Article Text

Abstract
Introduction Immuno-oncology therapies, including immune checkpoint inhibitors (ICIs), have transformed cancer care and have brought into question whether classic oncology efficacy assessments adequately describe the distinctive responses observed with these agents. With more ICI-based therapies being approved across multiple tumor types, it is essential to define unique clinical hallmarks of these agents and their associated assessments to better reflect the therapeutic impact for both patients and physicians. Long-term survival and objective responses, such as depth and durability of responses, treatment-free survival, efficacy in brain metastases, improved health-related quality of life, and unique safety profiles, are among the hallmarks that have emerged for ICI therapies. An established clinical hallmark is a sustained long-term survival, as evidenced by a delayed separation of Kaplan-Meier survival curves, and a plateau at ~3 years. Combination ICI therapies provide the opportunity to raise this plateau, thereby affording durable survival benefits to more patients. Deepening of responses over time is a unique clinical ICI hallmark, with patients responding long term and with more durable complete responses. Depth of response has demonstrated prognostic value for long-term survival in some cancers, and several ICI studies have shown sustained responses even after discontinuing ICI therapy, offering the potential for treatment-free intervals. Although clinical evidence supporting efficacy in brain metastases is limited, favorable ICI intracranial responses have been seen that are largely concordant with extracranial responses. While patient outcomes can be significantly improved with ICIs, they are associated with unique immune-mediated adverse reactions (IMARs), including delayed ICI toxicities, and may require multidisciplinary management for optimal care. Interestingly, patients discontinuing ICIs for IMARs may maintain responses similar to patients who did not discontinue for an IMAR, whether they restarted ICI therapy or not.
Conclusion Herein, we comprehensively review and refine the clinical hallmarks uniquely associated with ICI therapies, which not only will rejuvenate our assessment of ICI therapeutic outcomes but also will lead to a greater appreciation of the effectiveness of ICI therapies.
- immunotherapy
- CTLA-4 antigen
- programmed cell death 1 receptor
- drug therapy
- combination
- review
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immuno-oncology (I-O) therapy has revolutionized cancer treatment and has led to clinical responses unique to this class of agents. Contemporary I-O treatments include a range of therapies, such as immune checkpoint inhibitors (ICIs) and chimeric antigen receptor T-cell immunotherapies.1 With the discovery of checkpoint receptors involved in immune response regulation,2 ICIs have changed the landscape of immune activation and cancer treatment. Single-agent ICIs, including inhibitors of cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death-1 (PD-1), and programmed death ligand 1 (PD-L1), have demonstrated significant antitumor activity and are being combined with other agents to increase clinical benefit.3 4
