Article Text

Abstract
Background Recombinant human interleukin-2 (rhIL-2, aldesleukin) is Food and Drug Administration approved for the treatment of metastatic melanoma and renal cell carcinoma and has achieved durable response in a subset of patients. However, its utility as an immunotherapeutic drug is limited by undesirable activation of immune suppressive regulatory T cells (Tregs) and a short half-life requiring frequent high dose administration, leading to unacceptable toxicities. We have engineered MDNA11, a long-acting IL-2 superkine, to overcome these limitations by (1) modifying receptor selectivity in favor of anti-cancer immune cells to increase therapeutic efficacy and (2) fusion to human albumin to extend the pharmacokinetic (PK) profile, circumventing the need for frequent dosing.
Methods MDNA11 was evaluated using in vitro and in vivo studies including: binding analyses to measure receptor affinity, IL-2 pathway signaling, PK studies in mice, and efficacy studies in syngeneic tumor models as single agent and in combination with immune checkpoint inhibitors. Finally, the safety and pharmacodynamic profile of MDNA11 was assessed in non-human primate (NHP).
Results Binding studies with MDNA11 demonstrated increased affinity for IL-2Rβ (CD122) and no binding to IL-2Rα (CD25). As a result, MDNA11 exhibits reduced/limited Treg stimulation while triggering an enhanced activation of natural killer and naïve CD8 T cells compared with rhIL-2. When administered to animals with pre-established tumors, MDNA11 controlled tumor growth in a monotherapy setting and in combination with anti-PD1 or anti-CTLA4 to induce durable tumor clearance with a once weekly dosing regimen. In a NHP model, MDNA11 was well tolerated while triggering durable and potent immune responses including expansion of lymphocytes without significant effect on Tregs and eosinophils, the latter been linked to an increased risk of vascular leak syndrome.
Conclusion MDNA11 is a next generation long-acting IL-2 immunotherapeutic with a highly favorable pharmacodynamic profile that translates to a strong therapeutic efficacy in preclinical tumor models and a strong and durable immune response in NHP.
- immunotherapy
- CD8-positive T-lymphocytes
- CD4-positive T-lymphocytes
- cytokines
Data availability statement
Data are available upon reasonable request. Data sets and material/reagents analyzed and/or used in this study are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Interleukin-2 (IL-2) plays pivotal roles in supporting immune responses by regulating lymphocyte activity.1 2 Commercial recombinant human (rh)IL-2 (aldesleukin) was one of the first immunotherapeutic cytokines approved for the treatment of metastatic melanoma and renal cell cancer.3–5 While it demonstrated objective responses in 10%–15% of patients, its application in immunotherapy is limited by a short in vivo half-life and its adverse side effect profile, which often requires intensive care during treatment.6 7 In addition, rhIL-2 is particularly potent at expanding regulatory T cells (Tregs), which act to dampen the antitumor immunity needed to achieve meaningful clinical responses.1 6 7
At the molecular level, IL-2 can engage, with limited capacity, the intermediate-affinity IL-2 receptor (IL-2Rβγc or CD122/CD132) expressed on natural killer (NK) and naïve CD8 T cells. Conversely, IL-2 binds with high affinity to the high-affinity IL-2R (IL-2Rαβγc or CD25/CD122/CD132) constitutively expressed on Tregs, which explains the hyper-responsiveness of these suppressive cells to even low levels of IL-2. Thus, frequent administration of high dose rhIL-2 is required to achieve therapeutic response in patients; however, this high exposure also leads to onset of adverse events such as pulmonary edema and vascular leak syndrome (VLS).8 To circumvent some of these obstacles, a number of strategies were developed including post-translational modification of rhIL-2 (eg, PEGylation),9 design of an IL-2/CD25 fusion protein,10 or development of an IL-2/IL-15 mimic,11 to mitigate CD25 binding and improve on safety. Although these strategies were successful at limiting Treg expansion by reducing binding to the high-affinity IL-2R, most of these molecules displayed reduced activity on CD8 T and NK cells compared with rhIL-2 due to decreased affinity for CD122.9 10 Thus, there is a growing interest to harness the therapeutic functions of IL-2 in a more selective and controlled manner.
We report here the characterization of MDNA11, a long-acting IL-2 superkine with selectively enhanced binding to CD122 and no interaction with CD25 (ie, Beta-only), resulting in potent and durable immunological and antitumor activity in preclinical models supporting prospective therapeutic benefit in future clinical trials.
Methods
All mice used in the study were housed, managed, and cared for in accordance with the guidelines approved by the Association and Accreditation of Laboratory Animal Care. Studies with non-human primate (NHP) were performed in accordance with the guidelines approved by Institutional Care and Use Committee.
BLI Octet study
Experiments were conducted using Ni-NTA biosensors in a 96-well plate in the Octet RED96 bio-layer interferometry (BLI) instrument (ForteBio). Human, mouse and cynomolgus IL-2Rα or IL-2Rβ (online supplemental table S1) were immobilized to the Ni-NTA biosensors at 200 nM and then dipped into a titration of selected protein analytes in phosphate-buffered saline (PBS) buffer containing Kinetics reagent (ForteBio). Assay volume was 200 µL per well and reactions were performed at 25°C. The assay baseline drift was subtracted out using a protein-bound biosensor dipped into buffer alone. Negative control was included in each experiment where no protein was loaded onto the Ni-NTA biosensor and a mid-range concentration of protein analyte was used in order to determine ‘bare’ biosensor binding profile. A blocking step was added using Superblock (Thermo Fisher).
Supplemental material
STAT5 signaling
Human peripheral blood mononuclear cells (PBMCs) were isolated over a density gradient, allowed to rest in complete media, and then stimulated for 15 min with MDNA11 or rhIL-2. To determine EC50, MDNA11 or rhIL-2 were subjected to 10-point fivefold dilutions from 25 nM in order to generate dose-response curves. Cells were fixed immediately following stimulation, and samples analyzed by flow cytometry after intracellular staining for phosphorylated STAT5 (pSTAT5) in naïve CD8+ T cells, NK cells and Tregs (online supplemental table S2)
PK studies in mice
Female C57Bl/6 mice (6–8 weeks old) were dosed with rhIL-2 or MDNA11 by intravenous injection. Blood samples were collected at different time points from three animals. Plasma samples were analyzed using a commercial ELISA kit (Invitrogen) to measure levels of rhIL-2. Levels of MDNA11 were quantified using a custom ELISA with MAB202 (R & D systems) as capture antibody and P600 rabbit polyclonal antibodies (Thermo Fisher) for detection. Pharmacokinetic (PK) parameters were calculated by a standard non-compartmental analysis method using the Phoenix WinNonlin software.
