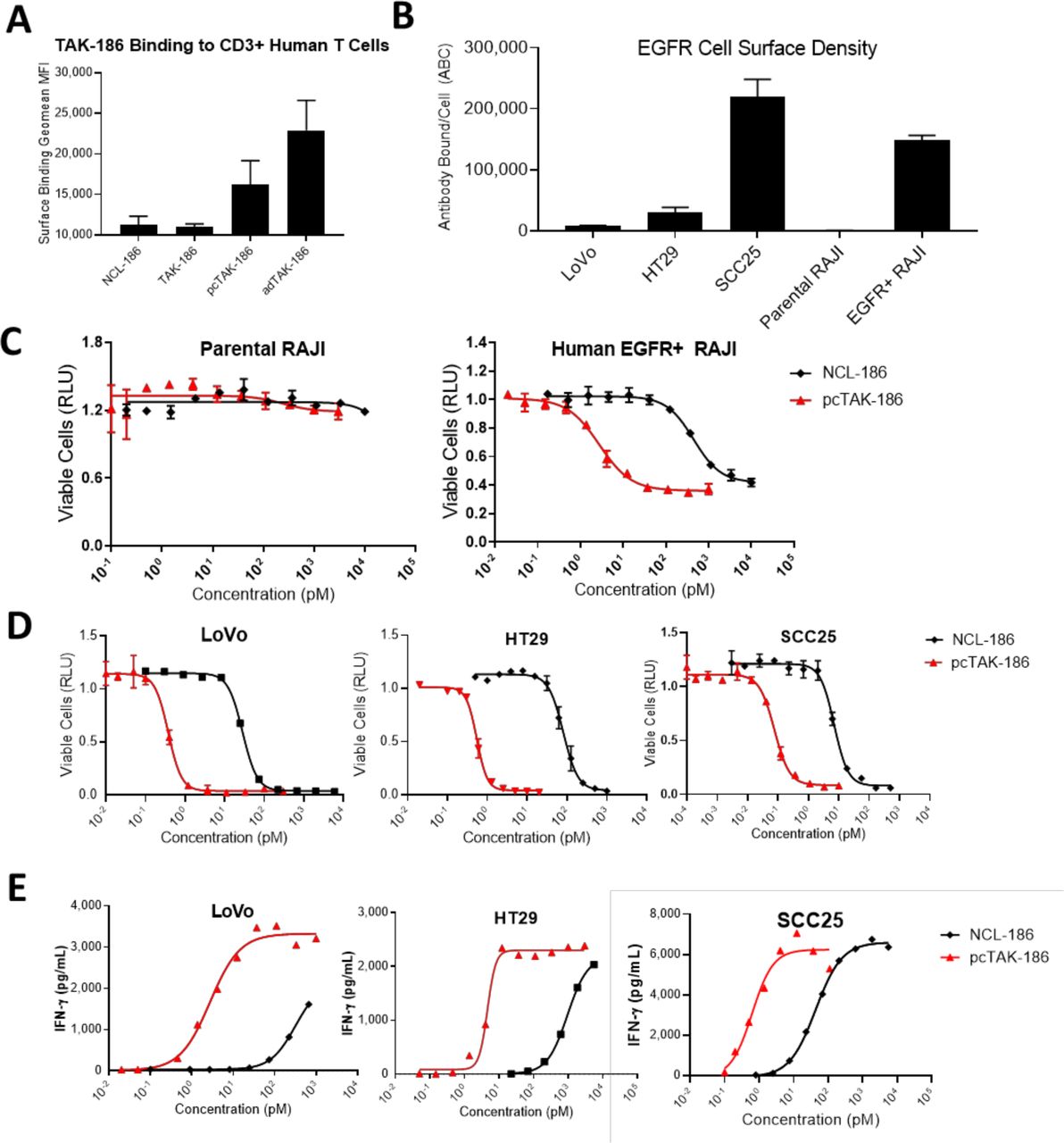
TAK-186 demonstrates potent in vitro activity following proteolytic activation. The binding of NCL-186, TAK-186, pcTAK-186 and adTAK-186 to human T cells was assessed by flow-cytometry. Data are presented as MFI±SD, n=2 (representative of two independent experiments) (A). The density of cell surface EGFR on human tumor cell lines and human EGFR transgenic RAJI cells was assessed by quantitative flow-cytometry. Data are presented as MFI±SD, n=2 (representative of two or more independent experiments) (B). NCL-186 and pcTAK-186 were tested in TDCC (C, D) and cytokine release (E) assays by coincubating EGFR-expressing human tumor cell lines LoVo, HT29 SCC25, EGFR transgenic RAJI, and EGFR negative parental RAJI with human T cells (data are mean±SD). Equivalent results were obtained from three independent experiments conducted with this effector cell donor, and additionally when tested in the presence of additional effector cell donors (online supplemental table 1). RAJI, Burkitts Lymphoma; MFI, mean fluorescence intensity; EGFR, epidermal growth factor receptor; TDCC: T cell dependent cellular cytotoxicity.