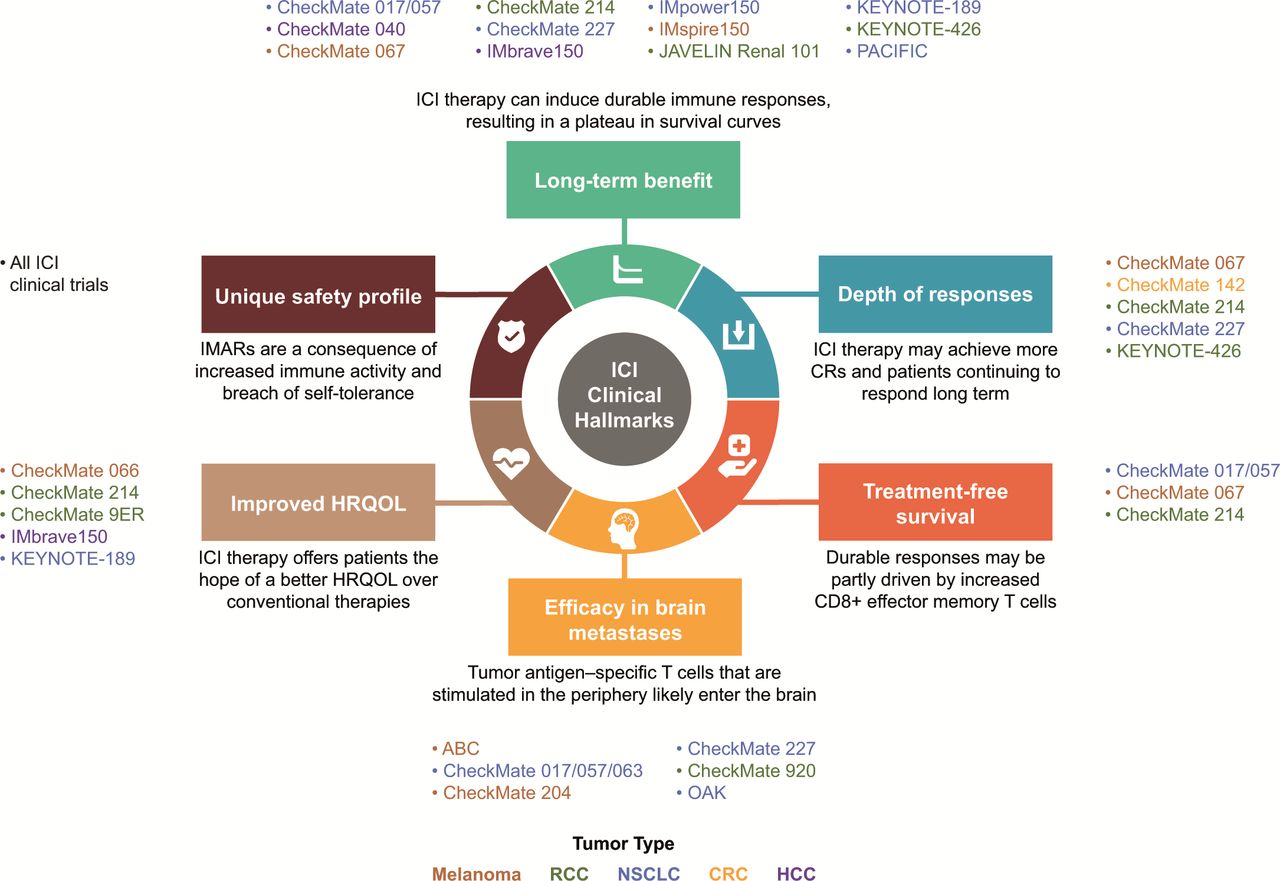
With the first ICI clinical trials of the CTLA-4 inhibitor ipilimumab in melanoma,5 clinical hallmarks have emerged that are unique to ICI therapies (online supplemental graphical abstract, figure 1). Understanding clinical benefits associated with ICI now goes beyond traditional outcomes of survival and objective responses, though overall survival (OS) remains the gold standard endpoint.6 Other long-term measures, such as depth of response (DepOR),7 treatment-free survival (TFS),8 9 efficacy in brain metastases,10–15 improved health-related quality of life (HRQOL),16 and durable responses, have evolved as additional hallmarks associated with ICI-based therapies, with the possibility for a cure in some patients.5
Supplemental material
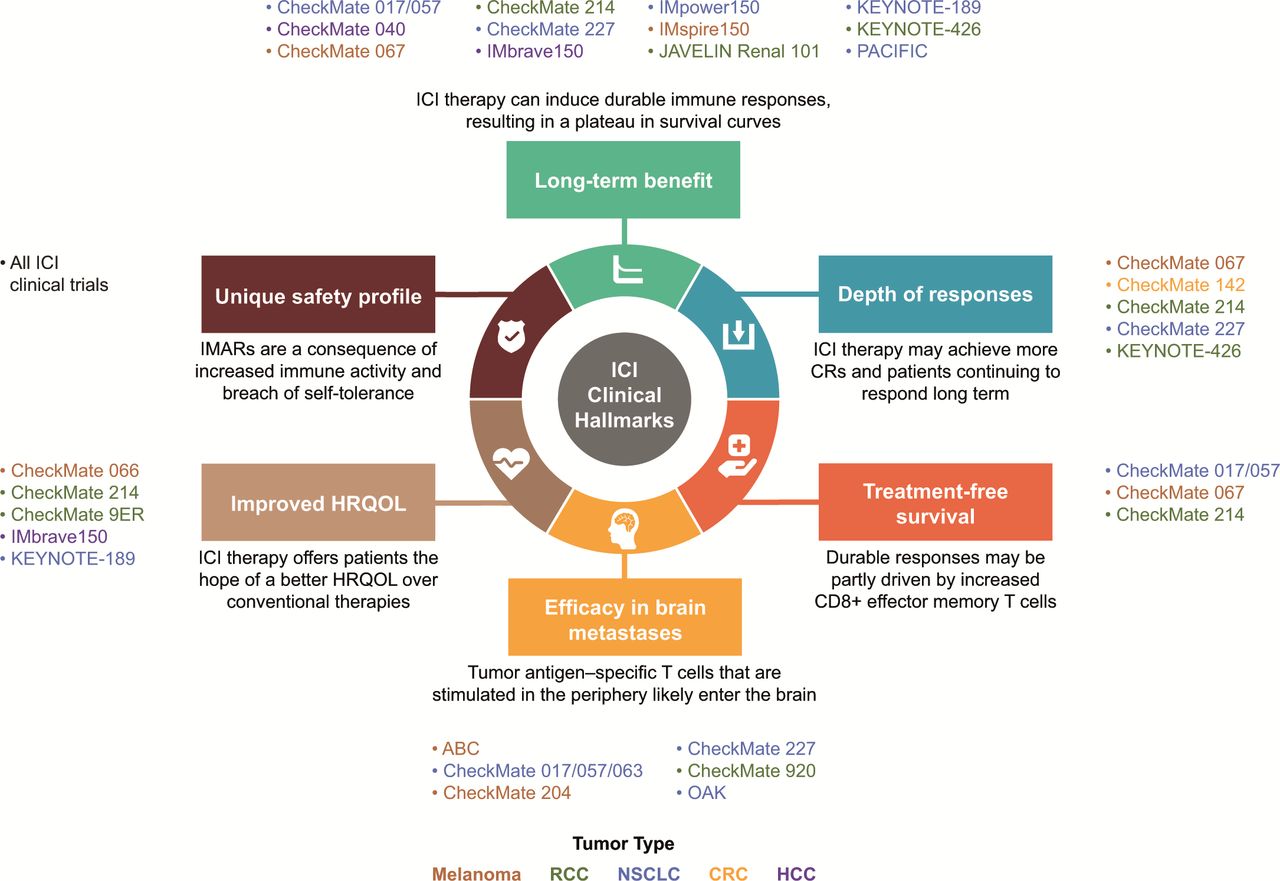
Clinical hallmarks of ICI-based therapies and representative studies.9 10 12 36 41 42 44 50 51 57 58 66 67 77 81 82 87 88 92 93 137–154 CR, complete response; CRC, colorectal cancer; HCC, hepatocellular carcinoma; HRQOL, health-related quality of life; ICI, immune checkpoint inhibitor; IMAR, immune-mediated adverse reaction; NSCLC, non-small cell lung cancer; RCC, renal cell carcinoma.
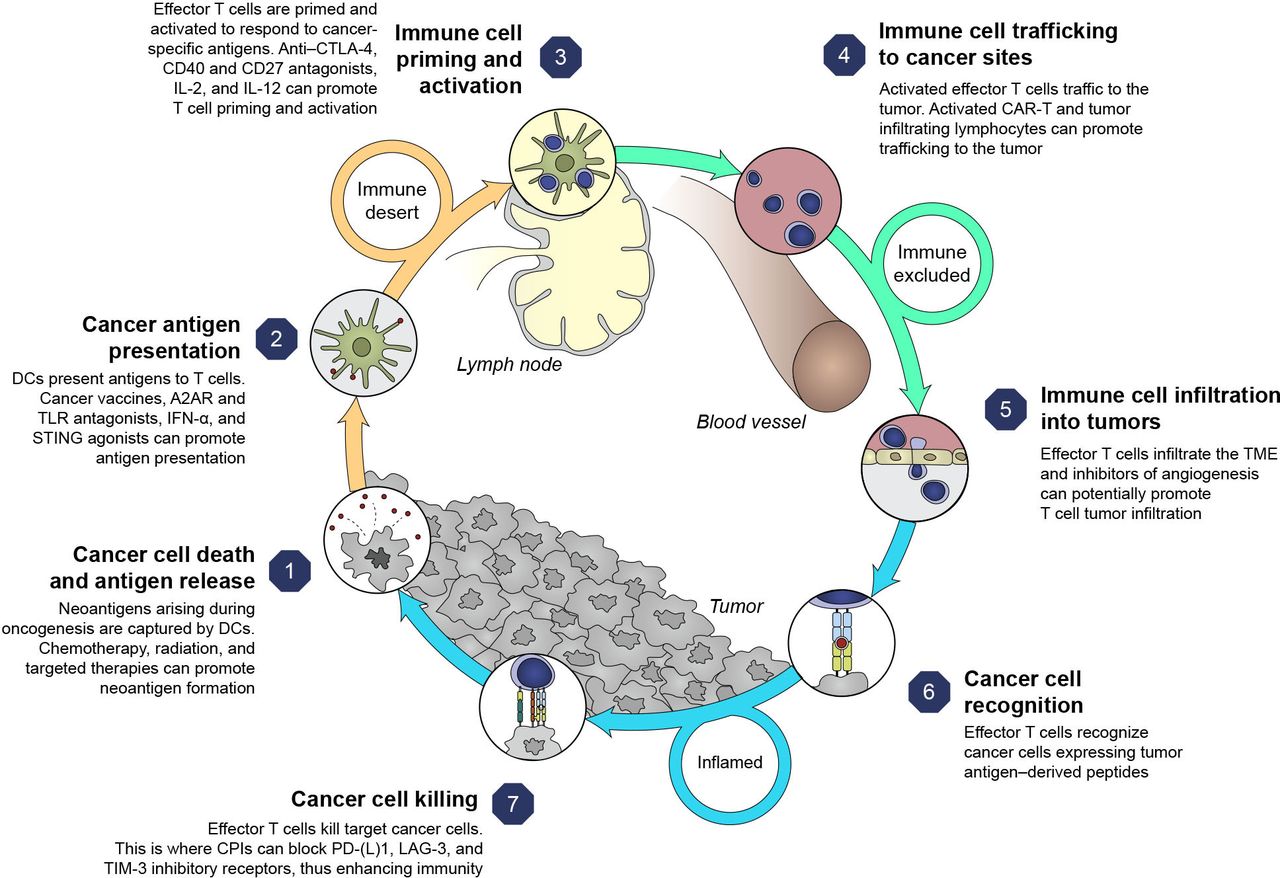
ICI combinations bring the prospect of improved outcomes to specific tumor types and disease settings. Rationales for multimodal approaches can be conceptualized based on the cancer immunity cycle that highlights both the stepwise series of events that contribute to anticancer effects and opportunities to optimize clinical responses (figure 2).17 18 However, responses to therapy across solid tumors are not uniform,18 and using a combination approach targeting multiple steps in the cycle may circumvent suboptimal responses. This emphasizes the rationale for combination therapies to address resistance mechanisms, the potential to target less immunogenic tumors (changing tumors from ‘cold’ (T-cell absence) to ‘hot’ (T-cell infiltration)), and to achieve greater clinical benefit than ICI monotherapy.19
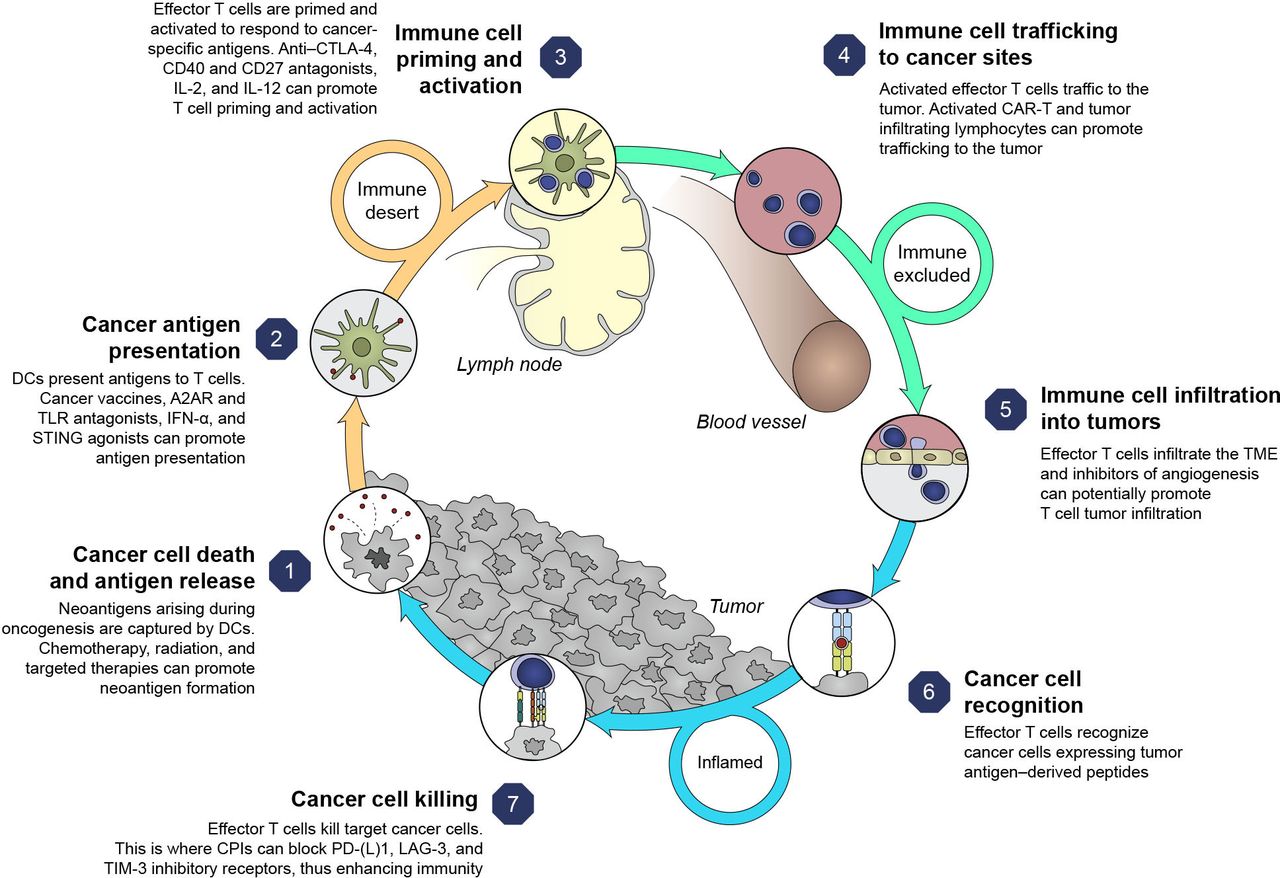
Cancer immunity cycle and therapy targets.1 17 Adapted from Chen and Mellman,17 with permission from Elsevier. A2AR, adenosine A2A receptor; CAR-T, chimeric antigen receptor T cell therapy; CD, cluster of differentiation; CTLA-4, cytotoxic T-lymphocyte antigen 4; DC, dendritic cell; ICI, immune checkpoint inhibitor; IFN-α, interferon alpha; IL, interleukin; LAG-3, lymphocyte-activation gene three protein; PD-1, programmed death-1; PD-L1, programmed death ligand 1; STING, stimulator of interferon genes; TIM-3, T-cell immunoglobulin domain and mucin domain-3; TLR, toll-like receptor; TME, tumor microenvironment.
In 2019, 76% of active PD-(L)1 trials were testing combination regimens, including combination with other ICI therapies, targeted therapies, chemotherapies, and radiotherapy.20–24 Several ICIs are approved in combination with chemotherapies, including nivolumab plus ipilimumab for metastatic non-small cell lung cancer (NSCLC) in the USA and the European Union (EU), nivolumab for gastric and gastroesophageal junction cancers and esophageal adenocarcinoma in the USA, atezolizumab for metastatic non-squamous NSCLC, triple-negative breast cancer (TNBC), and small