B16F10 melanoma model
Acclimatized female C57BL/6 mice (6–8 weeks of age) were implanted subcutaneously (SC) with 3×105 B16F10 melanoma cells and allowed to grow into established tumors. Animals were randomized into groups of 8 or 10 based on tumor volume. TA99 monoclonal antibody (mAb) (BioXCell) was dosed at 150 µg two times per week for 3 weeks; MDNA11 was dosed at 5 mg/kg one time per week for 3 weeks by intraperitoneal (IP) injection for both monotherapy and combination treatments.
To evaluate tumor-infiltrating lymphocytes (TILs), new groups of B16F10 tumor bearing mice were treated by IP injection with vehicle (PBS) or MDNA11 (5 mg/kg), and tumors harvested 3, 6 or 9 days following the treatment for analysis by flow cytometry (online supplemental table S3). For analysis of intracellular interferon-γ (IFNγ), tumors were harvested 6 days following treatment, processed into single cell suspension for ex vivo incubation with Brefeldin A, and analyzed by flow cytometry using anti-CD3, anti-CD8 and anti-IFNγ (clone XMG1.2).
CT26 colon cancer model
Acclimatized female BALB/c mice (6–8 weeks old) were implanted SC with 2×106 CT26 cells in their flanks and allowed to grow into established tumors. Mice were randomized into groups of 8 based on tumor size; average tumor size of each treatment group on dosing initiation was ~60 mm3. Mice were dosed by IP injection with MDNA11 (5 mg/kg; one time per week for 2 weeks) as monotherapy or in combination with anti-CTLA4 antibody 9D9 (200 µg two times per week for 2 weeks). Mice that showed complete tumor regression (CR) were rechallenged with CT26 tumor cells on days 49, 116, and 165. Blood samples (~200 µL) from rechallenged mice were collected on day 97 for analysis of effector (CD44hi/CD62Llo/−) and central (CD44hi/CD62Lhi) memory T cells in both CD4 and CD8 lineages by flow cytometry (online supplemental table S4). Mice were rechallenged SC on day 265 with CT26 tumor cells and sacrificed on day 270. Blood and spleens were collected for analysis of antigen-specific CD8 T cells by flow cytometry using anti-mouse CD3ε (Biolegend), anti-CD8 clone KT15 (MBL International) and H-2Ld MuLV gp70 Tetramer-PE (MBL International).
Immune cell depletion was performed by IP administration of anti-CD8 (BioXcell, clone 2.43; 400 µg per dose) and anti-asialo GM1 (Biolegend; 50 µL per dose) to deplete mice of CD8 T cells and NK cells, respectively. Multiple doses (10–12 days apart) of the antibodies were administered during the study to maintain immune cell depletion. Cell depletion was confirm by flow cytometry.
MC38 melanoma model
Acclimatized female C57BL/6 mice (6–8 weeks of age) were implanted SC with 5×105 MC38 cells and allowed to grow into established tumors. Animals were randomized into groups of 10 based on their tumor volume. Mice were dosed by IP injection with MDNA11 one time per week for 2 weeks at 2 or 5 mg/kg as monotherapy or in combination anti-PD1 antibody at 10 mg/kg two times per week for 3 weeks. Mice showing CR were rechallenged with MC38 tumor cells on Study Day 41.
NHP study
Male and female cynomolgus monkeys (Macaca fascicularis), aged 2–4 years, were dosed with MDNA11 by bolus intravenous injection on Study Days 1, 15 and 29. Treatment groups comprised of equal number of male and female animals. Immunophenotyping on whole blood was performed by flow cytometry at different time points following treatment using different marker panels (online supplemental table S5).
Statistical analysis
GraphPad Prism V.9.1 was used for graphical representations and statistical analyses. Relevant statistical tests are indicated in figure legends.
Results
MDNA11 binds IL-2Rβ (CD122) with high affinity without interacting with IL-2Rα (CD25)
Therapeutic efficacy of rhIL-2 is limited by a low potency on resting NK and naïve CD8 T cells while preferentially stimulating Tregs expressing the high-affinity IL-2R.1 6 7 Additionally, it has a short half-life owing to a low molecular weight (15 KDa),12 necessitating frequent dosing at a high dose to achieve any clinical response. To overcome these limitations, the IL-2 superkine H9,13 originally selected based on directed evolution for enhanced binding to CD122, was engineered to include two additional changes: (i) mutations (F42A and E62A) known to block CD25 binding,14 and (ii) fusion with human albumin to increase molecular size and improve in vivo half-life15 (figure 1A). To assess how these modifications in MDNA11 affect interaction with the IL-2R, a BLI study was carried out using human CD25 or CD122 chain as a binding receptor. While rhIL-2 displayed high affinity towards human CD25 (KD=24±1 nM), no binding could be detected with MDNA11 up to a concentration of 2000 nM (figure 1B). This is attributed to addition of the F42A and E62A mutations since a version of MDNA11 lacking these mutations was able to bind CD25 with similar affinity as rhIL-2 (online supplemental figure S1). Importantly, MDNA11 retains its enhanced selectivity for CD122 with a 30-fold higher binding affinity (KD=6.6±0.1 nM) compared with rhIL-2 (210±30 nM) (figure 1B). MDNA11 also did not bind cynomolgus and mouse CD25 while exhibiting similar affinity for cynomolgus CD122 (online supplemental figure S2 and table 1). Affinity of MDNA11 to mouse CD122 is reduced but remains superior over rhIL-2 (table 1). These data demonstrate the receptor selectivity of MDNA11 among different species and identifies the cynomolgus monkey as a highly relevant model for safety and pharmacodynamic (PD) assessments ahead of first in human (FIH) study.

