Article Text

Abstract
Background Neutrophils kill antibody-opsonized tumor cells using trogocytosis, a unique mechanism of destruction of the target plasma. This previously unknown cytotoxic process of neutrophils is dependent on antibody opsonization, Fcγ receptors and CD11b/CD18 integrins. Here, we demonstrate that tumor cells can escape neutrophil-mediated cytotoxicity by calcium (Ca2+)-dependent and exocyst complex-dependent plasma membrane repair.
Methods We knocked down EXOC7 or EXOC4, two exocyst components, to evaluate their involvement in tumor cell membrane repair after neutrophil-induced trogocytosis. We used live cell microscopy and flow cytometry for visualization of the host and tumor cell interaction and tumor cell membrane repair. Last, we reported the mRNA levels of exocyst in breast cancer tumors in correlation to the response in trastuzumab-treated patients.
Results We found that tumor cells can evade neutrophil antibody-dependent cellular cytotoxicity (ADCC) by Ca2+-dependent cell membrane repair, a process induced upon neutrophil trogocytosis. Absence of exocyst components EXOC7 or EXOC4 rendered tumor cells vulnerable to neutrophil-mediated ADCC (but not natural killer cell-mediated killing), while neutrophil trogocytosis remained unaltered. Finally, mRNA levels of exocyst components in trastuzumab-treated patients were inversely correlated to complete response to therapy.
Conclusions Our results support that neutrophil attack towards antibody-opsonized cancer cells by trogocytosis induces an active repair process by the exocyst complex in vitro. Our findings provide insight to the possible contribution of neutrophils in current antibody therapies and the tolerance mechanism of tumor cells and support further studies for potential use of the exocyst components as clinical biomarkers.
- immune evation
- immunity, innate
- immunotherapy
- tumor escape
- cytotoxicity, immunologic
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Neutrophils kill antibody-opsonized tumor cells by taking up pieces of their cell membrane, a process called trogocytosis. Still, tumor cells can resist this trogocytosis-mediated cell death. This study therefore aims to dissect the mechanism of tumor cell death evasion.
What this study adds
Although previous literature now shows that neutrophils can kill antibody-opsonized cancer cells through trogocytosis, cancer cells can resist this by inducing membrane repair. This not only provides evidence for the contribution of neutrophils in antibody therapy against cancer, but also shows that components of the exocyst complex may be considered as potential biomarkers to predict efficacy of antibody therapy in the context of neutrophil trogocytosis.
Introduction
Neutrophils are able to kill antibody-opsonized cancer cells using antibody-dependent cellular cytotoxicity (ADCC). The cytotoxic mechanism of action has for long remained unclear, but recently, we described that neutrophils actively tear off pieces of membrane from antibody-opsonized solid tumor cells. This process involves trogocytosis, and repeated trogocytosis can lead to the lytic death of the cancer cell, which we have coined trogoptosis.1 Neutrophil trogocytosis and ADCC strictly depend on Fcγ receptors and the presence of high-affinity CD11b/CD18 integrins.1–3 In addition, trogocytosis and killing can be enhanced on inhibition of CD47–signal regulatory protein-α (SIRPα) interactions.4 The myeloid inhibitory receptor SIRPα, expressed on innate immune cells including neutrophils, binds the ligand CD47 on tumor cells that acts as a ‘don’t eat me’ signal. As SIRPα dampens immune cell effector functions, it has become an interesting and specific target in immunotherapy already showing promising responses with minimal toxicity in initial clinical studies.5–7 Still, neutrophil trogocytosis of tumor cells does not induce tumor cell death in all cases, even with CD47–SIRPα interference. We hypothesized that when neutrophil trogocytosis induces plasma membrane damage, tumor cells might prevent subsequent cell death by repairing the induced membrane lesions. Membrane repair is a vital process during the life span of any cell and disruptions to the plasma membrane occur often, especially in cells that are subject to mechanic forces.8 Different mechanisms for membrane repair have been described, depending on the size and type of injury,9–12 yet these all depend on the presence of extracellular calcium (Ca2+) and are initiated on Ca2+ influx into the cell at the site of damage.8 The repair of cellular injuries of a mechanical nature is specifically mediated by the exocyst complex.13 This octameric protein complex, including EXOC7 and EXOC4, orchestrates the recruitment, docking, and fusion of intracellular vesicles to the inner plasma membrane, thereby patching the membrane from the inside-out, a dynamic process taking less than seconds.13–15
In this study, we show that solid tumor cells can indeed resist neutrophil trogoptosis by plasma membrane repair mediated by the exocyst complex. Depletion of extracellular Ca2+ increased neutrophil-mediated tumor cell death, while trogocytosis remained unaltered. Knocking down either of the two exocyst components EXOC7 and EXOC4 in different tumor cell lines caused a lower tumor cell resistance towards neutrophil ADCC. The clinical importance of the exocyst complex and, consequently, neutrophil anticancer activity in vivo was demonstrated by the inverse correlation between tumor exocyst complex mRNA expression and the pathological response in patients with breast cancer treated with therapeutic antibody trastuzumab in the NeoALTTO (Neoadjuvant Lapatinib and/or Trastuzumab Treatment Optimisation (NeoALTTO) trial.
Materials and methods
Isolation of effector cells from whole blood
Primary cells were isolated from blood collected from healthy donors and neutrophils and natural killer (NK) cells were isolated as previously described16 by isotonic Percoll density gradient centrifugation. NK cells were obtained from the peripheral blood mononuclear cell (PBMC) fraction using anti-CD56 magnetic-activated cell sorting (MACS) bead selection, according to instructions provided by the supplier (Miltenyi Biotec), and isolated NK cells were cultured overnight in Roswell Park Memorial Institute (RPMI) medium (Gibco) supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C and 5% CO2. Neutrophils were obtained from the pellet after lysis of the erythrocytes, and to achieve neutrophil activation, cells were cultured for 4 hours or overnight with 50 ng/mL interferon gamma (Peprotech) and 10 ng/mL granulocyte-colony stimulating factor (G-CSF, Neupogen) or, for live cell imaging experiments, for 30 min 10 ng/mL with granulocyte macrophage-colony stimulating factor (GM-CSF, Peprotech).
Cell lines and genetic modifications
Human epidermal growth factor receptor (EGFR)+ epidermoid carcinoma cell line A431 (American Type Culture Collection (ATCC)) and HER2/neu+ breast cancer cell line SKBR3 (ATCC) were cultured at 37°C and 5% CO2 in RPMI (Gibco) with 10% (v/v) fetal calf serum or Iscove modified Dulbecco media (IMDM, Gibco) with 20% (v/v) fetal calf serum, respectively, both supplemented with 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin. To generate EXOC7 knockdown SKBR3 and A431 cells using shRNA (hereafter named SKBR3 EXOC7 shRNA-mediated knockdown (EXOC7KD) and A431 EXOC7KD, respectively), we used the pLKO.1puro-EXOC7KD constructs (TRCN0000137284 and TRCN0000137267, Sigma MISSION). As control, we used pLKO.1puro-scrambled constructs (SHC002, Sigma MISSION). Lentiviral particles were generated by transient cotransfection of 293 T cells with pLKO.1puro, pMDLg/pRRE, pRSV-rev, and pCMV-VSVg. On days 1 and 2 after transfection, the cells were put on 293T medium with 100 mg/L butyrate. After overnight incubation, virus-containing supernatant was collected and filtered through 0.45 µM. Viral particles were concentrated by ultracentrifugation for 2 hours at 20 000 rpm at 4°C; pellets were resuspended in phosphate buffered saline (PBS) and stored in 50 µL aliquots at −80°C. A431 and SKBR3 cells were transduced with a 50 µL aliquot of concentrated EXOC7KD lentivirus, and transduced cells were selected on 1 µg/mL puromycin (Invivogen). For each experiment, cells were freshly transduced and the resulting knockdown was examined by western blot.
We used CRISPOR (crispor.tefor.net/17) to determine the Cas9 target sites present in the coding sequences of EXOC7 and EXOC4, in order to create EXOC7 and EXOC4 knockout A431 cells. A ds oligo (ThermoFisher Scientific) was generated for EXOC7: 5′ AATTTCGCAGCCTGATGACG 3′ and EXOC4: 5′ GGGATGAGCTTCGGAAACTG 3′, both with a specific BsmBI overhang. These oligos were cloned into the BsmBI sites of pLentiCRISPRv2. The constructs were grown in Escherichia coli Stbl3 and sequences were verified. Lentiviral particles were generated by transient cotransfection of 293 T cells with pLentiCRISPRv2-EXOC7 or pLentiCRISPRv2-EXOC4, psPAX2 and pCMV-VSVg. The day after transfection, cells were put on A431 culture medium. Virus-containing supernatant was harvested at days 2 and 3 after transfection and filtered through 0.45 µM, prior to addition to A431 cells on two successive days. Transduced A431 cells were selected with 1 µg/mL puromycin (Invivogen). Surviving cells were put on limiting dilution in a 24-well plate at 1 cell/mL, and growing clones were routinely maintained. Expression of EXOC7 or EXOC4 proteins, respectively, in these clones was determined by western blot: we obtained A431 cells with reduced but stable EXOC4 expression (hereafter referred to as A431 EXOC4KD cells) and A431 cells with EXOC7 reduced in size (hereafter referred to as A431 EXOC7Δ157–190 cells).