cell lung cancer in the USA and EU, and pembrolizumab for NSCLC, head and neck squamous cell carcinoma (HNSCC), esophageal and gastroesophageal junction cancer, and TNBC in the USA as well as NSCLC and HNSCC in the EU.21–23 25–27 ICI combinations with targeted agents—for example, tyrosine kinase inhibitors (TKIs) or vascular endothelial growth factor (VEGF) inhibitors—have demonstrated impressive outcomes, resulting in Food and Drug Administration (FDA) approvals for advanced renal cell carcinoma (RCC) and HCC.21 23 24 28 The rationale for combining anti-PD-1 and anti-CTLA-4 therapies is strong, as both antibodies employ distinct but complementary mechanisms to activate T-cell function and induce an antitumor effect.22 29 30 The combination of nivolumab and ipilimumab is now FDA-approved for the treatment of melanoma, RCC, HCC, NSCLC, mesothelioma, and microsatellite instability-high/mismatch repair-deficient colorectal cancer (CRC).22 To improve clinical benefits for more patients, rational combination therapies will need to be designed based on an understanding of the mechanisms of action for each agent and its impact on the cancer immunity cycle.31
To comprehensively assess the effectiveness and outcomes of ICI therapies, we sought to refine the clinical hallmarks attributed to the ICI component of combination strategies (figure 1). There, indeed, is a need for increased awareness and broader inclusion of these hallmarks in clinical trial design. Investigation of measures inclusive of and beyond long-term survival, such as DepOR, TFS, efficacy in brain metastases, improved HRQOL, and a unique safety profile, have evolved as additional hallmarks of ICI-based therapies to take into consideration.
Clinical hallmarks of ICI therapies
Long-term survival benefits
Compared with standard cytotoxic chemotherapy, ICIs restore active T-cell infiltration, increase T-cell repertoire diversification, and stimulate cancer-specific immune responses.32 33 New benchmarks have been established with improved long-term survival and durable responses in significant proportions of patients,34–37 with the potential for TFS.
Survival benefits can be challenging to quantify, especially in trials with initial reporting based on short follow-up. However, with longer follow-up, an emerging clinical hallmark of ICI-based therapies is a delayed separation of Kaplan-Meier survival curves with an observed plateau beginning at ~3 years, demonstrating long-term survival benefits.5 This contrasts with chemotherapy, where early clinical responses are often seen but are short-lived.38 The first long-term ICI outcomes were reported for ipilimumab in advanced melanoma; despite low objective response rates (ORR), responses were durable, as shown by a plateau in the OS curve at ~20% around 3 years, and the plateau was sustained at a 10-year follow-up.5 This brings the promise of a potential ‘functional cure’ in some patients with ongoing, long-term responses, even after treatment discontinuation.39 Several early ICI trials are now reporting data for up to 5 years or more, with similar plateaus seen at year 3.36 40–44 Combination ICI therapies provide the opportunity to raise the plateau, thereby affording more patients durable OS benefits (figure 3).