MDNA11 is a long-acting IL-2 superkine with enhanced receptor selectivity that preferentially stimulates effector immune cells. (A) Schematic diagram representing the MDNA11 protein. The bioactive component is human IL-2 with seven mutations (L80F, R81D, L85V, I86V and I92F to enhance binding to IL-2Rβ; and F42A and E62A to block binding to IL-2Rα) for improving receptor selectivity, denoted as MDNA109FEAA. (B) Representative bio-layer interferometry Octet analysis of rhIL-2 and MDNA11 binding to human CD25 (IL-2Rα) and CD122 (IL-2Rβ). KD values are represented as mean±SD. (C) Representative dose-response curves of pSTAT5 induced by rhIL-2 and MDNA11 in naïve CD8 T cells, NK cells and Tregs. (D) EC50 values are represented as mean±SD across multiple peripheral blood mononuclear cell donors. For this experiment, N=3–4/group with *p<0.05 and **p<0.01 compared with rhIL-2 (Mann-Whitney non-parametric test). (E) CD8/Treg and NK/Treg EC50 ratios comparing potency in pSTAT5 signaling. NK, natural killer; pSTAT, phosphorylated STAT; rhIL, recombinant human interleukin.
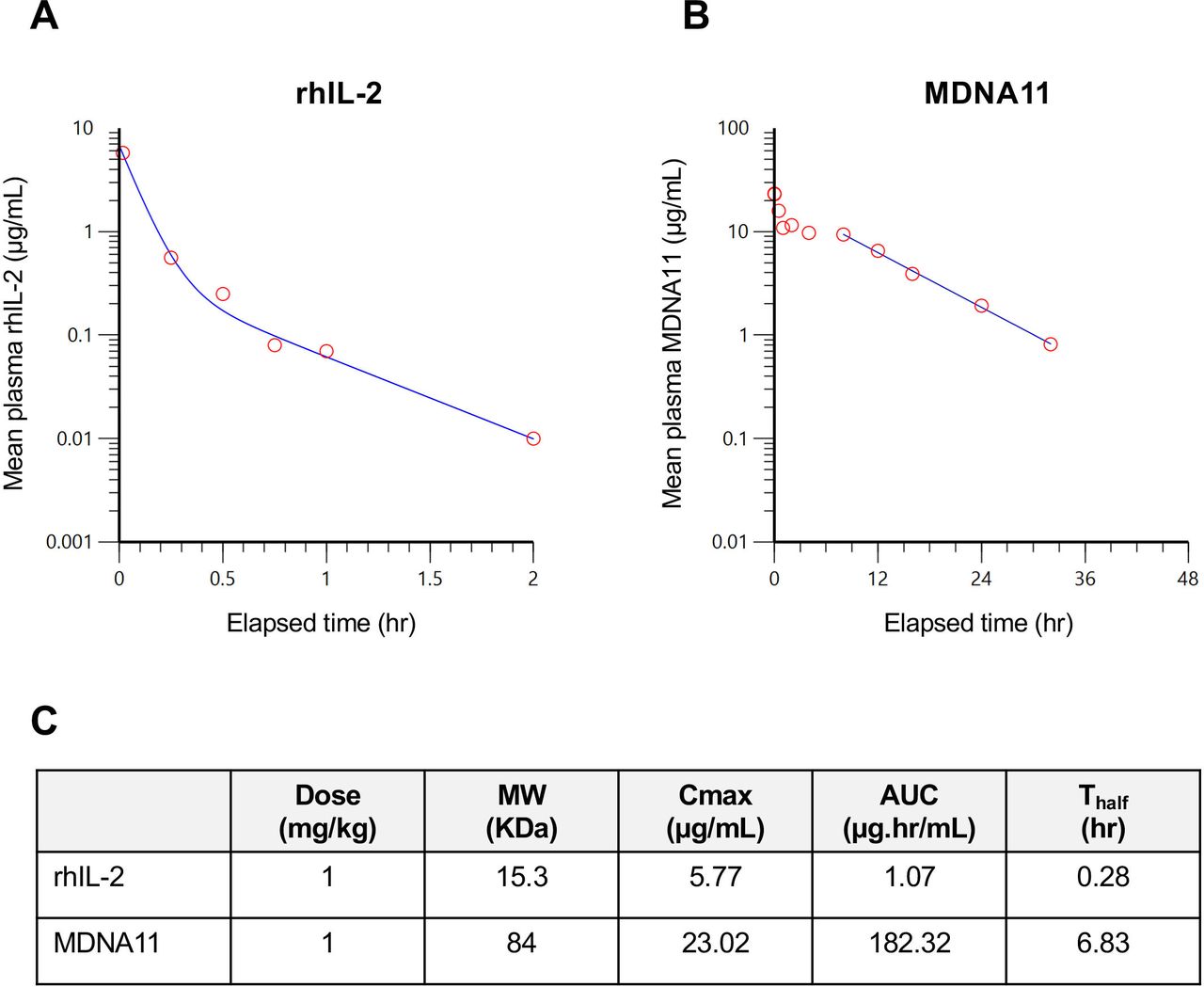
MDNA11 exhibits an improved PK profile in mice. (A) Plot of plasma levels of rhIL-2 vs time. (B) Plot of plasma levels of MDNA11 vs time. Each time point is the mean level measured in three mice. (C) Summary of PK parameters comparing rhIL-2 and MDNA11 based on non-compartmental analysis. AUC, area under the curve; PK, pharmacokinetic; rhIL, recombinant human interleukin.
MDNA11 binding to human, cynomolgus or murine IL-2 receptors
MDNA11 favors activation of naïve CD8 T cells over Tregs
To confirm the impact of improving receptor selectivity of MDNA11 on immune cell activation, we assessed signaling through the STAT5 pathway in resting NK and naïve CD8 T cells expressing the intermediate-affinity IL-2R (IL-2Rβγc) versus Tregs, which express the high-affinity IL-2R (IL-2Rαβγc). Flow-cytometry analysis of pSTAT5 conducted on multiple human PBMCs treated with MDNA11 showed a 28-fold increase in EC50 (160±21.7 pM) to activate Tregs compared with rhIL-2 (5.6±3.1 pM), indicating a statistically significant (p<0.01) reduction in potency in this suppressive immune population (figure 1C–D). On the other hand, the potency of MDNA11 on naïve CD8 T cells was significantly enhanced with more than a sevenfold decrease (p<0.05) in EC50 values (463.8±141.6 pM) compared with rhIL-2 (3389.5±1571.7 pM). The potency of MDNA11 on NK cells also increased (EC50=68.9±9.3 pM) compared with that of rhIL-2 (EC50=201.5±175.6 pM). Accordingly, the EC50 ratios for pSTAT5 activation in naïve CD8/Tregs or NK cells/Treg were dramatically reduced due to a combination of increased potency on CD8 and NK cells and reduced activity on Tregs (figure 1E). These highly favorable changes in immune cell response are consistent with a shift in preferential binding of MDNA11 to intermediate-affinity IL-2R expressed by effector immune cells.