Antibodies and reagents
All antibody-dependent cellular assays were performed in the presence or absence of 5 µg/mL cetuximab (anti-EGFR, Merck KGaA) or trastuzumab (anti-HER2/neu, Roche) and with or without addition of 5 µg/mL SIRPα blocking antibody (clone 12C44). For the experiments with or without extracellular Ca2+, neutrophils and tumor cells were taken up in HEPES buffer (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) supplemented with 5 g/L human albumin (Albuman, Sanquin Plasma Products) and 5.5 mM glucose or Minimal Essential Medium (MEM, Gibco), both with or without 1 mM CaCl2. For the experiments with the cell permeant chelator BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid), tumor cells were pretreated with BAPTA (ThermoFisher) for 30 min at 37°C and washed before coincubation with neutrophils. For the membrane-cholesterol depletion experiments, tumor cells were preincubated with 2.5 mM methyl-β-cyclodextrin (MβCD) (Sigma) for 20 min at 37°C and washed before coincubation with neutrophils.
Scratch assay
SKBR3 and A431 wild type cells were seeded on glass coverslips (14 mm diameter) until 80%–90% confluent. Culture medium was gently washed away from the coverslips and MEM (Gibco), with or without 1 mM CaCl2, was added. The confluent monolayers were scratched with a needle, and after a 5 min recovery period at 37°C, cells were washed and stained with propidium iodide (PI). The cells were then fixed with 3.7% (w/v) paraformaldehyde (PFA) and stained with 4’,6’-diamidino-2-phenylindole (DAPI) before imaging (Leica SP8 microscope).
Live cell imaging
1×104 SKBR3 wild-type cells were overnight seeded on an eight-well μ-slide (Ibidi) with polymer glass coverslips (25 or 30 mm diameter). Cells were labeled for 30 min at 37°C with lipophilic membrane dye (DiD, 5 µM; Invitrogen) or, in case of Ca2+ INflux experiments, also with Fluo-4-AM (1:500, ThermoFisher). Tumor cells were thoroughly washed after. Neutrophils were prestimulated for 30 min with 10 ng/mL GM-CSF (Peprotech), and both cells were resuspended in HEPES buffer and incubated for 90 min at a target:effector ratio of 1:10 in the presence of the appropriate antibodies. Imaging started as soon as possible after coincubation, and images were captured every 3 s using a Leica TCM SP8 confocal microscope (Leica). For the live cell imaging of neutrophil trogoptosis (figure 1), neutrophils were stained with Hoechst (1:10 000), and TO-PRO-3 Iodide (1:4000, ThermoFisher) was added to the medium as a live/dead indicator. Neutrophil trogocytosis was quantified by counting the amount of neutrophils participating in trogocytosis of a single tumor cell, as well as the occurrence of tumor cell death throughout imaging. Interaction of neutrophils and a tumor cell is defined as active adhesion of effector cells to the tumor cell (figure 1B,C). For the live cell imaging of Ca2+ influx (figure 2B,C), images were quantified by measuring the mean fluorescence intensity (MFI) of Fluo-4-AM in the tumor cells, and trogocytic events were scored manually using FIJI software.
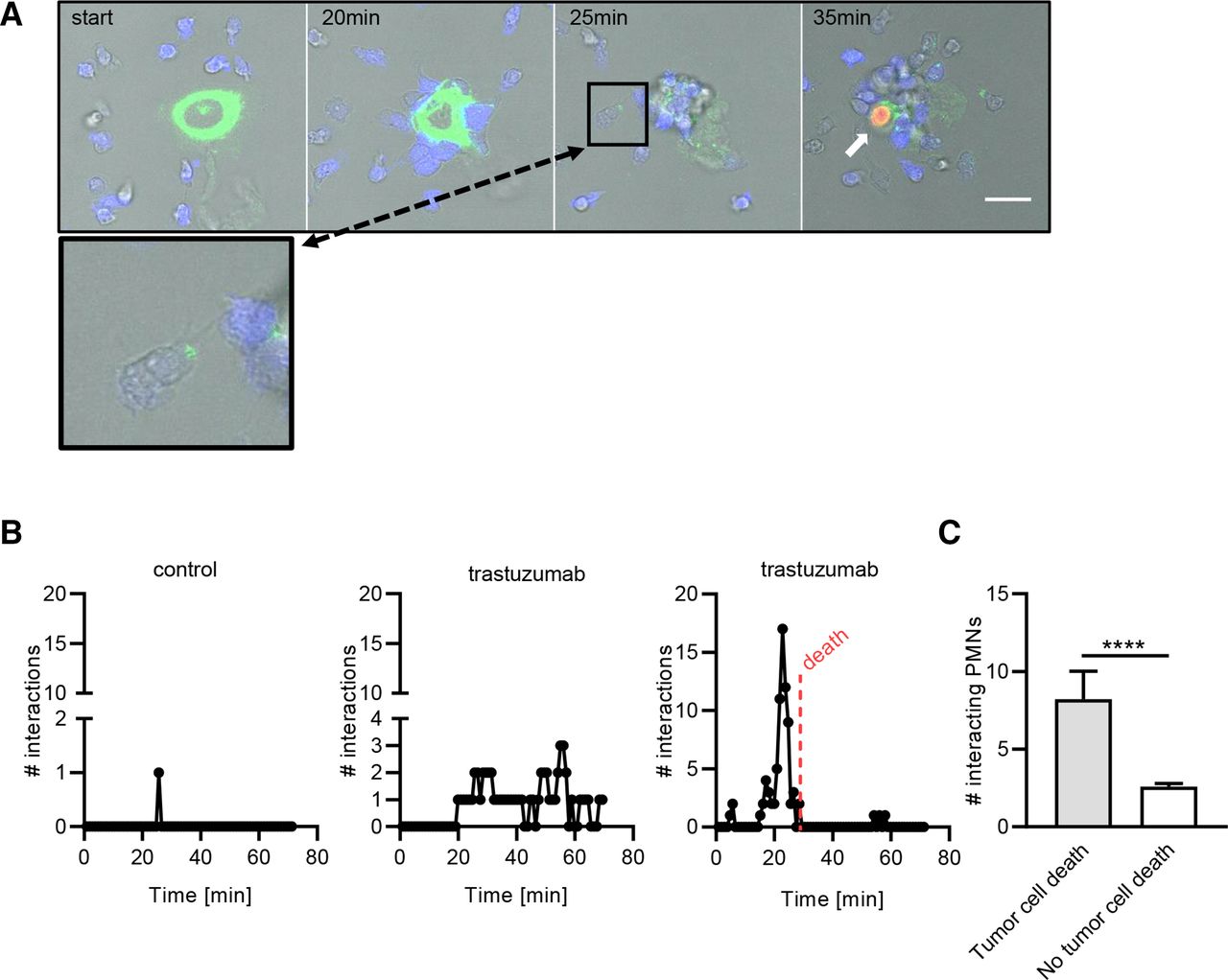
Multiple iterative trogocytic events are needed to induce cancer cell death. (A) Tmab-opsonized SKBR3 cells and neutrophils were coincubated and followed over time using live cell imaging. SKBR3 cells were labeled with membrane dye DiO (green) and neutrophils with Hoechst (blue). The white arrow points at a lysed tumor cell, indicated by DNA-binding dye TO-PRO-3 Iodide (orange). The scale bar represents 20 µM. The inset shows a close-up of a neutrophil interacting with a tumor cell at t=25 min, containing at least one DiO+ fragment. (B) The amount of trogocytic interactions between neutrophils and one non-opsonized SKBR3 cell (control, left graph) and two Tmab-opsonized SKBR3 cells (middle and right graph; each graph depicts one SKBR3 cell) during a representative live cell imaging experiment. Every dot represents the amount of trogocytic interactions by neutrophils (y-axis) at t=x min. Tumor cell death is indicated by the red dotted line. (C) Quantified results of four live cell imaging experiments of SKBR3 cell trogocytosis by neutrophils, showing the amount of trogocytic interactions in relation to the occurrence of tumor cell death. n=25 tumor cells assessed from four independent live cell imaging experiments. Statistics: (C) unpaired t-test; ****, p<0.001. Tmab, trastuzumab.