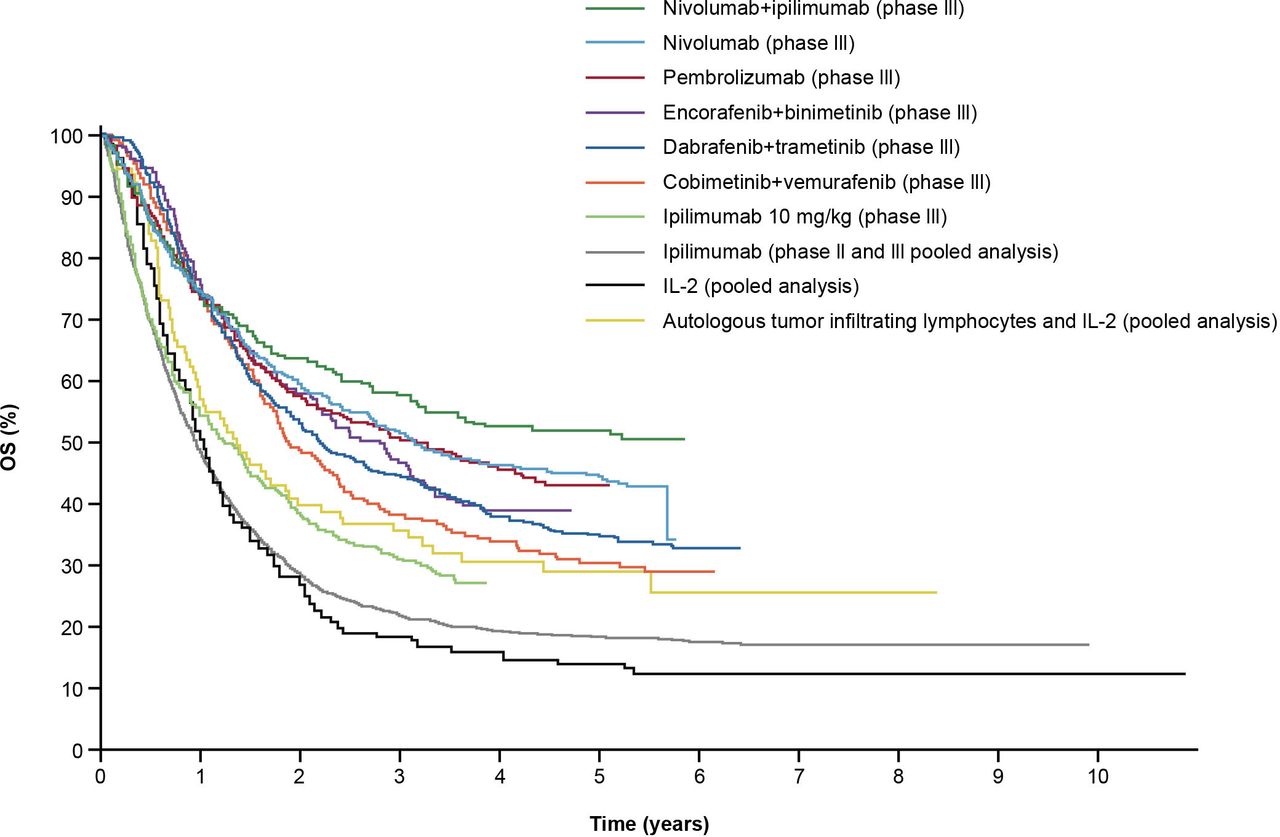
Long-term OS in clinical trials with immune checkpoint inhibitor and targeted therapies in patients with advanced melanoma.39 Data presented represent first-line treatment options, with the exception of those for IL-2 and pooled ipilimumab, for patients with melanoma; results will vary by tumor type. Permissions to use figures: licensed under the Creative Commons Attribution V.4.0 License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by/4.0/). IL, interleukin; OS, overall survival.
Nivolumab plus ipilimumab has demonstrated improved survival and/or durable responses across multiple advanced tumors, including melanoma,36 HCC,45 DNA microsatellite instability-high/mismatch repair-deficient CRC,46 RCC,42 43 and NSCLC.41 47 For example, landmark survival rates at 5 years showed that 52% of patients with advanced melanoma treated with nivolumab plus ipilimumab were alive, vs 44% and 26% receiving nivolumab or ipilimumab monotherapy, respectively.36 With a median follow-up of 55 months, superior OS, progression-free survival (PFS), and ORR benefits were maintained with nivolumab plus ipilimumab versus sunitinib for advanced RCC.43 The probability of response lasting ≥4 years was higher with nivolumab plus ipilimumab versus sunitinib (59% vs 30%, respectively).43 Median OS with nivolumab plus ipilimumab was not reached (NR) vs 38.4 months with sunitinib, and 4-year probabilities were 53.4% vs 43.3%, respectively.43 Nivolumab plus ipilimumab also demonstrated delayed PFS benefit, with curve separation favoring nivolumab plus ipilimumab in the tails of the curves; a plateau emerged at ~30 months with nivolumab plus ipilimumab, with the tails showing ≥10% separation between treatments.42 43 Pembrolizumab plus ipilimumab demonstrated favorable survival in patients with melanoma at a 3-year follow-up, where 36-month PFS and OS were 59% and 73%, respectively.48 Similarly, the combination of pembrolizumab plus axitinib in patients with RCC demonstrated significant OS (median NR vs 35.7 months, respectively; p=0.0003) and PFS (median 15.4 vs 11.1 months, respectively; p<0.0001) advantages over sunitinib.49 The OS curves for combination therapy separated early, and the risk of death was ~32% lower with pembrolizumab plus axitinib versus sunitinib at a median follow-up of 30.6 months.49
After a minimum follow-up of 4 years in patients with NSCLC and PD-L1 ≥1%, the median OS was longer with nivolumab plus ipilimumab (17.1 months) versus nivolumab (15.7 months) or chemotherapy (14.9 months).47 Similar results were seen in patients with a PD-L1 <1%: median OS was 17.2 months with nivolumab plus ipilimumab vs 12.2 months with chemotherapy.41 After a minimum follow-up of 32.4 months, the median OS in patients with NSCLC was longer with atezolizumab plus bevacizumab plus chemotherapy versus bevacizumab plus chemotherapy: 19.8 vs 15 months, respectively.50 Pembrolizumab showed survival improvements with chemotherapy for advanced NSCLC.51 52 Pembrolizumab plus chemotherapy demonstrated favorable OS after a median follow-up of ~4 years: median OS was 22 months vs 10.6 months with chemotherapy.52 While cross-trial comparisons should not be made, many ICI combination therapies show improved efficacy outcomes versus monotherapies. Multimodal approaches that combine ICIs with agents such as chemotherapy, radiotherapy, TKIs, and ICIs have provided significant improvements in patient outcomes in some cases. Increasing baseline tumor burden was associated with significantly shorter OS and PFS.53 This supports the combined use of ICI therapies with debulking strategies, such as chemotherapy and radiotherapy. Combining two ICI therapies with differing modes of action may also allow for improved responses. For example, the roles of PD-1 and CTLA-4 in inhibiting immune responses, including antitumor responses, are largely distinct. CTLA-4 likely regulates T-cell proliferation early in an immune response, primarily in lymph nodes, whereas PD-1 suppresses T cells later in an immune response, primarily in peripheral tissues.54
Increased DepOR over time
DepOR is defined as the maximum percent tumor reduction from baseline and may prove useful as a measure of benefit that could provide an earlier read-out than time-to-event endpoints, providing an additional method of comparing treatment responses.55 Unlike conventional anticancer treatments, a hallmark of ICI-based therapies is a deepening of response over time, with more durable complete responses (CRs) and patients continuing to respond long-term.