MDNA11 exhibits superior PK profile in mice
The overall molecular size of MDNA11 was increased approximately fivefold, from 15 KDa to 83 KDa, by fusion to human albumin (figure 1A) to overcome rapid clearance via kidney filtration.16 We determined the PK profile of MDNA11 in comparison to rhIL-2 by measuring the plasma levels in mice of these molecules at different time points following a single intravenous administration. As anticipated, a rapid clearance of rhIL-2 was observed with a terminal half-life of 0.28 hour (figure 2A). In contrast, MDNA11 demonstrated a slower rate of clearance (figure 2B), resulting in a 25-fold increase in terminal half-life (T1/2=6.83 hours). Maximal cytokine concentration in the blood (Cmax) and the area under the curve reflecting exposure over time were also higher in plasma samples collected from MDNA11 treated mice compared with rhIL-2 (figure 2C). These data support the possibility to achieve in vivo therapeutic efficacy without the need for frequent administration of MDNA11.
MDNA11 delays B16F10 tumor growth through enhanced NK and CD8 T cell recruitment
Therapeutic efficacy of MDNA11 was evaluated in an aggressive B16F10 melanoma model, in which we also investigated potential synergy with the therapeutic TA99 mAb targeting the tyrosinase-related protein 1 (TYRP-1) expressed by B16F10 tumors.17 Although administration of MDNA11 monotherapy to mice with pre-established B16F10 tumors delayed growth and extended overall survival, administration of the TA99 mAb alone showed no benefit nor prolonged animal survival (figure 3A–B). Therapeutic efficacy and survival benefit were observed with the combination of MDNA11 and TA99, but these effects appeared to be driven mostly by MDNA11.
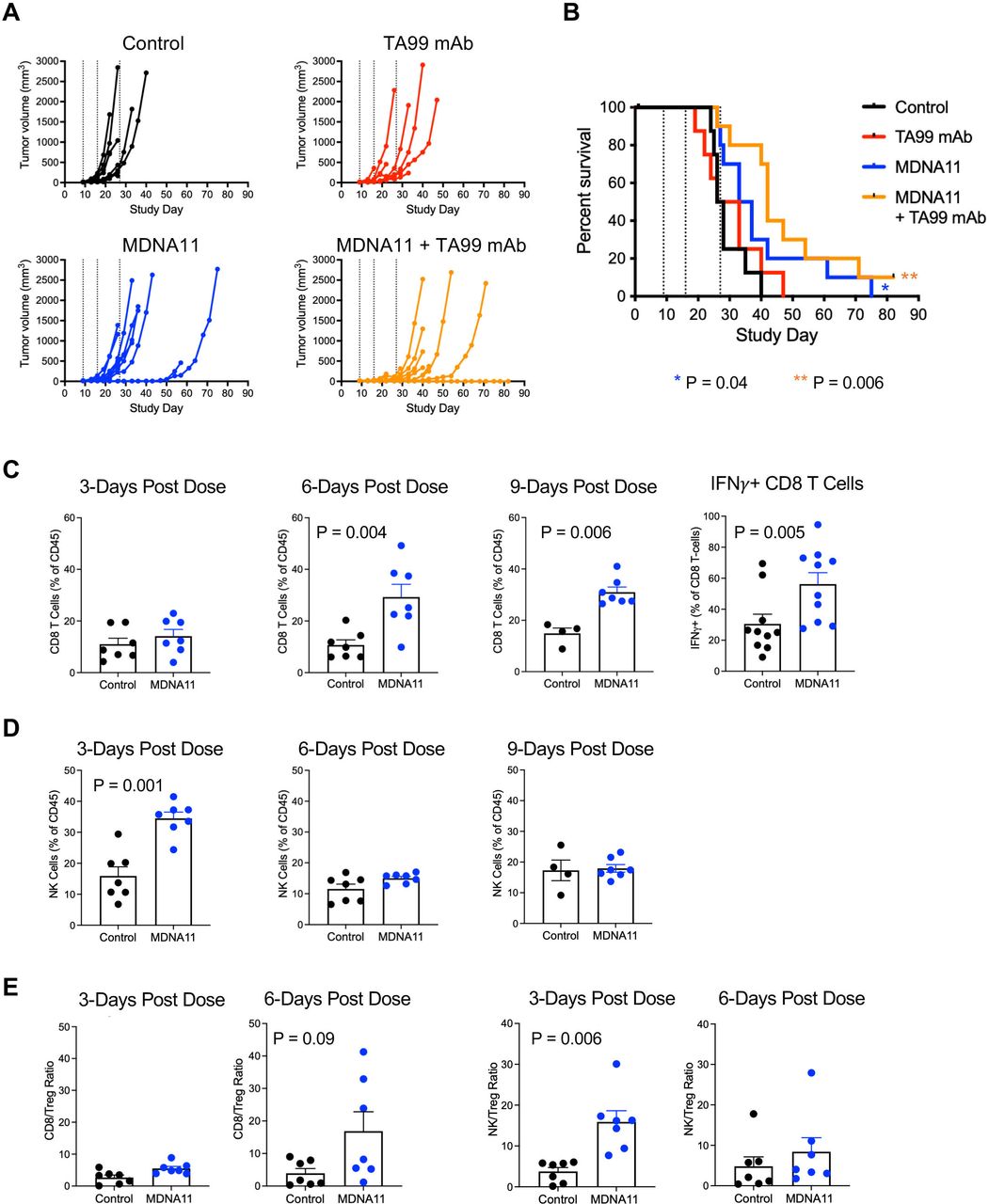
Therapeutic activity of MDNA11 as a monotherapy or in combination with the TA99 mAb in the B16F10 melanoma model. (A) Individual tumor growth of each treatment group (N=8–10 per group). Dotted lines indicate MDNA11 treatment. (B) Kaplan-Meier survival curves of different treatment groups followed for 82 days. P values for comparison to control using the Mantel-Cox test. (C) Analysis of tumor-infiltrating CD8 T cells at 3, 6 and 9 days post treatment, including intracellular IFNγ expression at 6 days post treatment. (D) Analysis of tumor-infiltrating NK cells at 3, 6 and 9 days post treatment. (E) Ratio of CD8 T cells/Tregs (left two panels) and NK cells/Tregs (right two panels). Data are represented as mean±SEM. Statistical analysis performed using the Mann-Whitney non-parametric test. IFN, interferon; mAb, monoclonal antibody; NK, natural killer; Tregs, regulatory T cells.