Supplemental material
EGFR expression was next assessed on a panel of cell lines by quantitative flow cytometry. The cell lines tested included the EGFR negative cell line Raji (parental Raji), RAJI engineered to express EGFR (EGFR +Raji), the colorectal adenocarcinoma cell lines LoVo and HT29, and the head and neck squamous cell carcinoma cell line SCC25. As expected, the parental Raji cell line demonstrated low background levels of signal (661), while the engineered EGFR +RAJI cells measured 147 982 antibodies bound per cell (ABC (figure 2B)). The tumor cells lines LoVo, HT29 and SCC25 measured increasing levels of endogenous EGFR, specifically 8728, 29 769 and 219 309 ABC respectively (figure 2B).
The potency of TAK-186 was measured in vitro by culturing human T cells with the cell lines noted above. Using a standard TDCC assay, we titrated the concentration of pcTAK-186 and NCL-186 to measure the EC50 and determine the window of activity of the intact prodrug relative to the activated form of TAK-186 on each cell line. We observed depletion of human EGFR +RAJI cells, absent on the parental RAJI cells (figure 2C), demonstrating EGFR-dependent cell killing. We observed high potency of pcTAK-186 depletion of LoVo (EC50 0.37 pM), HT29 (EC50 0.54 pM) and SCC25 (EC50 0.07pM) cell lines (figure 2D), 76–148-fold increased relative to TAK-186 in prodrug form (figure 2D). Consistent in vitro activity was demonstrated in the presence of multiple effector cell donors (online supplemental table 1). We additionally measured the in vitro cytokine release mediated by pcTAK-186 by incubating tumors cells in combination with human PBMC, where we measured the potency of IFN-γ release in LoVo (EC50 4.3 pM), HT29 (EC50 4.2 pM) and SCC25 (EC50 0.6 pM) (figure 2E). The cytokine release was observed similarly across the cytokine panel measured, and over multiple effector cell donors (online supplemental table 1). The potency of pcTAK-186 observed correlated with EGFR surface expression (figure 2B), and the in vitro activity demonstrated toward SCC25 was consistent among cell lines expressing high levels of EGFR (data not reported).