DepOR has shown prognostic value for long-term survival in some cancers. In an FDA analysis of DepOR and survival in patients with advanced melanoma, a larger DepOR with ICI therapy correlated with a longer and higher rate of OS.7 Another analysis of DepOR in patients with advanced NSCLC treated with a PD-1 inhibitor found that patients with higher percentages of maximal tumor shrinkage achieved higher OS benefits.55 Similarly, a combination of nivolumab plus ipilimumab in patients with advanced RCC also demonstrated a correlation between DepOR and OS.56
In patients treated with ICI combinations versus monotherapies, the CR rates are greater in general. With longer follow-up, higher CR rates are noted (figure 4), as seen with nivolumab plus ipilimumab in patients with melanoma,36 40 57 58 RCC,43 59 and CRC.46 60 In patients with advanced melanoma, after a minimum follow-up of 9 months, the CR rate was higher with nivolumab plus ipilimumab versus either monotherapy, as was the ORR.57 After a follow-up of 5 years, ORR with nivolumab plus ipilimumab had not changed, but the percentage of CRs was greater, indicating an evolution of response as more patients with a partial response (PR) had converted to a CR over time.36 57 In a pooled post hoc analysis of 5-year data for nivolumab plus ipilimumab versus nivolumab monotherapy for advanced melanoma, patients with a CR in either arm at 12 months had an 85%–86% chance of being alive at 5 years, even without subsequent systemic therapy.61 An increasing proportion of patients with CRs was also observed with pembrolizumab in combination with ipilimumab in patients with advanced melanoma.48 62 While the ORR was preserved from a median follow-up of 17 months to 36.8 months, the CR rate almost doubled.48 62 This conversion of PRs to CRs appears more pronounced in patients on dual ICIs, where this hallmark is possibly driven by activation of a broad repertoire of T-cell receptors able to recognize unique tumor antigens63 and increased CD4+/CD8+ effector memory T cells,64 reflecting a larger activated/memory T-cell diversity after ICI therapy.

Patients with melanoma in CR over time. CR rates among patients undergoing immune checkpoint inhibitor therapy steadily increased, indicating that the best response can improve over time.36 40 57 58 CR, complete response; IPI, ipilimumab; NIVO, nivolumab.
Treatment-free survival
Several ICI studies demonstrate durable responses even after discontinuing combination therapy, resulting in prolonged treatment-free intervals, which may lessen potential for new or further toxicities and may have a cost-saving impact.42–44 65–67 TFS is a novel endpoint that has been defined as the time from treatment discontinuation to subsequent systemic therapy (treatment-free interval) or death (TFS, figure 5A).8 9 Patients who discontinue ICI therapy may experience durable disease control without need for subsequent systemic therapy.68 However, thus far, for combination ICI therapy, this has only been published with nivolumab plus ipilimumab data, but not for ICIs plus TKIs or ICIs plus chemotherapy.

(A) Illustration of TFS assessment and OS curve partitioning. TFS estimates for (B) NIVO plus IPI, (C) nivolumab, and (D) ipilimumab in patients with melanoma in CheckMate 067 and 069. Data labels represent the mean number of months and the percentage of time in a treatment state during the 60-month period.8 9 Areas are restricted mean times. aTime after cessation of protocol therapy without toxicity before initiation of subsequent systemic anticancer therapy or death. bTime after cessation of protocol therapy with toxicity while treatment-free. Permissions to use figures: licensed under the Creative Commons Attribution V.4.0 License (CC BY 4.0) (https://creativecommonsorg/licenses/by/40/). IPI, ipilimumab; NIVO, nivolumab; OS, overall survival; TFS, treatment-free survival.
A study of nivolumab plus ipilimumab versus either monotherapy in advanced melanoma defined TFS as the area between Kaplan-Meier curves.8 9 To use these endpoints, the curves were divided into three phases: time on protocol therapy, TFS, and survival after subsequent therapy initiation (figure 5A). Curves were further subdivided to summarize the survival experience, free of anticancer therapy, with integration of toxicity data to characterize the patient’s well-being.8 9 As illustrated in figure 5, a greater proportion of patients who had received nivolumab plus ipilimumab versus nivolumab or ipilimumab were alive and in TFS for up to 5 years (33%, 17%, and 20%, respectively).9 Furthermore, the restricted mean TFS was longer for nivolumab plus ipilimumab versus either monotherapy (19.7, 9.9, and 11.9 months, respectively).8 9 Mean TFS with grade ≥3 treatment-related adverse events (AEs) represented only a small proportion of the 60-month period.9
In a 5-year analysis of patients with advanced melanoma, the treatment-free interval was 18.1 months with nivolumab plus ipilimumab but was only 1.8 months with nivolumab and 1.9 months with ipilimumab.36 Of patients alive at 5 years, 74% who received combination therapy were not receiving study drug or subsequent systemic therapy vs 58% with nivolumab and 45% with ipilimumab monotherapies.36 Among responders who discontinued study treatment for advanced RCC, a greater proportion who received nivolumab plus ipilimumab versus sunitinib did not require next-line therapy, including more with ongoing TFS.43 67 These results demonstrate the long-term TFS benefits of ICI therapy. Experience is currently limited to nivolumab plus ipilimumab and may be worthy of study with other combinations. Treatment-free intervals may factor in the shared decision making between patients and oncologists, as well as payers. Notably, the American Society of Clinical Oncology (ASCO) included treatment-free interval in the ASCO value framework, highlighting its importance as a key measure when assessing the value of cancer treatments.69
Efficacy in brain metastases
Brain metastases are a common complication of advanced malignancies that are difficult to treat and associated with poor outcomes.14 The role of ICIs in brain metastases is largely unknown, as these patients have traditionally been excluded from ICI clinical trials. The inflammatory tumor microenvironment (TME) of brain metastases is highly complex and immunosuppressive, and is comprised of interactions between diverse immune and cancer cells that may contribute to metastatic progression and impair therapeutic responses.70–74 Myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages are recruited to the brain and play a negative role in antitumor immune responses.70 71 There can be PD-L1 expressed on cancer cells, T-cell activity downregulated due to tumor-induced T-cell exhaustion, and fewer tumor-infiltrating lymphocytes in brain metastases leading to the immunosuppressive environment.71–74 Historically, few chemotherapies and targeted therapies have shown efficacy in brain metastases.75 PD-(L)1 therapies have been shown to reduce T-cell exhaustion in the brain.14 As intracranial tumor blood vessels are less permeable than extracranial vessels,76 ICI drugs likely cannot cross the blood–brain barrier; instead, tumor antigen-specific T cells are stimulated in the periphery and enter the brain subsequently.77
Clinical evidence supporting ICI efficacy in brain metastases is limited but has demonstrated favorable intracranial responses and survival, showing ICIs can have activity in brain metastases resulting from various solid tumors.10–15 Efficacy in brain metastases is being explored with pembrolizumab, nivolumab, atezolizumab, and durvalumab as monotherapies, which show promising activity.12 13 15 78 79 The first study of PD-1 inhibitors in untreated brain metastases was with pembrolizumab. Intracranial response rates were 22% and 33% for melanoma and NSCLC, respectively.15 Central nervous system responses were durable and were strongly concordant with systemic responses.15 Across initial single-agent ICI studies in patients with melanoma, NSCLC, or RCC, the median time from treatment onset to intracranial response was ~2 months.11 For patients with untreated brain metastases from RCC, intracranial response rates were 12% with nivolumab monotherapy; however, no objective response was reported in patients with brain lesions that were multiple or larger than 1 cm.80
Combination therapies demonstrate increased intracranial efficacy versus ICI monotherapies. Intracranial ORR and CR seen with nivolumab plus ipilimumab in patients with melanoma were around double that with nivolumab monotherapy.10 Intracranial responses with nivolumab plus ipilimumab for asymptomatic, untreated melanoma brain metastases were largely concordant with extracranial responses and developed rapidly (median 2.3 months).11 81 The intracranial ORR was 57% (with 26% being CRs), and the overall ORR was 53%.81 PFSs of 6 and 9 months were 64.2% and 59.5%, respectively, for intracranial responses, and 75.9% and 70.4%, respectively, for extracranial responses.81 OS was also maintained with ICI therapies in patients with brain metastases. For example, in patients with advanced NSCLC, survival and responses with nivolumab plus ipilimumab were comparable to patients without brain metastases.82
Ongoing trials are investigating ICI therapy with radiotherapies for brain metastases, and evidence points to better outcomes with concurrent therapies. An analysis of patients undergoing stereotactic radiation therapy or stereotactic radiosurgery plus concurrent ICIs found a significant OS improvement with concurrent therapy versus non-concurrent therapy or radiotherapy alone, which may be associated with a decreased incidence of AEs and/or development of subsequent brain metastases.83 An analysis of patients with brain metastases receiving stereotactic radiation therapy or stereotactic radiosurgery plus concurrent CTLA-4 or PD-(L)1 inhibition reported significantly improved 1-year local control versus radiotherapy alone without increased risk of radiation necrosis.84 Time to new metastases was also significantly longer with concurrent PD-1 and/or CTLA-4 therapy and Gamma Knife radiosurgery.85 While radiotherapy plus ICIs may improve local brain metastasis control, more studies are needed to determine optimal timing and sequencing of radiotherapy and ICIs.