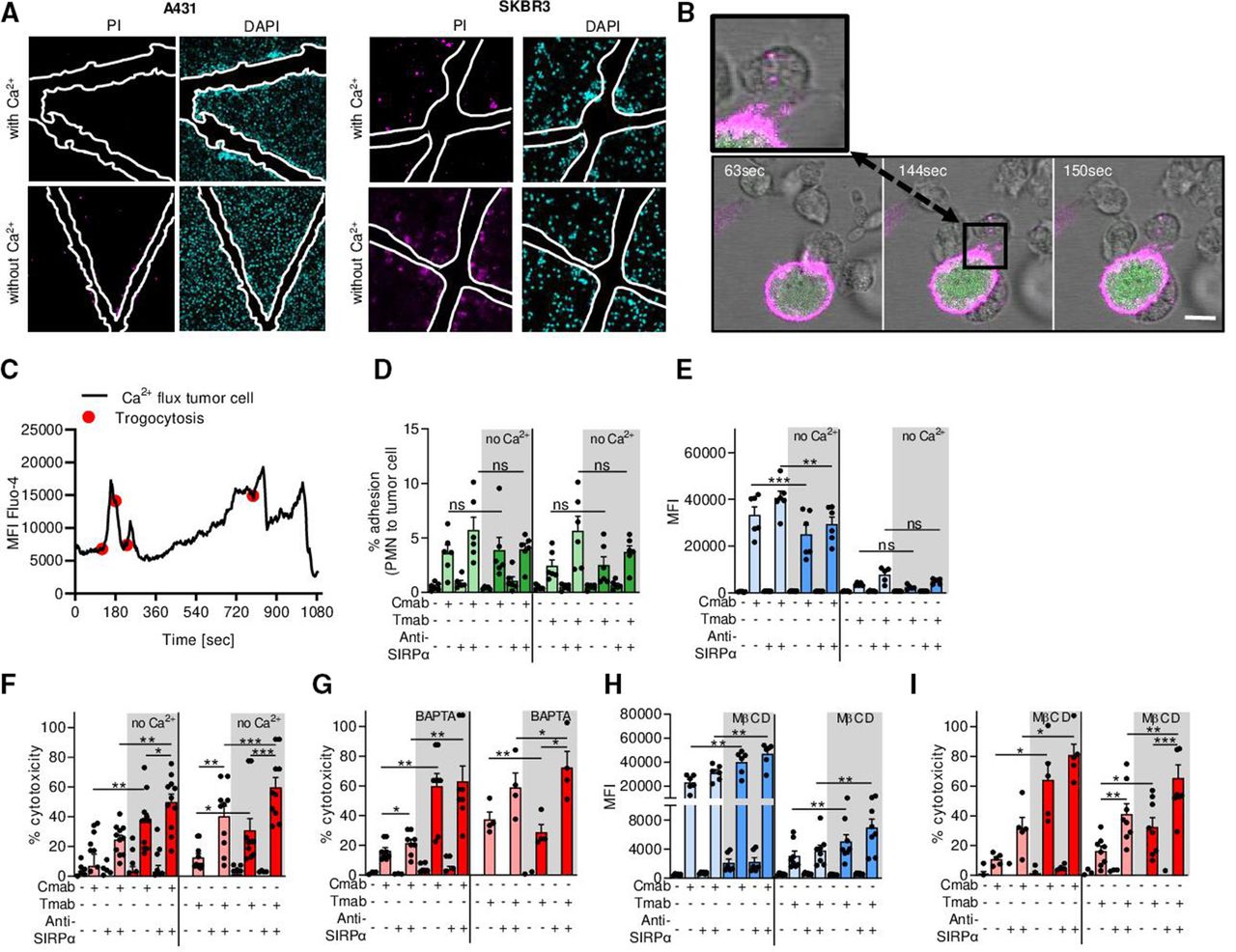
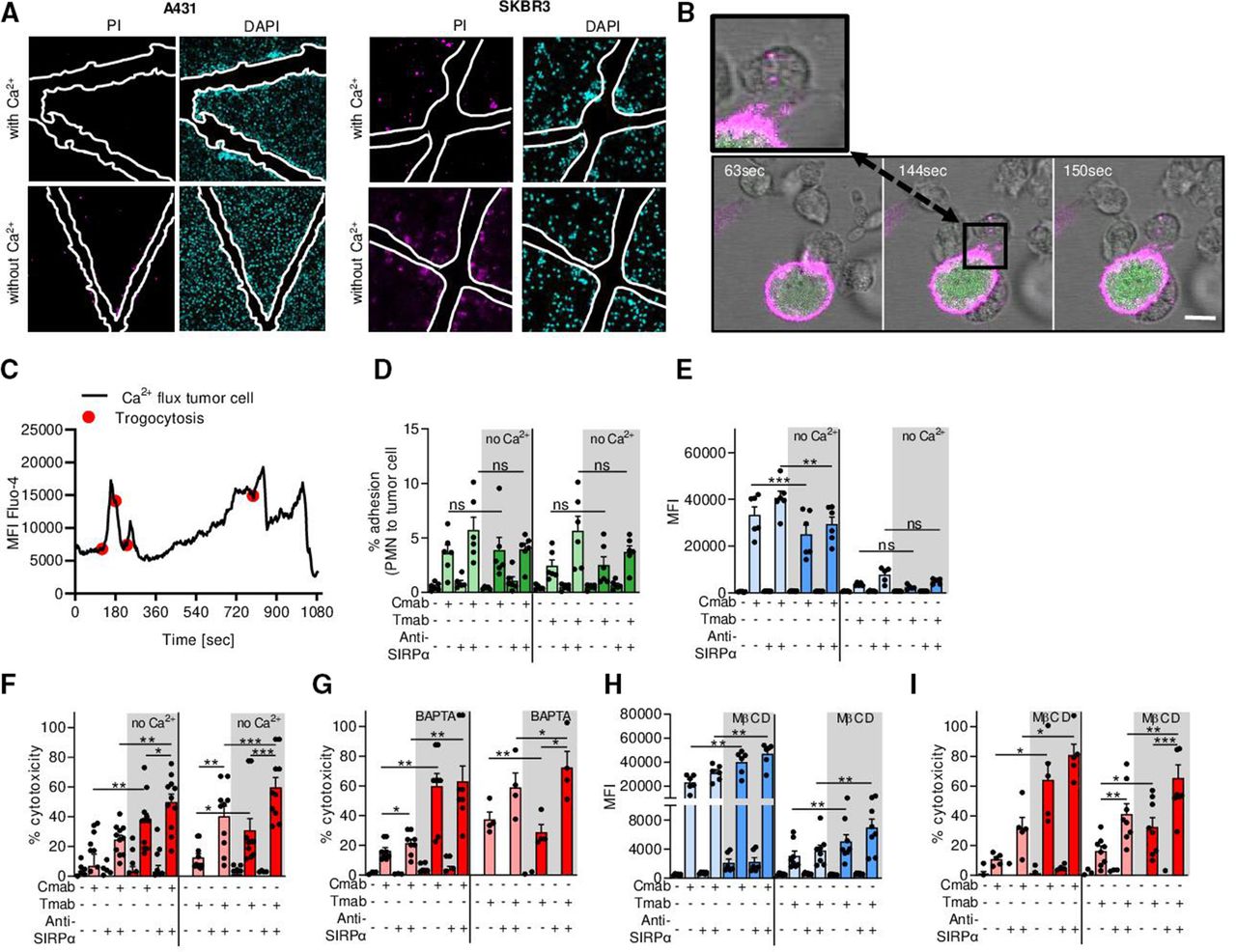
Cancer cells need extracellular Ca2+ for plasma membrane repair during neutrophil ADCC. (A) Scratch assay with A431 and SKBR3 cancer cells in the presence or absence of Ca2+. After scratching the cell monolayer, cells were treated with DAPI (cyan) to stain all nuclei and PI (magenta) to visualize cell death. The white outlines indicate the scratch area. (B) Live cell imaging of tumor cell Ca2+ influx during neutrophil trogocytosis. SKBR3 cells were labeled with DiO (magenta) and Fluo-4 (green). Live cell imaging shows neutrophils (unstained) interacting with tumor cells and taking up DiO+ tumor cell membrane (inset). The scale bar represents 15 µM and the time points are t=63, 144, and 150 s for frames 1, 2, and 3, respectively. (C) Quantification of live cell imaging from (B) shows that Ca2+ influx (shown by MFI of Fluo-4) follows neutrophil trogocytosis (indicated by red dots). (D–I) A431 cells (left) or SKBR3 cells (right) were opsonized with (+) or without (−) Cmab or Tmab, respectively. To interfere with inhibitory CD47–SIRPα interactions, anti-SIRPα antibodies were added to the neutrophils as indicated (+ or −). (D) Neutrophil–cancer cell conjugate formation (represented as the percentage of adhered neutrophils to cancer cells) with or without extracellular Ca2+. n=6 donors from two independent experiments. (E) Neutrophil trogocytosis with or without extracellular Ca2+. n=6 donors from three independent experiments for A431; n=5 donors from two independent experiments for SKBR3. (F) Neutrophil ADCC with or without extracellular Ca2+. n=11 donors from five independent A431 cell experiments and n=10 donors from five independent SKBR3 experiments. (G) Neutrophil ADCC in the presence of intracellular Ca2+ chelator BAPTA. n=8 donors from three independent experiments for431, n=4 donors from two independent experiments for SKBR3. (H) Neutrophil trogocytosis after pretreatment of target cells with MβCD. n=6 donors from three independent experiments for A431; n=8 donors from four independent experiments for SKBR3. (I) Neutrophil ADCC after pretreatment of target cells with MβCD. n=5 donors from three independent experiments for A431; n=8 donors from four independent experiments for SKBR3. Statistics: (D) ordinary one-way ANOVA with post hoc Sidak to correct for multiple comparisons; (E–I) repeated measures one-way ANOVA with post hoc Sidak to correct for multiple comparisons. *, p<0.05; **, p<0.01; ***, p< 0.001. ADCC, antibody-dependent cellular cytotoxicity; ANOVA, analysis of variance; Cmab, cetuximab; MβCD, methyl-β-cyclodextrin; MFI, mean fluorescence intensity; ns, not significant; PI, propidium iodide; PMN, polymorphonuclear cells (neutrophils); SIRPα, signal regulatory protein-α; Tmab, trastuzumab.