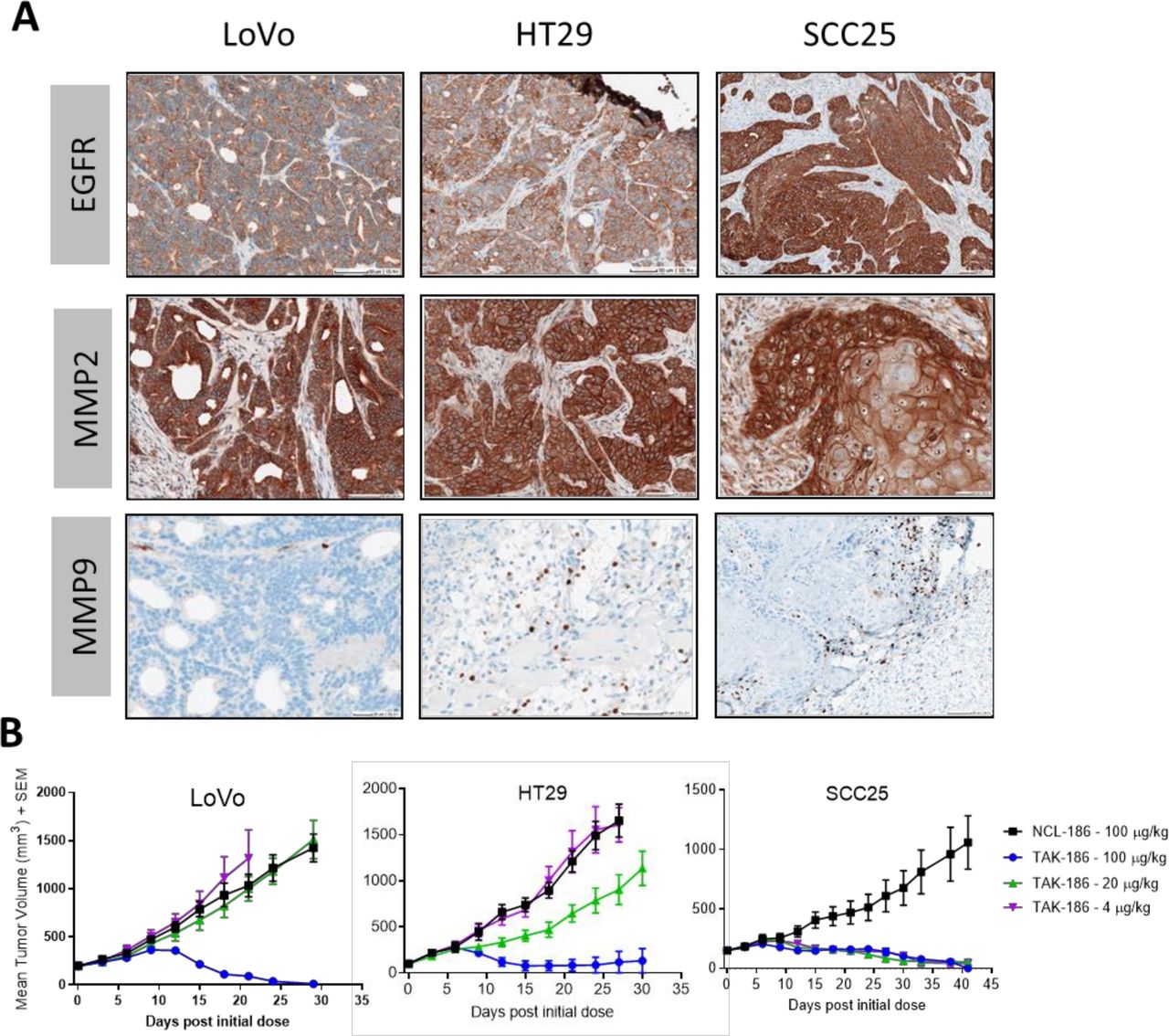
Tumor models express a range of EGFR, MMP2 and MMP9 expression as measured by IHC
The levels of EGFR, MMP-2 and MMP-9 expression were assessed on human tumor xenograft sections by IHC (figure 3A). Consistent with the in vitro measurements, EGFR expression levels varied with tumor type, with the highest expression in the head and neck squamous cell carcinoma model SCC25 relative to the colorectal cancer (CRC) tumor models LoVo and HT29, which expressed the lowest and second highest levels, respectively. Differential EGFR expression between SCCHN and CRC in patient tumors has also been noted in the literature.27 Expression of MMPs appeared to be relatively consistent among the three tumor types, with homogenous expression of MMP-2, while MMP-9 expression was detectable in a subset of cells that appear to be infiltrating the tumor.