Improved HRQOL
ICI therapy favorably impacts HRQOL, which is important to evaluate the full cancer impact. In a systematic review of PD-(L)1 therapies associated with consistent prolongation of time to symptomatic deterioration across therapies and solid tumors compared with cytotoxic therapy, PD-(L)1 was associated with better symptomatic control at various follow-up points.16 With combination therapies showing greater toxicity, there is concern that HRQOL may be diminished. However, nivolumab plus ipilimumab maintained HRQOL in a manner similar to nivolumab monotherapy in patients with advanced melanoma at a 5-year analysis, despite having marked differences in rates of immune-mediated adverse reactions (IMARs).36 86 There were no meaningful sustained HRQOL deteriorations during or after treatment with nivolumab plus ipilimumab or nivolumab monotherapy.36 86 Patients with advanced RCC receiving nivolumab plus cabozantinib reported better HRQOL than patients receiving sunitinib.87 HRQOL was improved with atezolizumab plus bevacizumab versus sorafenib for unresectable HCC, leading to a delay in time to deterioration of HRQOL, physical functioning, and role functioning.88 Median time to deterioration of disease-related symptoms also took longer with combination treatment, and a lower proportion of patients experienced clinically meaningful symptom deterioration.88 However, improved HRQOL was not seen with all ICI combinations; for example, pembrolizumab plus axitinib and sunitinib monotherapy demonstrated similar HRQOL benefits for advanced RCC.89 Though data are limited, ICI therapy offers patients the potential of a better HRQOL over conventional therapies, related to the long-term benefits as well as the favorable safety profiles of ICI therapies. Notably, the European Society for Medical Oncology (ESMO) and ASCO included improvement in HRQOL in the ESMO-Magnitude of Clinical Benefit Scale and ASCO value framework, respectively, highlighting its importance as a key measure when assessing the value of cancer treatments.69 90
Unique safety profile
While patient outcomes can be significantly improved with ICI therapies, they are also associated with the emergence of unique autoimmune-like toxicities known as IMARs.91 Mechanisms of IMARs differ from AEs of non-ICI agents and are a consequence of increased immune activity and possibly a breach of self-tolerance.92 93 The precise mechanisms of IMARs are not fully understood,92 93 but they are likely similar to those promoting antitumor responses.93 Studies have shown that ICIs can induce T-cell diversification, expansion, and infiltration, affect B-cell responses and induction of autoantibodies, and promote cytokine responses, all of which may be involved in the pathophysiology of IMARs, though to what extent is unknown and this may differ between toxicities.92–97
ICI monotherapy is associated with significantly lower risk of overall and chemotherapy-related AEs (eg, fatigue, nausea, diarrhea, and hematological toxicities) and is overall better tolerated versus chemotherapy, with a significantly lower risk of any all-grade and grade 3–4 AEs; however, it is also associated with an increase in IMARs, including rash, pruritus, colitis, hypothyroidism, hyperthyroidism, and pneumonitis.98 While the presence of specific IMARs varies based on malignancy and inhibitor used,91 the most commonly affected organs are skin, gastrointestinal, and endocrine99; however, any organ may be affected, multiple IMARs may develop, and severe or life-threatening IMARs can occur.65 91 100 Most IMARs manifest within the first 6 months of ICI therapy initiation65 100; however, late-onset IMARs may occur, even months after ICI discontinuation, and can render emerging diagnostics more complex.101 The term ‘delayed immune-related events’ was coined to characterize these late-onset events, defined as IMARs developing ≥90 days after ICI discontinuation.101
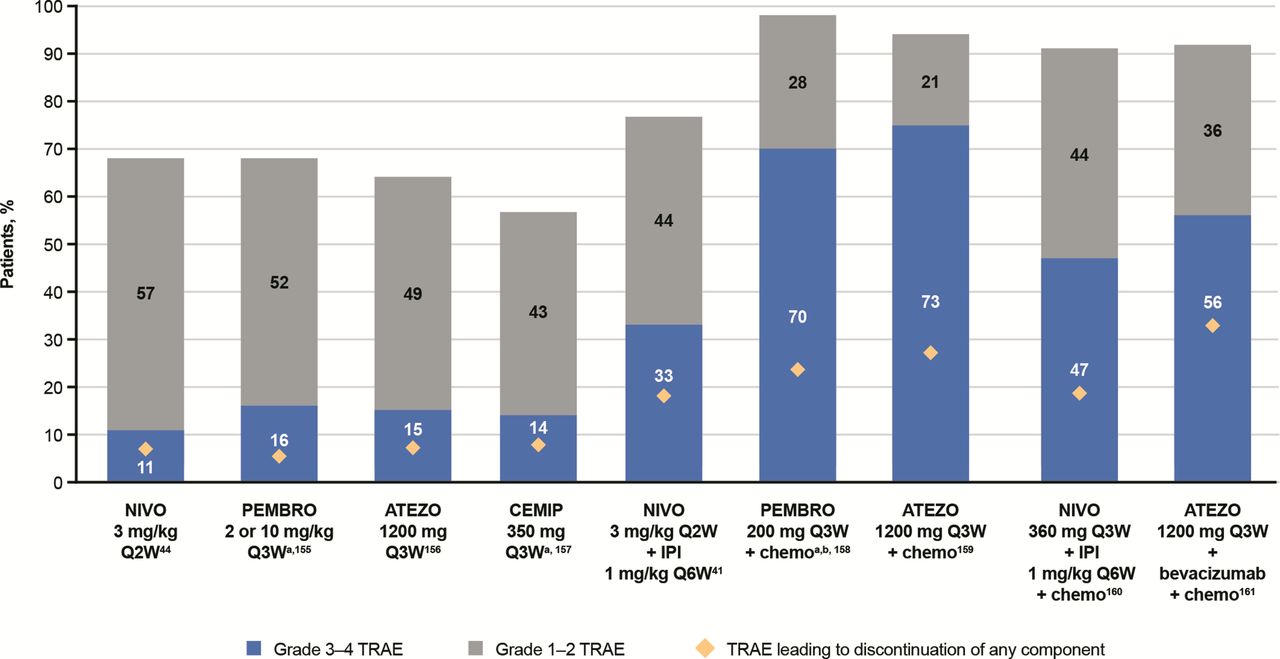
While the safety profile of ICI monotherapies is well established, additional complexities arise with combination therapies where toxicities are increased, and it becomes challenging to identify which therapy underlies the toxicity. Combination CTLA-4/PD-(L)1 results in a significantly higher frequency of IMARs with typically earlier onset versus monotherapies.65 102 103 Each combination has a characteristic safety profile and usually results from a combination of the AE profiles of each monotherapy (figure 6). It is important to properly assess the underlying cause of the toxicity, especially when therapies from different classes are used. For example, concomitant use of anti-PD-(L)1 with a TKI requires an understanding of potential overlapping toxicities; while the management of TKI-related toxicity could be a dose interruption or reduction, IMARs are typically managed with corticosteroids. Anytime ICIs are given as backbones for combination treatments, IMARs can occur and should be treated as immune-mediated reactions. There is a need to identify biomarkers for patients who may or may not respond to ICI combinations to avoid the higher toxicities associated with combination therapies. Recently, real-world data have highlighted the importance of late-onset and long-lasting IMARs for patients receiving ICIs.104
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
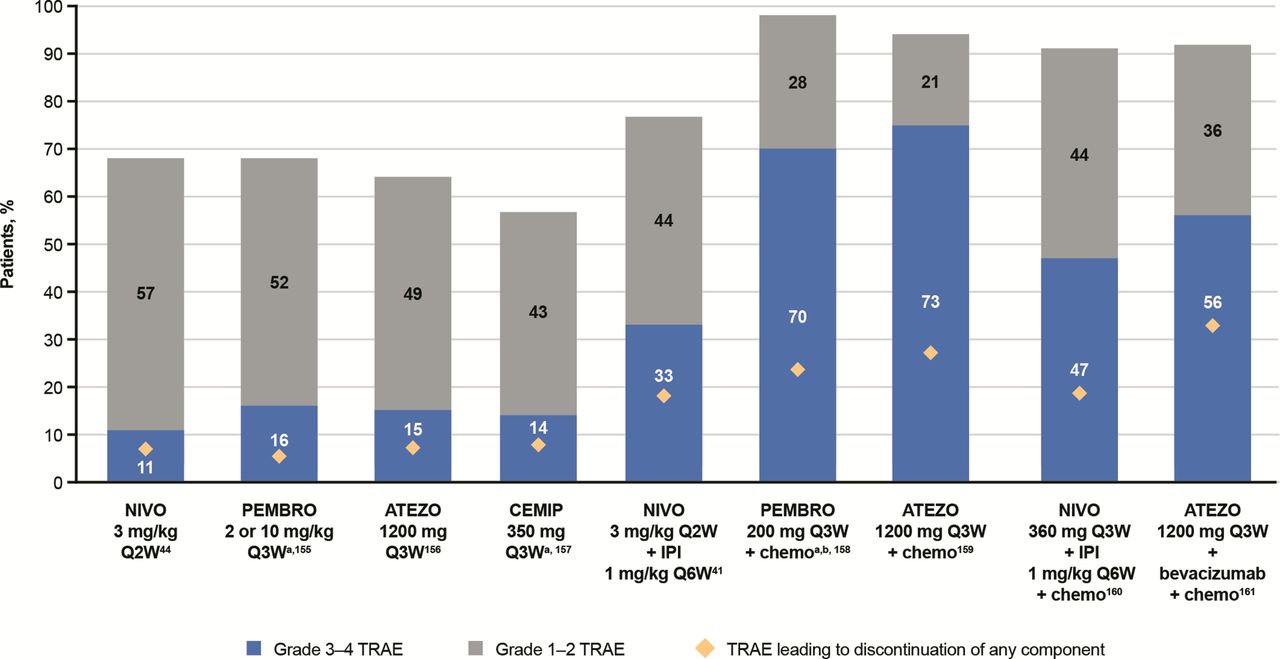