Conjugate formation using ImageStream
Using ImageStream flow cytometry, we quantified synapse formation between neutrophils and tumor cells, as previously described.1 Neutrophils were labeled for 30 min with Cell-Trace Calcein Violet-AM fluorescent dye (Invitrogen). Tumor cells were stained with CMTPX/cell-tracker red fluorescent dye (Invitrogen) for 30 min. Cells were washed and resuspended in the appropriate medium. Cells were coincubated at a target:effector ratio of 1:5 in the presence of the appropriate antibodies and reagents (described previously) at 37°C and 5% CO2. After 45 min, cells were fixed with 3.7% (w/v) PFA in PBS and samples were analyzed using the ImageStreamX flow cytometer (Amnis). Results were quantified using the IDEAS data analysis software (Amnis).
Quantification of trogocytosis using flow cytometry
Trogocytosis was measured using flow cytometry (Canto II and LSR II, BD) by analyzing the uptake of fluorescently labeled tumor cell membrane by the neutrophils. Tumor cells were labeled with lipophilic membrane dye (5 µM, Invitrogen) and opsonized with the appropriate therapeutic antibodies for the indicated conditions. Neutrophils were coincubated with the tumor cells in a 96-well round-bottom plate at a target:effector ratio of 1:5 at 37°C and 5% CO2. After 90 min, cells were fixed and measured on the flow cytometer. The neutrophil population was gated and evaluated on the uptake of tumor cell membrane by means of MFI.
Antibody-dependent cellular cytotoxicity
ADCC was measured in triplicates using the well-established 51Cr release assay.4 Tumor cells were labeled with 100 µCi 51Cr (PerkinElmer) for 90 min and afterwards washed and incubated with neutrophils or NK cells in a 96 round-bottom plates, in the presence of the appropriate antibodies and reagents (described previously). Neutrophil-mediated ADCC was performed at a target:effector ratio of 1:50; NK cell-mediated ADCC was performed at a target:effector ratio of 1:5. After 4-hour incubation, supernatant was harvested and radioactivity was measured in a gamma counter (Wallac) or a microbeta2 reader (PerkinElmer). Cytotoxicity was calculated as [(experimental release–spontaneous release)/(maximum release–spontaneous release)]×100%.
Imaging of lysosomal-associated membrane protein 1 (LAMP-1+) vesicles
2×104 SKBR3 cells were preseeded overnight in a 24-well plate on a glass coverslip and were labeled with DiO (ThermoFisher). After washing, tumor cells were coincubated with neutrophils at a target:effector ratio of 1:5 in complete IMDM medium for 2 hours at 37°C and 5% CO2. Trastuzumab was added where indicated. Cells were fixed with 3.7% (w/v) PFA and permeabilized with 0.1% triton (v/v)+0.5% (v/w) bovine serum albumin (BSA) in PBS blocking solution (2% BSA in PBS). After permeabilization, cells were stained with rabbit anti-LAMP-1 (Abcam) and secondary AF594 anti-rabbit antibody (Invitrogen). Vectashield including DAPI (Vector, H-1200) was used to mount the coverslips on the glass slides. The microscope images (Leica SP8 microscope) were quantified by measuring the intensity of LAMP-1 staining along the cross section of the tumor cell using ImageJ. The intensity of DiO labeling was used to determine the edges of the cell.
Western blot
1×106 SKBR3 WT, Scr, EXOC7KD and A431 WT, Scr, EXOC7KO and EXOC4KO cells were lysed in 100 µL complete Protease Inhibitor Cocktail (Roche) supplemented with 0.45 M EDTA diluted 1:1 with 2× sample buffer (25 mL Tris B; Invitrogen, 20 mL 100% glycerol; Sigma Aldrich, 5 g SDS; Serva, 1.54 g DTT; Sigma Aldrich, 20 mg bromophenol blue; Sigma Aldrich, 1.7 mL β-mercaptoethanol; BioRad in H2O Gibco) for 30 min at 95°C. 2.5×105 cells were ran on 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and proteins were transferred to nitrocellulose membrane (GE Healthcare Life Science). The membrane was blocked using 5% (v/v) skim milk powder (Campina, the Netherlands) for 1 hour at room temperature. Antibodies against EXOC7 (rabbit polyclonal, Abcam), EXOC4 (mouse clone 2E12, Millipore), and GAPDH (Millipore) were used. Secondary goat anti-mouse-IgG IRDye 800 or goat anti-rabbit-IgG IRDye 700 antibodies (LI-COR Biosciences) were used. Antibodies were incubated for 1 hour at room temperature and protein bands were visualized using Odyssey (LI-COR Biosciences).
Exocyst mRNA sequencing of tumor biopsies
RNA of 254 patients was sequenced as described by Fumagalli et al.18 Read pairs were trimmed using Trimmomatic.19 Alignment was performed using STAR.20 The number of reads mapping to each gene was then assessed with the R statistical software with the Rsamtools package. Gene expression levels were corrected for library batch effects using ComBat.21 All exocyst genes (EXOC1–EXOC8) were sufficiently well expressed for analysis (the least expressed was EXOC8, with an average of 19.5 reads per sample). The mean of all those genes was used as the signature of the exocyst complex (exocyst signature (sigExoc)). The signature value was compared with the pathological complete response (pCR) (defined as breast+axilla pCR) using two-sided Mann-Whitney U tests.
Data analysis and statistics
All statistical tests were analyzed using GraphPad Prism V.7 and V.8. Statistical differences between the two groups were tested by unpaired or paired t-tests; multiple comparisons were performed by one-way or repeated measures one-way analysis of variance with Sidak post hoc testing. The type of tests performed are indicated in the figure captions. Statistical significance is indicated with asterisks (*), where **** indicates p values of <0.0001; ***, p values of < 0.001; ** indicates p values of <0.01; and * indicates p values of <0.05. P values of > 0.05 were deemed not significant (ns).
Results
Multiple iterative neutrophil trogocytic events are needed to induce cancer cell death
Neutrophil trogocytosis of antibody-opsonized tumor cells can lead to cancer cell lysis, yet the exact mechanisms and kinetics leading to cancer cell death have remained largely unclear, to date. To investigate this, we followed the process of neutrophil trogocytosis through live cell imaging by labeling the membrane of HER2/neu+ SKBR3 breast cancer cells and opsonizing with the clinical anti-HER2/neu antibody trastuzumab. Neutrophils started to interact with the cancer cells 20 min after coincubation; after 25 min, neutrophils started becoming positive for the tumor cell membrane dye (indicated in green) (figure 1A, inset). After a subsequent 10 min and further accumulation of tumor cell membrane within the neutrophils, lysis of the tumor cell and subsequent nuclear staining (indicated by the white arrow) were observed (figure 1A). We quantified trogocytic events in relation to the occurrence of cell death for non-opsonized SKBR3 cells (figure 1B, left panel) and trastuzumab-opsonized SKBR3 cells (figure 1B, middle and right panels). We observed that for tumor cell death to occur, multiple iterative trogocytic events were required in a relatively short time frame of approximately 35 min. As shown in the middle panel of figure 1B, trogocytosis was not always followed by tumor cell death. Indeed, we found that while most neutrophils could successfully take up pieces of tumor cell membrane, a significant amount of neutrophil–tumor cell interactions were needed for tumor cell lysis to occur (figure 1C). Together, this indicates that tumor cells are able to resist neutrophil trogoptosis up to a certain threshold.