TAK-186 demonstrates dose-dependent tumor regression in established human tumor xenografts with a range of EGFR, MMP2 and MMP9 expression. EGFR (A–C), MMP2 (D–F), MMP9 (G–I) expression was assessed in human tumor xenografts LoVo, HT29 and SCC25 via IHC. Scale bars included in the images, with images representative of three independently stained samples. Established subcutaneous tumor xenografts were dosed at 4–100 µg/kg q3d×7; human T cells were adoptively transferred intravenously at dose initiation. Mean tumor volume (mm3 ±SEM) is plotted against time postdose initiation (n=6). Tumor regression was equivalent across 3 (HT29, LoVo) or 2 (SCC-25) independent studies. Tumor growth inhibition mediated by TAK-186 at 100 µg/kg was 100% in LoVo (p=0.0004) and 92% HT29 (p<0.0001), and 31% when dosed at 20 µg/kg in HT29 (p=0.1355). Tumor growth inhibition was 96% in SCC25 at 4 µg/kg (p<0.0001). Statistical analysis used an unpaired t-test with Welch’s correction. The dose schedule administered was consistent with the exposure of TAK-186 observed in mice (figure 4, online supplemental table 4). IHC, immunohistochemistry; MMP, matrix metalloproteinases; EGFR, epidermal growth factor receptor
Supplemental material
TAK-186 regresses established solid tumors in mice
The efficacy of TAK-186 was tested in EGFR-expressing human tumor xenografts in mice. Once tumors were established in immunodeficient NSG mice, 2.5×106 T human cells were adoptively transferred intravenously into each mouse, followed by an initial dose of TAK-186 or the negative control molecule NCL-186. TAK-186 or NCL-186 were subsequently administered every 3 days over a total of 7 doses (q3d×7). TAK-186 demonstrated tumor regression at doses≥4 µg/kg, in a dose and EGFR expression-dependent manner (figure 3B). Tumor regression was observed at 4 µg/kg in the SCCHN model SCC25 relative to both colorectal tumor models, LoVo and HT29 where TAK-186 regressed tumors at 100 µg/kg. Tumor growth inhibition was demonstrated in HT29 at 20 µg/kg, a model higher in EGFR expression relative to the LoVo model, where activity was not observed at this dose, further supporting the dependance of activity on EGFR level (figure 3A). In all studies NCL-186 resulted in activity similar to vehicle control (online supplemental figure 1).
Supplemental material
TAK-186 protease-mediated cleavage is required for tumor activity
TAK-186 was designed to mediate tumor-specific activity, dependent on protease-mediated cleavage in the TME. In prodrug form, TAK-186 is stable in circulation, demonstrating only slightly reduced exposure relative to NCL-186 (figure 4A). When dosed as either a precleaved molecule or recombinantly expressed as an active dimer (figure 1B), TAK-186 was rapidly cleared from circulation (figure 4A, online supplemental table 4). This clearance supports the safety mechanism designed into the COBRA platform reducing exposure of active drug to any normal target expressing tissue, should any escape the tumor. Treatment of LoVo tumor-bearing mice with pcTAK-186 resulted in a lack of tumor regression at 100 µg/kg, while the same dose of TAK-186 resulted in complete regressions (figure 4B). This lack of tumor regression mediated by pcTAK-186 is consistent with the shorter half-life of this molecule and supports that tumor-mediated cleavage of TAK-186 is required to elicit tumor regression.