To elucidate the mode of action of MDNA11 in this model, B16F10 tumors were harvested 3, 6 and 9 days after administering of MDNA11 to analyze TILs by flow cytometry. Tumors isolated from MDNA11-treated mice exhibited significantly higher infiltrating CD8 T cells at 6 and 9 days after treatment (figure 3C) with a limited increase in Tregs as reflected by the elevated CD8 T cell/Treg ratio compared with tumors from control mice (figure 3E). In addition, percentage of tumor infiltrating CD8 T cells with intracellular IFNγ was statistically higher in MDNA11 treated mice (figure 3C), consistent with enhanced CD8 T cell activation. Infiltration of NK cells peaked earlier at Day 3 (figure 3D) and similarly resulted in an increased NK/Treg ratio (figure 3E). Altogether, these data indicated that MDNA11 promotes CD8 T and NK cell recruitment resulting in delayed B16F10 tumor growth.
MDNA11 potently inhibits MC38 colon tumors as monotherapy and in combination with anti-PD1
Therapeutic effect of MDNA11 as monotherapy or in combination with an anti-PD-1 immune checkpoint inhibitor (ICI) was evaluated in the MC38 colon cancer model. Anti-PD1 alone had no effect on the growth of MC38 tumors, whereas MDNA11 demonstrated a potent inhibitory effect when administered once weekly for a total of two doses (figure 4A). Administration of a high dose MDNA11 (5 mg/kg) in fact resulted in tumor clearance in all treated mice, demonstrating strong therapeutic efficacy as monotherapy. A lower dose of MDNA11 (2 mg/kg) although inhibited tumor growth, did so to a lesser degree (7 of 10 CR). Combination of low dose MDNA11 (2 mg/kg) with anti-PD1 was able to achieve complete tumor regression (10 of 10), however, it was not clear whether this outcome was additive or synergistic.
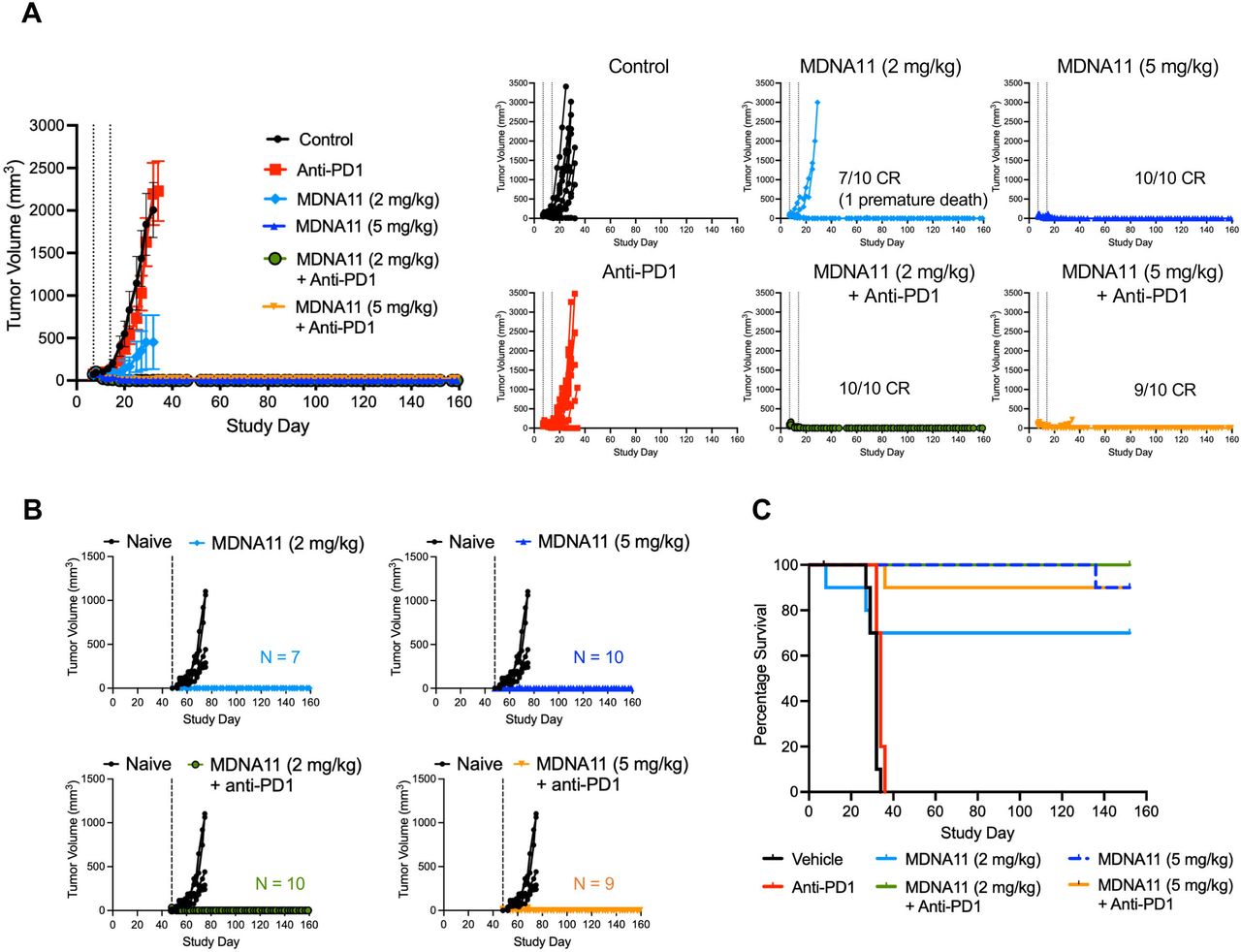
Therapeutic potency of MDNA11 as monotherapy and in combination with anti-PD1 in a MC38 colon cancer model. (A) Average tumor size per group (mean±SEM) with individual tumor growth of each treatment group represented on the right (N=10 per treatment group). CR indicates complete tumor regression. Dotted lines indicate MDNA11 treatment. (B) Mice with CR were rechallenged with MC38 tumor cells on Study Day 48. Naïve untreated mice were used as controls. Broken vertical lines indicate rechallenge. (C) Kaplan-Meier survival curves of different treatment groups up to Study Day 159.