Cancer cells need extracellular Ca2+ for plasma membrane repair during neutrophil ADCC
Plasma membrane repair is a vital process in the lifetime of any cell, including tumor cells, and is entirely dependent on the presence of extracellular Ca2+.8 22 To test our hypothesis that tumor cell membrane repair helps withstand neutrophil ADCC, we first explored plasma membrane repair in A431 carcinoma and SKBR3 breast cancer cell lines using a scratch assay. For the repair of such a mechanically induced wound, the presence of extracellular Ca2+ was crucial: in the absence of extracellular Ca2+, cells were not able to repair the damage. Especially at the borders of the area of the scratch (indicated in white), the injured cells became positive for the DNA-binding dye PI, indicative of cell death (figure 2A). Next, using live cell imaging, we explored the kinetics of Ca2+ influx into tumor cells during neutrophil trogocytosis. We labeled the membrane of trastuzumab-opsonized SKBR3 cells with DiO (shown in magenta) and used Fluo-4 (shown in green) as an intracellular Ca2+ indicator to measure and quantify the intensity of the Ca2+ influx over time (figure 2B,C). We observed that temporal rises in intracellular Ca2+ (indicated by intensity of Fluo-4 staining in figure 2B) directly followed trogocytic events indicated with the red dots in the quantification in figure 2C. When neutrophils did not interact with opsonized tumor cells, intracellular Ca2+ remained stable over time (online supplemental figure 1).
Supplemental material
To investigate the necessity of Ca2+ for tumor cell membrane repair during neutrophil trogocytosis and killing, we performed flow cytometry-based trogocytosis assays and ADCC experiments in the absence of extracellular Ca2+. Neutrophil ADCC requires initial conjugate formation with the tumor cells, which precedes neutrophil trogocytosis. This interaction is further enhanced by interfering with CD47–SIRPα interactions.1 2 To evaluate the conjugate formation in the presence or absence of extracellular Ca2+ between neutrophils and antibody-opsonized cancer cells, we used EGFR+ epidermoid carcinoma A431 cells and HER2/neu+ breast cancer SKBR3 cells opsonized with cetuximab or trastuzumab, respectively. We observed that both the synapse formation and trogocytosis were fully dependent on antibody opsonization, and the absence of extracellular Ca2+ did not increase the amount of conjugates nor the extent of trogocytosis (figure 2D,E). However, neutrophil-mediated ADCC of both SKBR3 and A431 cells was significantly enhanced in the absence of extracellular Ca2+ (figure 2F). Like trogocytosis, killing occurred only in opsonized conditions and was further enhanced after CD47–SIRPα blockade using anti-SIRPα antibodies (figure 2F). Similar results were obtained by preincubating the tumor cells with BAPTA, a cell-permeant chelator of intracellular Ca2+ (figure 2G).
Another way to impair plasma membrane repair is to manipulate the lipid composition of the plasma membrane by extracting cholesterol from the cancer cell membrane.23 Preincubating tumor cells with MβCD showed that neutrophil trogocytosis increased only modestly, whereas the killing of the tumor cells was again significantly enhanced (figure 2H,I). Together, these results show that while in the absence of Ca2+ the quantity of neutrophil trogocytosis remains largely unaffected, the efficacy of trogocytosis, hence the induction of target cell death, trogoptosis, increases. This indicates a crucial role for Ca2+ in tumor membrane cell repair during neutrophil ADCC.
The exocyst complex repairs plasma membrane mechanical damage after neutrophil trogocytosis
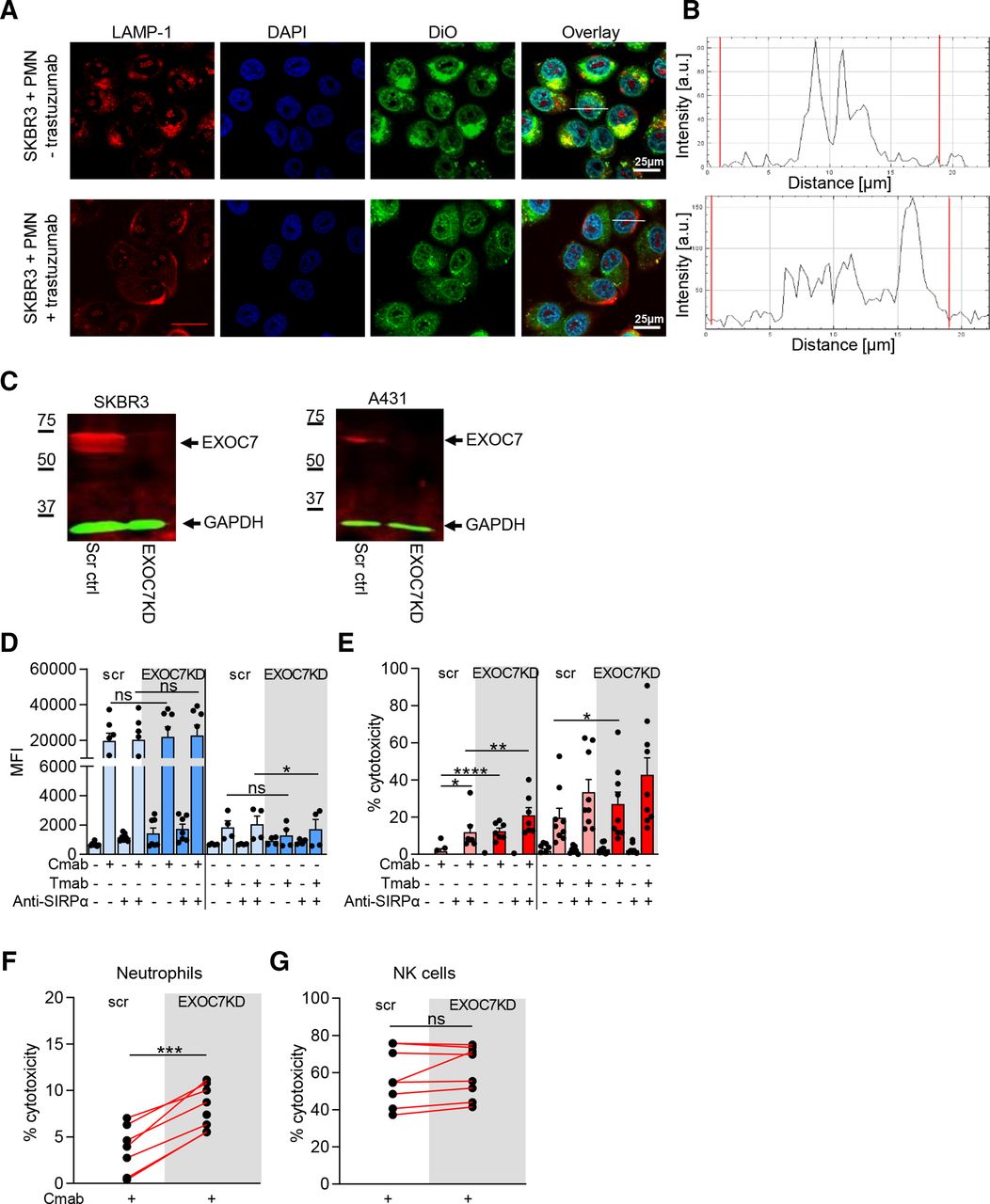
Membrane repair involves the recruitment of intracellular vesicles, including LAMP-1+-containing lysosomes, to the site of injury. Fusion of these lysosomes with the plasma membrane seals the lesion, and this can be detected by the presence of LAMP-1 on the cell surface.24 25 To investigate whether LAMP-1 translocates to the cancer cell plasma membrane after neutrophil trogocytosis, we incubated neutrophils with SKBR3 cells in the presence or absence of trastuzumab. We found that in antibody-opsonized conditions, LAMP-1 translocated towards the membrane of the tumor cells, as shown in the microscopy images (figure 3A) and the quantified results thereof (figure 3B). LAMP-1 translocation did not occur in the absence of trastuzumab (figure 3A,B) or in conditions without neutrophils (online supplemental figure 2A,B), indicating that antibody-dependent neutrophil-tumor cell interactions lead to LAMP-1 translocation to the plasma membrane.