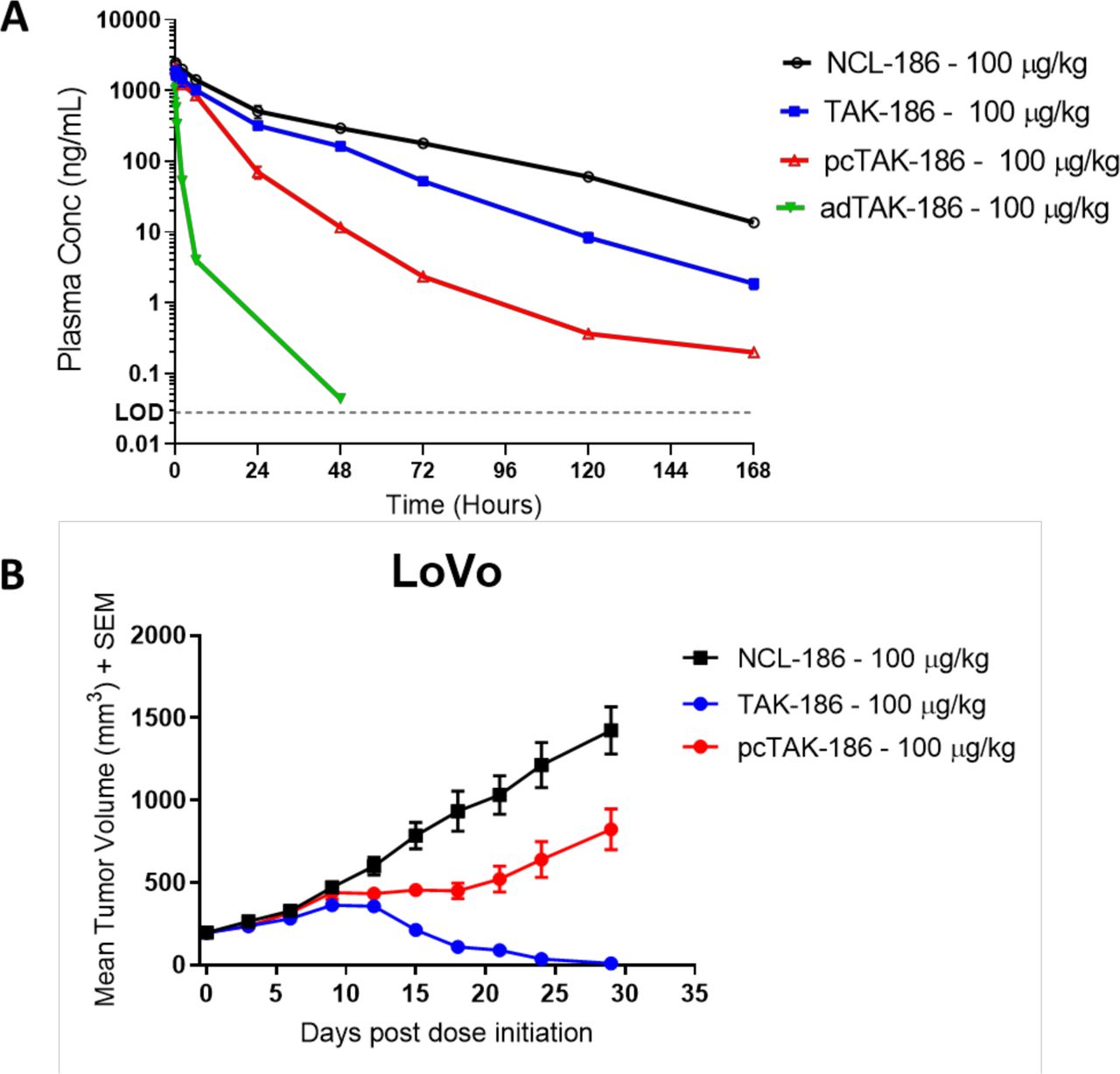
Activated TAK-186 is cleared rapidly anddoes not mediate tumor regressions. (A) NCL-186, TAK-186, pcTAK-186, adTAK-186 were dosed in NOD-SCID mice, and plasma concentration both adTAK-186 and pcTAK-186 was demonstrated to be reduced relative to TAK-186. TAK-186 had slightly reduced exposure relative to NCL-186. N=2, error bars represent SD. (B) Established LoVo tumors were dosed with NCL-186, TAK-186, or pcTAK-186 q3d×7. Consistent with clearance, pcTAK-186 did not induce tumor regression relative to TAK-186 at equivalent dose. Mean tumor volume (mm3 ±SEM) is plotted against time post dose initiation (n=6). Relative to NCL-186, TAK-186 inhibited tumor growth by 100% (p=0.0004) and pcTAK-186 by 42% (p=0.0217) at day 29. Statistical analysis used an unpaired t test with Welch’s correction. Tumor regression mediated by TAK-186 was equivalent across three independent studies.