MC38 tumor bearing mice that experienced tumor regression following treatment were subjected to a MC38 rechallenge to investigate whether they have developed an immunologic memory against these tumors. Whereas naïve mice experienced robust MC38 tumor growth, all previously treated mice showed no evidence of growth at the rechallenge site in the absence of any further treatments (figure 4B) and this was associated with significantly longer survival (figure 4C). These data indicated that treatment of mice with MDNA11 as a monotherapy or in combination with anti-PD1 antibody resulted in durable regression of their primary tumors and development of a memory response against these tumors.
MDNA11 synergizes with anti-CTLA4 to control CT26 tumors and induce long-term memory and antigen-specific CD8 T cells
We evaluated the potential synergy between MDNA11 and anti-CTLA4 in a syngeneic CT26 colon cancer model. Although MDNA11 and anti-CTLA4 administration to mice with pre-established CT26 tumors demonstrated effective tumor growth control as single agent (50% CR respective monotherapy arms), treatment with the combination cured all (100% CR) mice (figure 5A). To assess whether cured animals developed protective memory responses, several rechallenges were performed at independent sites on days 49, 116 and 165 of the study. As shown in figure 5B, there was no tumor growth following each rechallenge in contrast to the control non-treated naïve mice implanted with CT26 tumor cells. We next asked whether resistance to multiple rechallenges was driven by the development of memory T cells. Increased frequencies of central (CD44hiCD62Lhi) and effector (CD44hiCD62Llo) CD8 and CD4 memory T cells were indeed detected in all treatment groups but with highest frequencies in the MDNA11/anti-CTLA4 combination (online supplemental figure S3). At the end of the experiment, mice were rechallenged for a fourth time with CT26 tumor cells to boost pre-existing memory cells and sacrificed 5 days later (Study Day 270) to quantify CD8 T cells specific to the CT26 tumor-associated antigen gp70.18 As expected, higher frequencies of gp70-specific CD8 T cells were observed in the blood and spleen of mice previously treated with MDNA11 and were further enhanced when combined with anti-CTLA4 (figure 5C). Elevated level of memory T cells together with increased frequency of gp70-specific CD8 T cells in MDNA11-treated animals is consistent with their acquired resistance to multiple CT26 rechallenges and durable survival benefits (figure 5D).

MDNA11 with anti-PD1 to potently control CT26 tumors, induced memory response and antigen specific CD8 T cells. (A) Average tumor volume (±SEM) of treatment groups (N=8; left) and individual tumor growth curves (right). Dotted lines indicate MDNA11 treatment. CR indicates complete tumor regression. (B) Mice with CR were rechallenged with CT26 tumor cells on Day 49, 116 and 165 (broken vertical lines). Naïve untreated mice were used as controls. (C) Analysis of gp70-specific CD8 T cells in spleen and blood at study termination. *P<0.05 compared with control (Mann-Whitney non-parametric test). (D) Kaplan-Meier survival curves of different treatment groups followed for 270 days. (E) Effect of CD8 T and NK cell depletion on the therapeutic efficacy of MDNA11. Black arrows (Study Days 8, 20 and 30) indicate immune depletion and blue arrows (Study Days 14 and 24) indicate MDNA11 administration. Statistical analysis based on data at Study Day 40 was performed using the Mann-Whitney non-parametric test. CR, complete tumor regression; NK, natural killer.
To investigate the mechanism of MDNA11 mediated tumor growth control, CT26 tumor bearing mice were treated with antibodies to selectively deplete CD8 T cells or NK cells (online supplemental figure S4) while undergoing treatment with MDNA11. In both cases, tumor growth inhibition was significantly reduced compared with treatment with MDNA11 alone, indicating that both CD8 T cells and NK cells are involved in MDNA11 elicited antitumor efficacy (figure 5E).
MDNA11 induces durable immune response in NHP
Cynomolgus monkeys are highly suitable for testing immuno-oncological compounds19 20 and were used to evaluate the safety and PD profile of MDNA11. Binding studies showed MDNA11 binds human and cynomolgus IL-2 receptors with similar selectivity and affinity (table 1). Male and female monkeys (2–4 years of age) received vehicle (N=10) or MDNA11 by slow intravenous bolus injection at 0.15 mg/kg (N=6), 0.3 mg/kg (N=6) or 0.6 mg/kg (N=10) on Study Day 1 (first dose) and 15 (second dose) to closely mimic the planned clinical treatment regimen, and a third dose on Study Day 29 before undergoing necropsy 2 days later. MDNA11 was well tolerated with all animals exhibiting stable body weight during the study (online supplemental figure S5). Untoward clinical observations were noted but these occurred primarily in the highest dose group (0.6 mg/kg) following the first dose and were less prevalent after the second and third doses. The most common clinical observations included diarrhea and reduced activity that were generally transient and resolved within several days.