The exocyst complex repairs plasma membrane mechanical damage after neutrophil trogocytosis. (A) Confocal microscope images of SKBR3 cells incubated with neutrophils (+PMN) and opsonized with (+) or without (−) Tmab. Cells were stained with anti-LAMP-1 (red), DAPI (blue) and membrane dye DiO (green). (B) LAMP-1 fluorescence intensity across the cross section of a Tmab-opsonized tumor cell (white line in overlay image, A) after neutrophil coincubation. The red lines indicate the edge of the cell, as determined using DiO fluorescence. (C) Representative western blot of EXOC7 in SKBR3 (left) and A431 (right) scr ctrl and EXOC7KD cells. GAPDH expression was used as loading ctrl. (D,E) A431 cells (left) or SKBR3 cells (right) were opsonized with (+) or without (−) Cmab or Tmab, respectively. To interfere with inhibitory CD47–SIRPα interactions, anti-SIRPα antibodies were added to the neutrophils as indicated (+ or −). (D) Neutrophil trogocytosis of scr and EXOC7KD cells. n=7 donors from three independent experiments for A431; n=4 donors from two independent experiments for SKBR3. (E) Neutrophil ADCC of scr and EXOC7KD cells. n=7 donors from three independent experiments for A431; n=9 donors from five independent experiments for SKBR3. (F) Neutrophil ADCC and (G) NK cell ADCC of scr ctrl and EXOC7KD A431 cells opsonized (+) with Cmab. n=8 donors from three independent experiments. Statistics: (D,E) repeated measures one-way ANOVA with post-hoc Sidak to correct for multiple comparisons, (F,G) paired t-test; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. ADCC, antibody-dependent cellular cytotoxicity; ANOVA, analysis of variance; Cmab, cetuximab; ctrl, control; EXOC7KD, EXOC7 shRNA-mediated knockdown; LAMP-1, lysosomal-associated membrane protein 1; MFI, mean fluorescence intensity; NK, natural killer; ns, not significant; scr, scrambled; Tmab, trastuzumab.
We hypothesized that neutrophils tearing off pieces of tumor cell membrane causes mechanical damage to the tumor plasma membrane. It was recently shown that the exocyst complex is responsible for repairing, in particular, mechanical wounds of the plasma cell membrane.13 The human exocyst complex consists of eight proteins (EXOC1, EXOC2, EXOC3, EXOC4, EXOC5, EXOC6, EXOC7, and EXOC8) and mediates the docking and fusion of lysosomal vesicles to the plasma membrane. Of these proteins, EXOC7 (mouse Exo70) and EXOC1 (mouse Sec3) dock the assembled exocyst complex carrying the lysosomal vesicle to the inner plasma membrane by binding to phospholipids.26 To explore the role of the exocyst complex during human tumor cell membrane repair in the context of neutrophil trogocytosis, we generated a knockdown of EXOC7 in SKBR3 and A431 cells using shRNA (hereafter referred to as SKBR3 EXOC7KD and A431 EXOC7KD, respectively) (figure 3C and online supplemental figure 2C,D). SKBR3 and A431 EXOC7KD cells showed similar HER2/neu, EGFR or CD47 expression compared with the respective scrambled cells (online supplemental figure 3A). Neutrophil trogocytosis of both SKBR3 as A431 EXOC7KD cells was similar, or even somewhat lowered, when compared with scrambled control cells (figure 3D). In contrast, ADCC was significantly enhanced in both EXOC7KD cancer cell lines compared with scrambled control, implying that cancer cells need EXOC7 to resist neutrophil-mediated killing (figure 3E). To further investigate the exocyst complex and to specifically demonstrate the mechanical nature of neutrophil ADCC, we compared the killing of A431 EXOC7KD cells by either neutrophils or NK cells from the same donor. NK cell-mediated ADCC is induced by pore-forming toxins (perforin) that allow entry of granzyme B into the targeted cell to induce apoptosis. It was previously shown that the exocyst complex is not involved in repairing damage inflicted by pore-forming toxins.13 Indeed, we showed that NK cell ADCC, as opposed to neutrophil ADCC, was not enhanced in the EXOC7KD cells (figure 3F,G). This emphasizes the distinctive mechanical effector mechanism of neutrophil killing and demonstrates that repair of mechanically damaged tumor cell membrane involves the exocyst complex.
Both exocyst components EXOC7 and EXOC4 are important in resisting neutrophil ADCC
Among the proteins consisting the exocyst complex, Sec8 (human EXOC4) is shown to be crucial in tethering of the secretory vesicles to the plasma membrane.15 Using CRISPR-Cas9, we aimed to knock out human EXOC4 in A431 cells, another exocyst complex component. We reached an efficient and stable reduction of the EXOC4 protein (hereafter referred to as A431 EXOC4KD; figure 4A and online supplemental figure 2E) and observed a somewhat lowered EGFR but similar CD47 expression in the absence of EXOC4 (online supplemental figure 3B). With these A431 EXOC4KD cells, we obtained similar results as shown in figure 3: neutrophil ADCC was specifically enhanced in A431 EXOC4KD cells compared with scrambled control, confirming the importance of at least two separate components of the exocyst complex in resisting neutrophil killing through target cell membrane repair (figure 4B). We also aimed to generate an EXOC7 knockout in A431 cells using CRISPR-Cas9. This resulted in an in-frame deletion of 102 bp that led to a version of the EXOC7 protein (referred to as A431 EXOC7Δ157–190 cells), lacking a part covering amino acids 157–190, as identified by DNA sequencing (figure 4C and online supplemental figure 2F). This amino acid stretch encompasses α-helical region 4 (H4) and the loop between H4 and H5 located within the N-terminal region of the molecule that contains a binding site for the GTPase TC10.27 The expression of CD47 and EGFR was similar to the scrambled control A431 cells (online supplemental figure 3C). Similar to A431 EXOC7KD cells, trogocytosis of A431 EXOC7Δ157–190 cells was not enhanced (figure 4D). However, neutrophil ADCC was significantly increased compared with scrambled control A431 cells, both in presence and absence of CD47–SIRPα interactions, where the latter increased ADCC even further (figure 4E). As shown in figure 2, cancer cell plasma membrane repair relies heavily on the presence of extracellular Ca2+. If the exocyst complex would be the main mechanism that is active during the repair of mechanical wounds, absence of extracellular Ca2+ should not further enhance neutrophil-mediated ADCC of EXOC7Δ157–190 or EXOC4KD A431 cells. Indeed, depletion of extracellular Ca2+ did not significantly enhance ADCC of mutant EXOC7 or knocked down EXOC4 A431 cells. Lack of Ca2+, however, did increase the killing of A431 scrambled control cells (figure 4F), as demonstrated in figure 1F. Together, this demonstrates that the exocyst complex is the primary Ca2+-dependent repair mechanism in these antibody-opsonized solid cancer cells during neutrophil trogocytosis.

Both exocyst components EXOC7 and EXOC4 are important in resisting neutrophil ADCC. (A) Representative Western blot of EXOC4 in A431 scr control and EXOC4KD cells. GAPDH was used as loading control. (B) Neutrophil ADCC of A431 scr and EXOC4KD cells. Cmab and anti-SIRPα were added as indicated (+/-). n=9 donors from five independent experiments. (C) Representative Western blot of EXOC7 in A431 scr control and EXOC7Δ157–190 cells. Tubulin was used as loading control. (D) Neutrophil trogocytosis of A431 scr and EXOC7Δ157–190 cells. Cmab and anti-SIRPα were added as indicated (+/−). n=4 donors from two independent experiments. (E) Neutrophil ADCC of A431 scr and EXOC7Δ157–190 cells. Cmab and anti-SIRPα were added as indicated (+/−). n=10 donors from five independent experiments. (F) Neutrophil ADCC of A431 scr (left), EXOC7Δ157–190 (middle) and EXOC4KD cells (right) with or without extracellular Ca2+. Cmab and anti-SIRPα were added as indicated (+/−). n=3–5 donors from three independent experiments. Statistics: (B,F) ordinary one-way ANOVA with post hoc Sidak to correct for multiple comparisons, (D,E) Repeated measures one-way ANOVA with post hoc Sidak to correct for multiple comparisons. *, p<0.05; **, p<0.01; ****, p<0.0001. ADCC, antibody-dependent cellular cytotoxicity; ANOVA, analysis of variance; Cmab, cetuximab; ctrl, control; EXOC4KD, EXOC4 knockdown; ns, not significant; scr, scrambled; SIRPα, signal regulatory protein-α.
EXOC7 mRNA expression is related to the clinical response of trastuzumab-treated patients with breast cancer
We have shown until now that the exocyst complex—by means of its components, EXOC7 and EXOC4—play an essential role in resisting neutrophil-mediated killing through trogocytosis. Downregulation of EXOC7 or EXOC4 resulted in improved antibody-dependent neutrophil cytotoxicity toward cancer cells. However, whether the exocyst complex is also of importance and active during antibody therapy in cancer in vivo is unknown.