TAK-186 intratumoral protease-mediated cleavage and activation
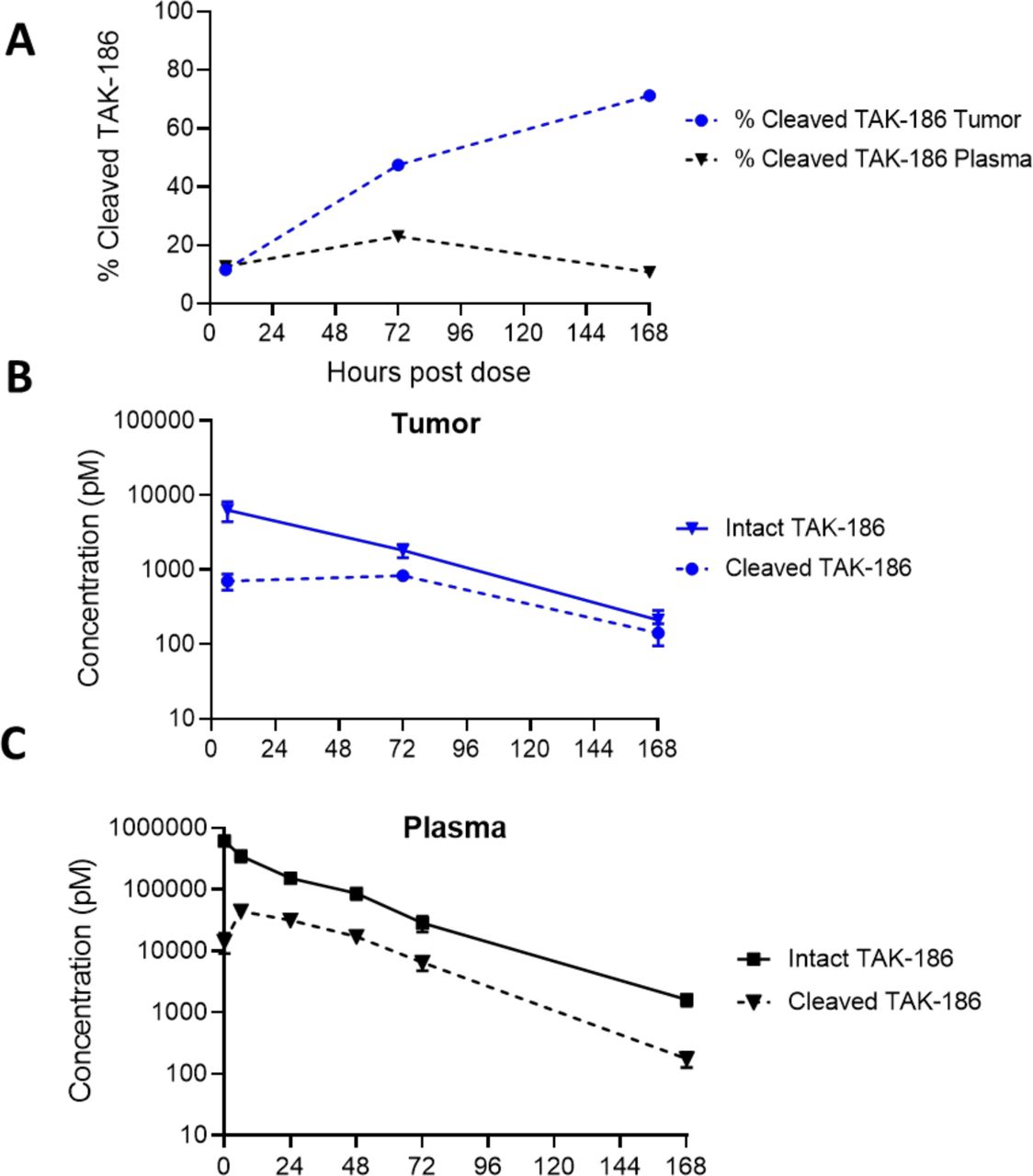
Given the observations above, we directly measured the amount of cleaved TAK-186 in the tumor relative to the plasma after administration. We developed a detection method utilizing a capillary-based immunoassay platform, which distinguishes TAK-186 in cleaved and intact forms concurrently based on differences in molecular weight between the species. The concentration of cleaved TAK-186 increased in the tumor relative to plasma initiating 72 hours post-dose measuring 48% and 23% cleaved, respectively, increasing to 71% in the tumor at 168 hours while decreasing in the plasma to 11%, as a percentage of intact (figure 5A). The concentration of cleaved TAK-186 remained constant in the tumor for the first 72 hours at ~750 pM, while intact TAK-186 decreased steadily over time (figure 5B). Both cleaved and intact forms of TAK-186 demonstrated similar rates of clearance from plasma after 6 hours, the relative fraction of cleaved versus intact TAK-186 consistent through 168 hours (figure 5C, online supplemental table 4). The percentage of cleaved TAK-186 in plasma is at maximum 20% intact, whereas in tumor cleaved TAK-186 reaches 71% of intact at 7 days as a result of the clearance rate of intact TAK-186 from plasma. The total TAK-186 in the tumor is consistent with HLE therapies of similar size (online supplemental figure 3).28
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intratumoral protease-mediated TAK-186 Cleavage. TAK-186 was dosed at 3 mg/kg in HT29 tumor-bearing mice. The concentration of cleaved and intact TAK-186 was assessed via Simple Western in plasma (A) or tumor lysates (B) concurrently via an anti-VHH antibody recognizing the sdAbs present in all forms of TAK-186, with cleaved TAK-186 differentiating from intact TAK-186 by size (online supplemental figure 4). The % cleaved relative to intact TAK-186 increases in tumor through 7d post dose, where the % cleaved in plasma is less than tumor at all timepoints beyond 72 hours. (n=3, error bars represent SD).