Safety profiles of ICI therapies in patients with non-small cell lung cancer (cross-trial comparisons cannot be made due to differences in dosing and methods used to assess TRAEs across studies). 41 44 155–161 aData for grade 3–4 TRAEs also includes grade 5 TRAEs. bRepresents total adverse events, not just TRAEs. ATEZO, atezolizumab; CEMIP, cemiplimab; chemo, chemotherapy; ICI, immune checkpoint inhibitor; IPI, ipilimumab; NIVO, nivolumab; PEMBRO, pembrolizumab; QxW, every x weeks; TRAE, treatment-related adverse event.
IMAR management guidelines
IMAR management guidelines include those provided by the National Comprehensive Cancer Network,105 the Society for Immunotherapy of Cancer,106 and ESMO,107 which provide treatment algorithms for common IMARs. Based on these guidelines, IMAR management requires close follow-up, patient engagement/self-management programs, and a multidisciplinary approach. Evidence-based IMAR management is limited and is an evolving area of research, with newer algorithms being proposed for severe and/or refractory cases.108 Using available management algorithms, we found that IMARs resolve in most patients, with the exception of endocrine IMARs.65 Common endocrine IMARs include thyroid dysfunction, hypophysitis, type 1 diabetes mellitus, and primary adrenal insufficiency, which, unlike non-endocrine IMARs, are often permanent and require lifelong hormone replacement.91
Notably, the presence of IMARs (skin and gastrointestinal toxicities in particular) has been associated with significantly improved survival and response outcomes with ICIs.94 103 109 110 However, results are conflicting,111 112 which may be related to a general lack of landmark analyses that can minimize the immortal time bias potentially associated with time-dependent factors, such as the development of IMARs.113 114
Studies have shown that patients who discontinue ICIs for IMARs maintain survival and response outcomes similar to patients who did not discontinue for an IMAR, whether they restarted ICI therapy or not.115 116 Whether or not to re-treat patients following an IMAR is a frequent clinical scenario. Data are limited, but an analysis of patients with NSCLC who were re-treated with ICIs found that survival was significantly improved, and while ~50% of patients developed a new or recurrent IMAR, they were mostly mild and manageable.116 The decision to re-treat patients is based on IMAR severity and sequelae, as supported by current guidelines,105–107 and should be made in consultation with patients and specialists.
Discussion
ICI hallmarks, including long-term survival, deepening of responses over time, promising treatment-free intervals, brain metastases activity, and improved HRQOL, are emerging and enriching the metrics of benefits in many advanced cancers. With ICI therapies, more patients are responding and living longer, with the potential for more CRs, treatment-free intervals, and better HRQOL, hence the need for hallmarks of ICI combinations to be used more often in study designs and at the patient and upstream healthcare system levels. With more patients surviving long term, there is need for robust survivorship care plans. The long-term survival hallmark of ICI-based therapies is an observed plateau of Kaplan-Meier survival curves beginning at ~3 years, demonstrating long-term benefits.5 Until long-term survival data are available, additional hallmarks are important inputs into shared clinical decision making. DepOR has shown prognostic value for long-term survival, and a hallmark of ICI-based therapies is a deepening of response over time, with more CRs and patients continuing to respond long term.36 48 57 62 Patients with advanced melanoma, CRC, or RCC receiving nivolumab plus ipilimumab demonstrated durable responses with longer treatment-free intervals, and a higher proportion of patients free of subsequent therapies were observed than patients receiving monotherapy.40 42 43 65–67 Along with these beneficial hallmarks, immune system activation leads to a safety profile unique to ICIs.91 Notably, IMARs may be a biomarker for patient outcomes, as they are associated with survival and responses.94 103 109 110 IMARs also bring a need for multidisciplinary team awareness and recognition of ICI AE profiles and management for these patients.
Combination ICI therapies promise further survival advantages and aim to raise the plateau of the curve so that more patients experience sustained OS benefits. The combination treatment arena is advancing, including numerous new ICI therapies with multiple targets, including but not limited to VEGF/tyrosine kinase,66 117 the enzyme poly (ADP-ribose) polymerase,118 other checkpoints such as lymphocyte-activation gene 3 (LAG-3),119 T-cell immunoglobulin and mucin domain-3 (TIM-3),120 T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif (ITIM) domain (TIGIT),121 and other immune-modulating pathways (eg, IL-2 pathway agonist and toll-like receptors). More approvals are expected with ICI combinations across tumor types, and the clinical hallmarks will need to adapt to the changing field. This includes the potential for new or updated endpoints, including DepOR, TFS, and HRQOL, and in earlier disease, pathological CRs in the neoadjuvant setting and disease-free survival in the adjuvant setting. There is a need to understand the determinants driving response, resistance, and AEs, and to identify potential predictive biomarkers to optimize treatment.