Using data from the NeoALTTO study (NCT00553358),18 28 we investigated the expression of exocyst complex mRNA (hereafter referred to as sigExoc) in correlation to the pCR of patients with breast cancer subjected to neoadjuvant treatment with trastuzumab (figure 5A) or the HER1/HER2 tyrosine kinase inhibitor lapatinib (figure 5B), the latter of which only causes direct growth inhibitory effects via HER1/HER2 and does not induce any immune cell-mediated killing. pCR for patients treated with combination of lapatinib and trastuzumab was not correlated with decreased exocyst mRNA expression. Together, our analysis indicates that in contrast to lapatinib, trastuzumab-treated patients with a pCR had significantly lower exocyst complex mRNA levels as compared with the patients with no pCR. These results imply an inverse relationship between exocyst complex mRNA levels and the clinical response to trastuzumab and provides further support for the contribution of Fc-receptor bearing cells including neutrophils, but also their lytic cytotoxic mechanism (ie, trogoptosis) during antibody therapy in cancer.

EXOC7 mRNA expression is related to the clinical response of Tmab-treated patients with breast cancer. Boxplots showing the comparison between the mean gene expression of the exocyst complex (sigExoc, left panel) and pCR in (A) Tmab-treated patients with breast cancer or (B) lapatinib-treated patients with breast cancer. Data are derived from 256 patients and the signature value was compared with the pCR (defined as breast+axilla pCR) using two-sided Mann-Whitney U tests. pCR, pathological complete response; sigExoc, exocyst signature; Tmab, trastuzumab.
Discussion
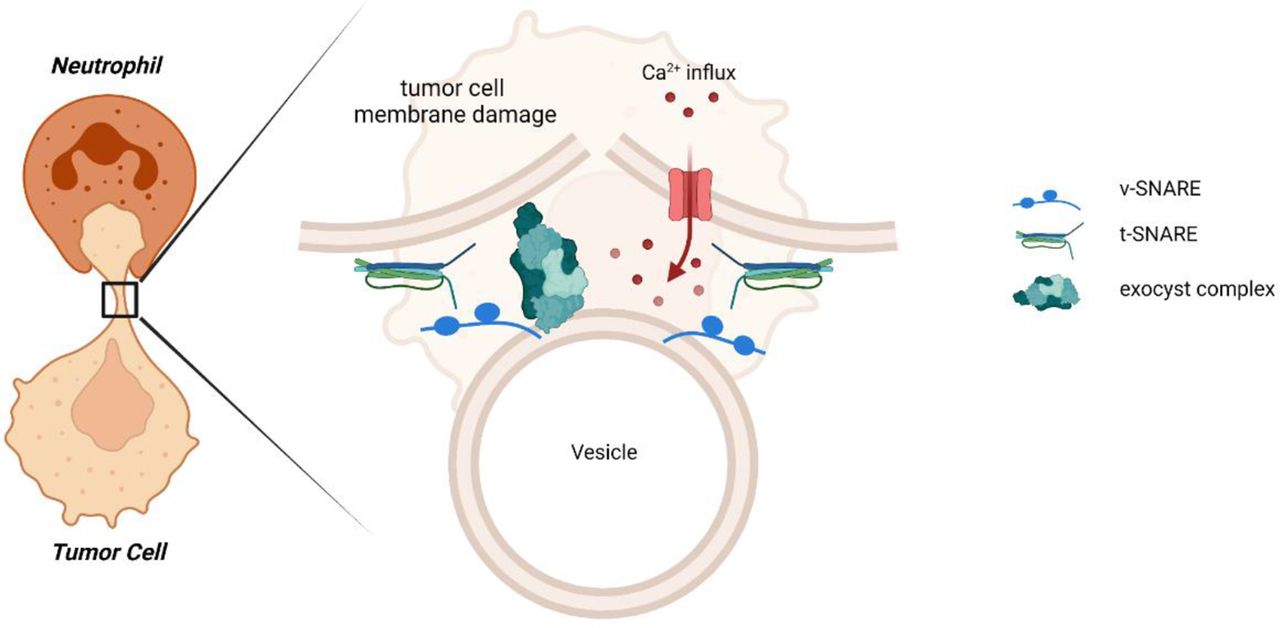
The mechanism by which neutrophils perform ADCC has for long been unclear; however, it has recently been discovered that neutrophils trogocytose antibody-opsonized tumor cells. Trogocytosis has been primarily known as a way of active membrane transfer between cells, but in the context of neutrophils, trogocytosis involves the mechanical disruption of the tumor cell membrane, resulting in a necrotic type of tumor cell death, which we previously coined trogoptosis.1 As we have demonstrated in this and previous studies, tumor cell killing is not a corollary of trogocytosis per se; in this study, we show that a certain number of sequential trogocytic events appear to be required in order for tumor cell death to occur. This indicates that tumor cells may resist neutrophil trogoptosis up to a certain threshold. Identification of the mechanisms that aid tumor cells to withstand neutrophil-mediated attack not only may enhance the efficacy of neutrophil antitumor therapies but also may reveal novel strategies to overcome tumor cell resistance. We hypothesized that tumor cells can resist neutrophil trogoptosis by inducing plasma membrane repair. For this, we looked into tumor cell membrane repair during neutrophil-mediated tumor cell destruction. By investigating the specifics of membrane repair, such as the universal requirement for extracellular Ca2+ and the contributions of EXOC7 and EXOC4, that is, essential components of the exocyst complex repair machinery, we were able to pinpoint the pivotal role of the exocyst complex, which repairs mechanical damage specifically (figure 6). These results not only reveal a way in which tumor cells resist neutrophil trogoptosis but also provide further support for the mechanical nature of neutrophil-mediated cytotoxicity that we previously proposed.1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
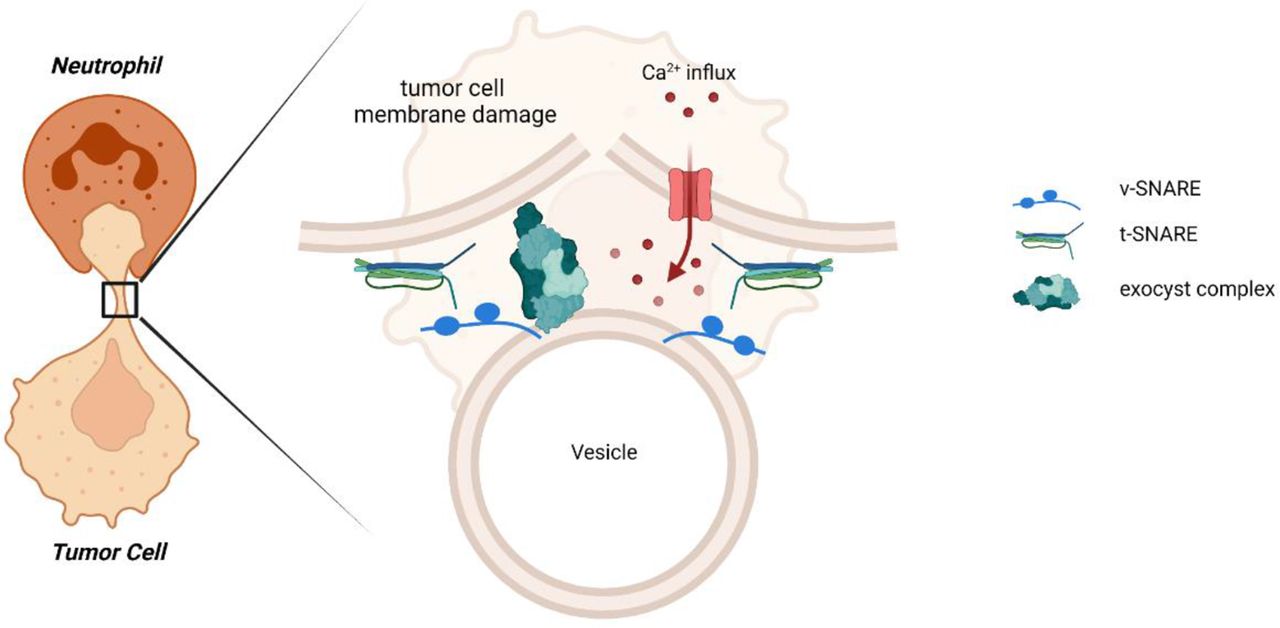
Tumor cells resist neutrophil-mediated killing by repairing their plasma membrane using the exocyst complex. In this report, we showed that tumor cells can escape neutrophil-mediated trogoptosis by Ca2+-dependent plasma membrane repair, both in vitro and in vivo. This injury of a mechanical nature induces an active repair mechanism in tumor cells, facilitated by the exocyst complex. The exocyst mediates the tethering and fusion of intracellular vesicles to the site of plasma membrane injury after neutrophil trogocytosis. t-SNARE, target-soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor; v-SNARE, vesicle soluble NSF attachment protein receptor .