Supplemental material
Discussion
A significant attribute of TCEs is their highly potent T cell-mediated killing relative to the low level of target coengagement required for activation.29 30 Because TCEs have the potential to be so potent, when left unchecked they run the risk of inducing harmful inflammatory responses in normal healthy tissues that express even very low levels of the target antigen, resulting in unmanageable toxicities to these tissues. Many approaches have relied on the differential level of target expression between tumor and normal tissues to allow for therapeutic exposures to be reached. However, as evidenced by the number of TCE programs that have been terminated early in clinic trials, and the lack of TCEs that have been approved for patients with solid tumors, reliance on differential antigen expression does not appear sufficient to administer safe and efficacious doses for these indications. The EpCAM targeted TCE catumaxomab was terminated as a result of DLT consistent with target expression in the liver, while none of the patients on treatment exhibited an objective response.31 Development of a TCE targeting the solid tumor antigen GPA33 was discontinued, where the target antigen was documented to have overexpression on tumor cells relative to normal tissue.32 Given the limitations demonstrated by these as well as other TCEs designed to target solid tumor antigens, novel TCE designs are required to overcome this problem.
In this set of preclinical studies, we demonstrate protease dependent activation of our EGFR targeting COBRA TAK-186. With repeat dosing of this highly potent, conditionally active COBRA, TAK-186 regressed established EGFR expressing tumors in both a dose-dependent and target density-dependent manner. Moreover, TAK-186-mediated tumor efficacy is driven by tumor dependent activation of the COBRA molecule. PK studies in mice reveal TAK-186 is stable on administration and following proteolytic linker cleavage and activation the active TCE is cleared rapidly relative to the intact prodrug, as designed.