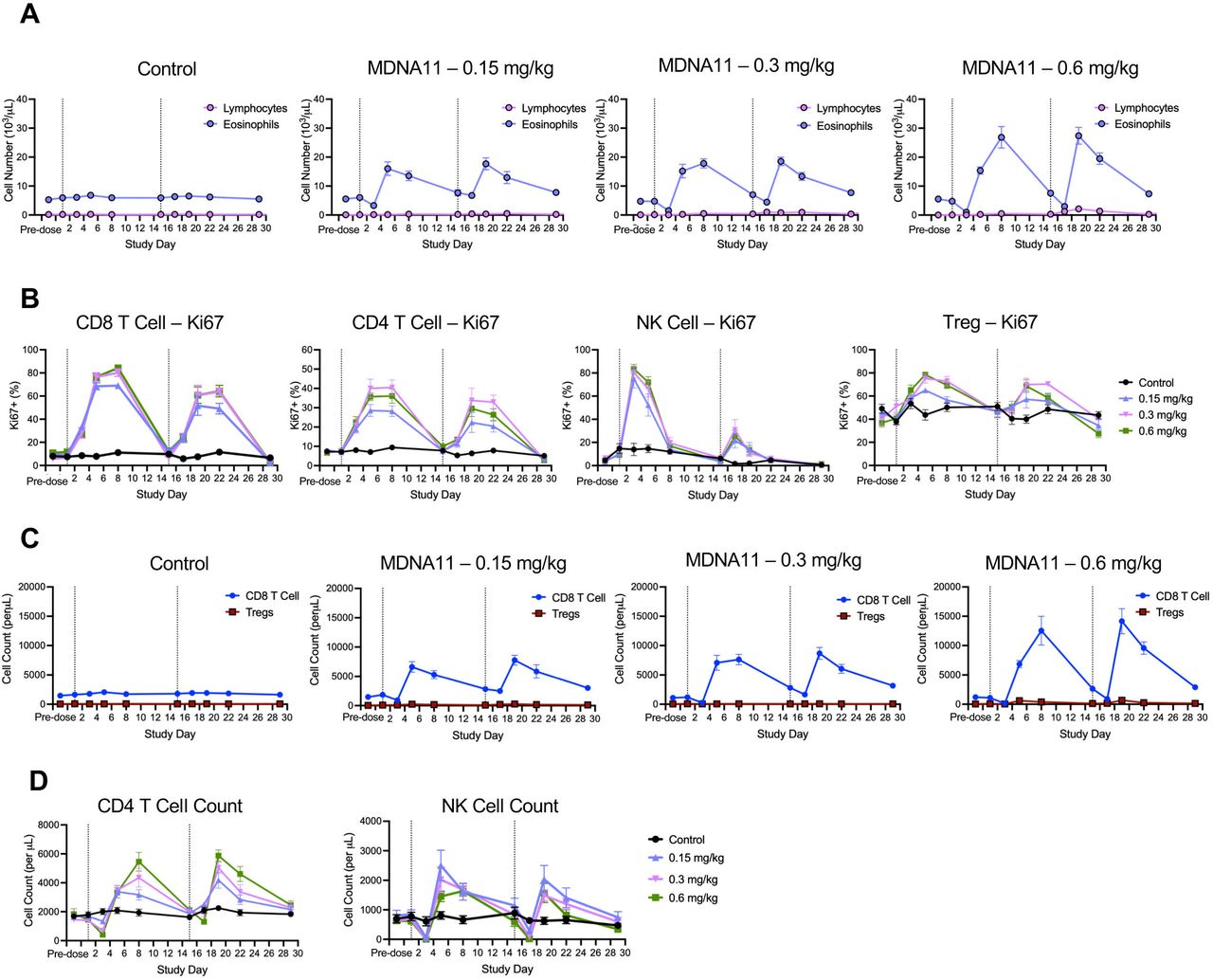
MDNA11 induced robust expansion of lymphocytes following administration on Study Days 1 and 15 (figure 6A). Little to no effect up to a dose of 0.3 mg/kg was observed in the eosinophil count, an innate immune subpopulation associated with the development of VLS21 22 (figure 6A). There was a more notable increase in eosinophil after the second dose among animals in the 0.6 mg/kg group; however, the cell count remained very low relative to lymphocytes. Detailed analysis of various immune subsets revealed a durable and dose-dependent effect of MDNA11 on CD8 and CD4 T cell proliferation as measured by Ki-67 expression (figure 6B). Proliferation remained elevated for at least 7 days following dosing in both of these immune populations. Effect of MDNA11 on NK cell proliferation appeared shorter and peak levels of Ki67 following the second dose (Day 15) were lower than those observed following the first dose (Day 1). Tregs showed high baseline levels of Ki67 that also increased following treatment with MDNA11 (figure 6B), but this translated to a limited increase in cell number (figure 6C). In contrast, a robust and more durable CD8 T cell expansion following both doses (figure 6C) was observed where majority were naïve CD8 T cells (CD25–) but the minor activated population (CD25+) was also responsive to MDNA11 (online supplemental table S6). Consistent with the increase in Ki67 expression, dose-dependent cell expansion was also observed with CD4 T cells and NK cells (figure 6D). Interestingly, the increase in NK cell number did not show a marked difference between the first and second dose compared with a much lower Ki67 response observed following the second dose. Altogether, these results demonstrate that MDNA11 induced a potent and durable immune response as prospective evidence for therapeutic efficacy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MDNA11 induces durable immune cell proliferation and expansion in non-human primate. (A) Kinetics of lymphocyte and eosinophil expansion following treatment with vehicle (control; N=10) and MDNA11 at 0.15 mg/kg (N=6), 0.3 mg/kg (N=6) and 0.6 mg/kg (N=10). Each treatment group is comprised of equal numbers of male and female animals. (B) Kinetics of proliferating CD8 T cells, CD4 T cells, NK cells and Tregs as measured by Ki67 expression. (C) Comparison of CD8 T cell and Treg expansion following administration of different doses of MDNA11. (D) Kinetics of CD4 T and NK cell expansion. All data represented as mean±SEM. Dotted vertical lines indicate treatment with MDNA11 (Days 1 and 15). NK, natural killer.
Discussion
While rhIL-2 has shown durable responses in a subset of metastatic melanoma and patients with renal cell carcinoma,3 5 its utility as an immunotherapeutic drug remains limited by a short half-life, undesirable activation of Tregs and toxicities including hypotension, capillary leak syndrome, cardiac complications, and renal failure.23–25 As a result, different strategies were explored to overcome these limitations, most of which involved post-translational modifications or structural re-engineering of rhIL-2 as well as de novo protein design, to mitigate CD25 binding and reduce clearance.9–11 However, in most cases binding to CD122 is either similar or reduced when compared with rhIL-2, resulting in no improvement or even a decrease in potency on effector immune cells that are required for therapeutic efficacy. In this study, we describe a long-acting IL-2 superkine with selectively enhanced binding to CD122 and no interaction with CD25 (ie, Beta-only), resulting in a potent and durable immunological and therapeutic responses in preclinical models.