First, we demonstrated the importance of Ca2+ with regard to tumor cell resistance against neutrophil killing. Depletion of Ca2+ enhanced neutrophil ADCC of cancer cells, while synapse formation and trogocytosis remained unchanged or in some cases even lowered. This indicates that the outcome of neutrophil trogocytosis, trogoptosis, is improved, either due to a strengthened neutrophil attack or a weakened resistance of tumor cells. We found that it was the weakened resistance that could explain the results best. Plasma membrane repair is vital for tumor cells to resist neutrophil trogoptosis, as was further supported by using MβCD, which depletes cholesterol from the membrane and impairs plasma membrane repair. Under these cholesterol-depleted conditions, tumor cell killing was enhanced as opposed to non-depleted conditions. This, combined with the visualization of LAMP-1+ intracellular vesicles translocating to the tumor cell plasma membrane on neutrophil incubation, demonstrated that tumor cell membrane repair acted downstream of the neutrophil trogocytosis process.
Knocking down EXOC7 and EXOC4 in different tumor cell lines, components of the exocyst complex which mediate membrane repair, demonstrated that tumor cells partially resist killing by repairing the plasma membrane lesions inflicted by neutrophils. These results are consistent with those of Fernandes et al, who demonstrated that the exocyst complex is required for the repair of Trypanosoma cruzi-induced plasma membrane lesions that are also of a mechanical nature.13 This finding was further confirmed using NK cells, ADCC of which occurs through the pore-forming perforin and granzymes expelled from the NK cell cytotoxic granules on FcR triggering. In this case, ADCC of EXOC7KD cells was not enhanced, signifying the mechanical nature of neutrophil trogoptosis and the specific conditions during which the exocyst complex mediates plasma membrane repair.
In absence of antibody opsonization, tumor cells were not killed by neutrophils, not even under conditions without Ca2+, EXOC7 or EXOC4, illustrating the specificity of neutrophil-mediated killing with regard to antibody opsonization. Moreover, our A431 EXOC4KD cells expressed lower EGFR compared with wild-type cells, but this can obviously not explain the higher neutrophil ADCC observed with these cells, which instead appears positively correlated to EGFR expression. Furthermore, neutrophil ADCC has been shown to be significantly inhibited by CD47–SIRPα interactions.4 29 Indeed, blocking this interaction using SIRPα blocking antibodies enhanced tumor cell killing. We continued to observe this enhancement even in the absence of extracellular Ca2+ (ie, by depletion or chelation) or in the absence of (intact) EXOC7 or EXOC4 in tumor cells. We recently identified the negative regulation of SIRPα on CD11b/CD18 integrin activation.2 Although the specific ligand(s) of CD11b/CD18 integrins during neutrophil ADCC remain unknown and possibly differ per tumor cell type, combination of impaired exocyst-mediated membrane repair and stronger neutrophil-to-tumor cell engagement due to SIRPα blockade could still further enhance neutrophil anticancer efficacy.
The exocyst complex mediates the exocytosis and tethering of secretory vesicles and is composed of eight proteins. Of these, Exo70 was first discovered in Saccharomyces cerevisiae30 and the crystal structure of Mus musculus and S. cerevisiae Exo70 revealed four domains (A–D) containing 19 α-helices.31–33 Our genetic modification of A431 tumor cells using CRISPR-Cas 9 led us to the discovery of a functional domain (amino acids 157–190) of EXOC7, the deletion of which mimicked the effect of Ca2+ depletion or EXOC7 or EXOC4 knockdown during neutrophil ADCC. As there is little knowledge of this region in human EXOC7, we can only speculate regarding its function. Based on the crystal structures of M. musculus Exo70, these missing amino acids comprise α-helix H4 completely and parts of α-helices H3 and H5, all located near the N-terminus.31 Exo70 is known to interact with different GTPases such as Rho3, Cdc42,34 35 as well as TC1027 and is thought to, together with Sec3, mark the site for recruitment of the other exocyst components.36 However, interaction with Rho3 and Cdc42 occurs around the C-terminal region, as is the site where Exo70 binds to the plasma membrane.26 34 37 Cryo-EM analysis and modeling of the whole exocyst complex suggests that the individual exocyst components bind in pairs using their N-terminal domains that are made up of long coiled coils that neatly intertwine. Exo70 pairs with Exo84 (human EXOC8), and all pairs subsequently assemble into the exocyst complex.38 Although our EXOC7Δ157–190 protein lacks a fragment of 150 amino acids away from the N-terminus, it might well be that the N-terminal coiled coil is disturbed, resulting in attenuated exocyst complex formation, even though EXOC7 is able to bind to the plasma membrane. Further studies into this, or the other EXOC7 domains during plasma membrane repair after neutrophil-mediated killing, could pave the way to understand and perhaps inhibit the function of the exocyst complex specifically in tumor cells during antibody therapy.
Finally, we demonstrated the clinical importance of the exocyst complex during antibody therapy in vivo. Using RNA expression data from patients with breast cancer, we showed that levels of exocyst complex mRNA were inversely related to a pCR when patients received neoadjuvant treatment with trastuzumab. For patients treated with the HER1/HER2 tyrosine kinase inhibitor lapatinib, differences in exocyst mRNA expression were not indicative for clinical outcome, in agreement with lapatinib typically interfering with HER2/neu and EGFR signaling pathways and not inflicting any type of mechanical or neutrophil-mediated (or any other type of immune-mediated) damage, which we did observe in the HER2/neu-directed trastuzumab-treated cohort, suggesting mechanical injury as a potential mechanism involved. pCR for patients treated with combination of lapatinib and trastuzumab was not correlated with decreased exocyst mRNA expression. Although in general the effect of combination of lapatinib+trastuzumab relates to higher pCR rates compared with either trastuzumab of lapatinib alone,39 the mechanism behind this synergy is not fully elucidated until now. Suggested mechanisms include an enhanced induction of apoptosis by a synergistic effect on HER2 signaling by the combination of lapatinib and trastuzumab, or an increased trastuzumab-induced ADCC when combined with lapatinib.18 28 In the latter case, the combination of both anti-HER2 drugs might shift the contribution of distinct immune cell compartments and overshadow the neutrophil-induced effect of trastuzumab. Also, we cannot fully exclude the contribution of macrophages in these findings: many immune cells, including NK cells and macrophages, can trogocytose.40 Here, trogocytosis is mainly used to mediate membrane transfer (to obtain new cell surface characteristics or to remove antigens from target cell membrane, often a immunomodulatory mechanism41 42) and has been described to result in the loss of target antigen and a consequential escape from antibody-dependent cell-mediated cytotoxicity.43 44 Human macrophages have been described to be capable of mediating killing through trogocytosis45 and via this mechanism could contribute to the exocyst-related outcomes in the trastuzumab-treated patient group, but since macrophages induce killing primarily via phagocytosis and not trogocytosis, we believe the contribution of neutrophils might dominate in this particular setting.
The contribution of neutrophils has been suggested also in other studies showing correlations between certain phagocyte-expressed FcγRIIa polymorphisms and immunotherapy treatment outcome.46–48 However, in contrary to the aforementioned studies, patients in the NeoALTTO study were subjected to neoadjuvant treatment with trastuzumab combined with paclitaxel, which, in contrary to many treatment regiments with heavy chemotherapeutics, preserves patients’ immunocompetence. This combination may therefore have provided more optimal circumstances to demonstrate the neutrophil contribution to the outcome of antibody treatment in patients with breast cancer.
Collectively, this study shows that while neutrophils can kill solid tumor cells, tumor cells consistently resist trogoptosis by means of plasma membrane repair. Our results demonstrate that neutrophil killing efficacy can be further improved with a two-hit punch: render tumor cells more vulnerable toward trogoptosis and relieving inhibition in neutrophils through SIRPα blockade. We also show that this involves the exocyst complex, which counteracts the mechanically inflicted damage to the tumor cell plasma membrane and appears also relevant during antibody therapy in patients with breast cancer.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Informed consent and approval were obtained from the healthy donors and the Sanquin Research institutional ethical committee, respectively.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DJvR and PB contributed equally to this paper (joint first authorship). HLM, DJvR, and PB contributed to the design and execution of the experiments and the design and evaluation of the data. MvH, PJHV, BK, ATJT, and KS performed experiments. DV, CSo, SE-A, MI, SG, CSa, SDC, JH, RR, and S-BK provided the patient material and clinical analysis. HLM, RvB, TWK, and TKvdB supervised the research. DJvR, PB, and HLM wrote the manuscript.
Funding This work was supported by the Dutch Cancer Society (grant numbers 10300 and 11537, awarded to TKvdB and HLM, respectively).
Competing interests TKvdB is the inventor of patent EP2282772, owned by Stichting Sanquin Bloedvoorziening, entitled ‘Compositions and Methods to Enhance the Immune System’, which describes targeting CD47–SIRPα interactions during antibody therapy in cancer.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.