In vitro characterization of TAK-186 demonstrates high potency of the active TCE, and conditionality relative to the NCL-186. These characteristics were consistent among tumor cell lines, and in all assays tested, including TDCC, cytokine release, and T cell activation. Additionally, the activity was present and replicated over multiple effector cell donors (online supplemental figure 2, online supplemental table 3).
Supplemental material
Supplemental material
To evaluate the efficacy of this highly potent COBRA TAK-186 in vivo, we developed established human tumor xenograft models in immunodeficient mice. Previous studies have demonstrated established tumor models as being a more rigorous method of measuring TCE-mediated efficacy than when compared with in vivo models where human tumor cells and effector cells are mixed and implanted at a set ratio, and then treated with TCE near to implantation; often referred to as Co-mix or Ad-mix Models.10 33 The three tumor models shown here expressed varying levels of EGFR, having relatively low, medium, and high levels of cell surface target. With repeat dosing, TAK-186 regressed these tumor xenograft models in a dose—and antigen expression level-dependent manner.
We next assessed stability of the prodrug relative to NCL-186. TAK-186 demonstrated only slightly reduced exposure relative to NCL-186 in non-tumor bearing mice, confirming minimal cleavage. In this same model, we observed that pcTAK-186 and adTAK-186 cleared rapidly relative to TAK-186, showing that the additional safety mechanism of loss of HLE on activation designed into the COBRA platform reduces exposure of the active TCE to any healthy tissue should any active molecule escape the tumor. The tumor-specific activation of TAK-186 was substantiated by the lack of tumor regression observed when established tumors were treated with pcTAK-186 when dosed equivalently to TAK-186.
We next developed an assay to directly measure cleavage of TAK-186. Given the combined potency and low dose required to mediate tumor regression, a sensitive detection method was established that concurrently measures intact and cleaved TAK-186 in plasma and tissue. TAK-186 dosed in mice with established tumors demonstrates accumulation of cleaved TAK-186 in tumor relative to plasma, supporting that activation is mediated specifically by proteases in the TME, confirming that cleaved TAK-186 is rapidly cleared from plasma, as designed. It should be noted this data represents an immunocompromised mouse with a co-opted murine stromal component, both of which contribute to the protease activity of the TME, thus, it might be anticipated that patient tumors may mediate enhanced cleavage, and subsequent response.34 35
Therapies targeted to EGFR have significant potential for improvement in a range of solid tumor indications, including head and neck squamous cell carcinoma, CRC, and NSCLC. EGFR as a target is widely expressed in normal tissues as well as tumors. Skin toxicities have been observed across EGFR inhibitors, impacting the quality of life of patients on these therapies, while also limiting the efficacy by restricting the dose that can be safely administered.36 37 The potency of TCEs would be anticipated to elicit additional toxicities, requiring new modalities with enhanced specificity. TAK-186 will bind to and target EGFR-expressing cells irrespective of mutational status, and additionally exhibits binding to EGFRviii (online supplemental table 2) demonstrating a broad potential for all EGFR-expressing tumor types.
Supplemental material
The combined data shown here, and the equivalent binding to human and cynomolgus monkey EGFR, CD3ε and serum albumin (online supplemental table 2) supports toxicological assessment in advancement of TAK-186 into clinical development. Additionally, this validation of the COBRA design using TAK-186 supports development of additional tumor-targeted therapies based on the novel COBRA platform.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DED wrote the paper, and is responsible for overall content. JDD, CM reviewed and edited the paper. Experiments/methodology: DED, JDD, LQ, AD, EK, PS, RW, BJH, YZ, JLK and MV. Conceptualization: RBD and CM.
Funding All authors are funded by and employees of Takeda Pharmaceutical company.
Competing interests All authors are employees of Takeda Pharmaceuticals.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.