Modulation of immune response for immuno-oncological application is achieved through fine tuning the differential signaling through the IL-2 receptors. Therapeutics targeting this pathway must provide sufficient activation to expand CD8 T cells without triggering a compensatory stimulation of Tregs. This was achieved with MDNA11 by enhancing affinity for CD122 while mitigating binding to CD25. Enhancement in receptor selectivity culminates in preferential signaling through the STAT5 pathway in resting NK and naïve CD8 T cells while achieving reduced Treg stimulation. In vivo, the therapeutic activity of MDNA11 is improved by extension of half-life through fusion with human albumin to increase molecular weight and reduce clearance via renal filtration and benefiting from recycling via the neonatal Fc receptor.15 Although beyond the scope of this study, the presence of human albumin in MDNA11 may also improve the pharmacologic activity of MDNA11 due to albumin’s natural propensity to accumulate in highly vascularized sites including solid tumors as previously shown with chemotherapeutic drugs.26
Therapeutic activity of MDNA11 was investigated in three murine cancer models. Treatment of the B16F10 melanoma model with MDNA11 resulted in a delay in tumor growth and extended survival due to preferential infiltration of NK and CD8 T cells. There was also evidence of enhanced activation of CD8 T cells within the tumors based on intracellular IFNγ expression. Interestingly, no additional therapeutic benefits were observed by combining MDNA11 with the TYRP-1-specific TA99 mAb. This is in line with another study reporting an absence of therapeutic benefits with infrequent dosing of TA99 mAb (~4–5 doses) as opposed to extended daily dosing (>20 doses),17 presumably reflecting the need for sustained exposure of tumors to this therapeutic mAb.
Treatment with MDNA11 alone induced tumor clearance in 50% of CT26 tumor bearing animals and 100% of tumors with MC38 tumors, with no evidence of relapse months after treatment had ended. The fact that this potent therapeutic efficacy was achieved with only two weekly doses suggests that the pharmacological effect of MDNA11 extends well beyond its already prolonged half-life. Notably, cell depletion studies showed that both CD8 T cells and NK cells are important for efficacy in the CT26 tumor model. Combination of MDNA11 with anti-PD1 (in MC38 model) or anti-CTLA4 (in CT26 model) demonstrated the potential for combination of MDNA11 with ICIs. Due to the therapeutic efficacy of MDNA11 monotherapy at the tested dose levels, it was not clear whether benefits derived from these combination treatments were additive or synergistic.
Induction of cell death can trigger the immune system to specifically target antigens from dead cells in a process known as immunologic cell death.27 Mutations in actively expressed genes can trigger the adaptive immune system to target neoplastic cells. Overexpression of IL-2 enhances a tumor vaccine by activating cytotoxic T lymphocytes and eliciting a memory response.28 MDNA11 monotherapy or in combination with ICI induced durable tumor regression of CT26 and MC38 tumors and therefore enabled these mice to undergo additional tumor rechallenges to determine whether they developed a memory response against these tumors. In both models, all previously cured animals rejected tumor growth at the re-challenge site, indicating that they have indeed developed a potent memory response. In the CT26 tumor model in particular, mice were resistance against three different rechallenges, with the last occurring more than 3 months after treatment with MDNA11. These animals also exhibited increased number of circulating memory T cells of both CD4 and CD8 lineages. Following a short-term exposure to CT26 tumor cells, these mice were found to have increased frequency of CD8 T cells against the CT26 tumor-associated antigen gp70, particularly those that had been treated with the combination of MDNA11 and anti-CTLA4. In summary, treatment with MDNA11 with or without an ICI was effective at not only controlling growth of primary tumors but also induced an antigen-specific memory response to protect against tumor re-exposure. A published report has also demonstrated the therapeutic benefit of combining an IL-2 superkine with a tyrosine kinase inhibitor to treat immunological cold tumors,29 underscoring the potential therapeutic versatility of MDNA11.
The safety and PD profile of MDNA11 were investigated in NHP to support the FIH study. MDNA11 was well tolerated up to 0.3 mg/kg on a bi-weekly dosing regimen with untoward clinical signs (ie, diarrhea and reduced activity) observed in animals receiving 0.6 mg/kg and particularly after the first dose. These observations were transient and suggested that 0.6 mg/kg was nearing the maximum tolerated dose in NHP. Treatment with MDNA11 induced robust proliferation and expansion of immune effector lymphocytes without significant effects on Tregs and eosinophilia. The latter observation has important safety implication as eosinophilia is linked to the development of VLS, one of the most common adverse side effects observed following rhIL-2 therapy.22 30 31 Tissue resident group 2 innate lymphoid cells (ILC2) express the CD25 chain and respond to rhIL-2 stimulation, resulting in sustained IL-5 production and eosinophil expansion.32 This effect has been mitigated with MDNA11 by the absence of CD25 binding; consequently MDNA11 is not expected to activate the ILC2/eosinophil axis nor trigger pulmonary edema through activation of CD25-expressing endothelial cells.33
Response of effector immune cells, particularly CD4 and CD8 T cells, to MDNA11 was durable and dose dependent, with proliferation, as measured by Ki67 expression, remaining at or near peak levels for at least 7 days following dosing. Cell counts remained above baseline on Day 15 prior to animals receiving their second dose, indicating that an elevated adaptive immune response could be sustained on a biweekly dosing regimen. While an increase in Ki67 expression was observed in the Treg population, the change in cell number was relatively small when compared with the expansion observed with effector immune cells.
In summary, a durable and potent efficacy together with a favorable safety profile positions MDNA11 as a potential best-in-class long-acting next generation IL-2 therapeutic for cancer immunotherapy. This can also provide a framework for broader applications including vaccination, adoptive transfer of CAR-T or T cell receptor-engineered T cells, combination therapy with other therapeutic antibodies, and development of bispecific molecules to target multiple cytokine pathways.
Data availability statement
Data are available upon reasonable request. Data sets and material/reagents analyzed and/or used in this study are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
The authors would like to thank John Chicca and his team at Molecular Diagnostic Services (San Diego, USA) for their work on in vivo studies; Alex Okyay Kilinc and his team at Morpheus BioLabs (San Diego, USA) for their work on ex vivo studies; Greg Wasney at SickKids’ Structural & Biophysical Core Facility (Toronto, Canada) for his work on BLI Octet. We also thank Chesarahmia Soeandy and Aanchal Sharma for their critical assessment of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RM, MAM, FM, LBP, PL, PS and MDT designed studies and performed data analysis. CG performed data analysis. MDT and CG wrote the paper with contribution from all authors. MDT is acting guarantor.
Funding Studies were funded by Medicenna Therapeutics, Toronto, Ontario, Canada.
Competing interests RM, CG, MAM, FM and MDT are employees of Medicenna and declare competing financial interest. LBP, PL and PS are consultants and were compensated by Medicenna for their work on the studies in this paper.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.