ICI therapies display different response patterns and tumor response kinetics compared with cytotoxic and molecularly targeted agents, necessitating the use of alternative endpoints. In light of this, the FDA recognizes the use of traditional and surrogate endpoints to support accelerated approval of cancer therapies.122 In these settings, the FDA may grant approval based on a surrogate endpoint that is ‘reasonably likely to predict clinical benefit’.122 In their 2018 guidance for industry, the FDA provides a robust discussion of oncology clinical trial endpoints and their limitations, including OS, PFS, ORR, CR, and those involving symptom assessment (HRQOL).122 The use of specific endpoints is guided by factors such as specific cancer, disease setting and location, size of effect and duration, DepOR, and available therapeutic options.122
As discussed by the FDA and other publications, there remain limitations with the clinical trial endpoints associated with the hallmarks of ICI therapy. For example, OS requires long follow-up periods in large trials and may be affected by crossover and subsequent therapies.122 With OS, there may also be a delay in benefit with ICI agents that may lead to a loss in statistical power, and conventional proportional hazards models may not appropriately capture ICI survival kinetics.6 Limitations associated with PFS include varying definitions among studies and being subject to assessment bias, measurement error, and missing data.6 122 PFS also requires frequent assessments and balanced timing of assessments among treatment arms and may not always correlate with survival.122 Similarly, definitions for ORR vary among studies, and ORR requires frequent assessments and balanced timing of assessments among treatment arms.122 ORR does not differentiate outcomes based on DepOR (CR vs PR), does not capture patients with durable stable disease, and may not always correlate with survival.6 122 A limitation associated with DepOR is that its predictive value remains uncertain.123Non-measurable lesions are not considered in the calculation of DepOR, which is based on measurement of tumor diameter. Additionally, reductions in target lesions do not always result in a diameter reduction, and tumor shrinkage may not always be symmetrical.123 For HRQOL, instruments currently used were designed for earlier chemotherapies and may not be sufficiently sensitive to capture important symptomatology unique to ICIs.122 124 Challenges with generalizing HRQOL data from prospective studies include limited data, heterogeneity in definitions and measurement tools, time point and duration of measurements, poor compliance, and missed data points between intervention and control arms.6 122 124 Despite these limitations, the endpoints discussed for ICI trials are valuable for assessing the benefits of these therapies.
While there are encouraging responses in a subset of patients, many patients experience either primary refractory or acquired resistance after a response, in part because immune escape mechanisms can arise from defects in any or multiple steps of the cancer immunity cycle.18 125 126 Resistance mechanisms are complex, involving tumor cell-intrinsic and cell-extrinsic factors. Intrinsic factors include expression of tumor genes and pathways related to immune recognition, cell signaling, and expression of factors, preventing immune cell infiltration and/or function within the TME.127–129 Extrinsic mechanisms involve non-tumor components, including Tregs, MDSCs, macrophages, and inhibitory checkpoints.130–132 One goal of developing combination ICI therapies is to circumvent these resistance mechanisms and to achieve better outcomes for patients.
A major breakthrough with ICIs is the potential to achieve durable responses in some patients that can be maintained even years after ending treatment. A challenge with ICI response durability as an endpoint is that a unique definition is not available, and the optimal treatment duration in case of a durable response is unknown. Another challenge is whether ICI therapy should be given until disease progression or AEs, or whether treatment could be interrupted following a certain duration and/or achieving a radiological response. In metastatic melanoma, stopping ICI therapy before 2 years can be considered after ≥6 months of treatment in patients with confirmed radiological control (stable disease or PR) in case of a complete pathological and/or metabolic response, according to the ESMO Guidelines Committee.133 Case reports of patients with advanced NSCLC who had an early response and received ICI therapy for <2 years reported ongoing benefits following discontinuation.134 A further challenge is that clinical trials using time-to-event endpoints are commonly designed with the assumption that the HRs between groups remain constant. The non-proportional hazards and delayed survival curve separation associated with ICI therapy reduces the ability for conventional statistical models to differentiate between them.135 Therefore, alternative methods are needed to shorten the study duration and/or patient numbers needed to mitigate the effects.135 For survival curves with a plateau, cure models may be used to investigate heterogeneity between long-term survivors and patients not surviving long term.136
Notably, ASCO and ESMO included HRQOL in the ASCO value framework, and ESMO-Magnitude of Clinical Benefit Scale and ASCO also included TFS, emphasizing their importance as key measures when assessing the value of cancer treatments.69 90 Evaluating the distinct hallmarks of ICI therapies and characterizing how they influence patients will inform future treatments. With ICI therapies being used in more patients and patients living longer, it is critical for regulatory bodies and treating physicians to recognize associated hallmarks to ensure appropriate outcomes and implement system-wide changes to address immune-mediated toxicities that require multidisciplinary management, including delayed ICI toxicities.
In conclusion, ICI-based therapies provide unique hallmarks that are associated with significantly improved clinical outcomes and a unique safety profile. Future research should focus on better exploiting these hallmarks with optimized dosing sequencing as we move into a treatment arena with new dual and triple combinations.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
The sponsor was involved in the review design and in the analysis and interpretation of data and information provided in the manuscript. However, the ultimate responsibility for opinions, data interpretation, and conclusions lies with the authors. Professional medical writing and editorial assistance were provided by Katherine Groschwitz, PhD, and Matthew Weddig of Spark Medica Inc, funded by Bristol Myers Squibb, according to Good Publication Practice 3 guidelines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published online. Figures 1 and 6 have been updated. References 155 and 157 have been updated.
Contributors All authors were responsible for the opinions, data interpretation, and conclusions in this study.
Funding This work was supported by Bristol Myers Squibb.
Competing interests SP received education grants and honoraria from and provided consultation, attended advisory boards, and/or provided lectures for AbbVie, Amgen, AstraZeneca, Bayer, Biocartis, Boehringer Ingelheim, Bristol Myers Squibb, Clovis, Daiichi Sankyo, Debiopharm, Eli Lilly, F. Hoffmann-La Roche, Foundation Medicine, Illumina, Incyte, Janssen, Merck Sharp and Dohme, Merck Serono, Merrimack, Novartis, Pharma Mar, Pfizer, Regeneron, Sanofi, Seattle Genetics, and Takeda. SP also talked at an organized public event for AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, F. Hoffmann-La Roche, Illumina, Merck Sharp and Dohme, Novartis, Pfizer, Sanofi, and Takeda, and received grants and research supports as a (sub)investigator in trials (institutional financial support for clinical trials) sponsored by Amgen, AstraZeneca, Biodesix, Boehringer Ingelheim, Bristol Myers Squibb, Clovis, F. Hoffmann-La Roche, Illumina, Merck Sharp and Dohme, Merck Serono, Novartis, and Pfizer. A-KL received grants or research support, institutionally, from Bristol Myers Squibb, BioCanRx, Novartis, Roche, Ipsen, and EMD Serono, and was in an advisory or consulting role for AbbVie, Astellas, Bristol Myers Squibb, Eisai, Ipsen, Janssen, Merck, Novartis, Pfizer, Roche, and TerSera. OM received grants and personal fees from Bristol Myers Squibb and Merck Sharp and Dohme, Pierre-Fabre and Amgen, and personal fees from Roche, Novartis, GSK, and Merck, outside the submitted work. CR was a consultant for Amgen, Bristol Myers Squibb, Novartis, Roche, Merck Sharp and Dohme, Pierre Fabre, and AstraZeneca. PS has received consulting fees or stock ownership or attended advisory boards for Achelois, Adaptive Biotechnologies, Affini-T, Apricity, BioAtla, BioNTech, Codiak, Constellation, Dragonfly, Earli, Glympse, Hummingbird, ImaginAb, Infinity Pharma, Jounce, JSL Health, Lava Therapeutics, Lytix, Marker, Oncolytics, PBM Capital, Phenomics, Polaris, Sporos, Time BioVentures, and Venn Biosciences.
Provenance and peer review Commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.