Article Text

Abstract
Several therapeutic monoclonal antibodies (mAbs), including those targeting epidermal growth factor receptor, human epidermal growth factor receptor 2 (HER2), and CD20, mediate fragment crystallizable gamma receptor (FcγR)–dependent activities as part of their mechanism of action. These activities include induction of antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), which are innate immune mechanisms of cancer cell elimination. FcγRs are distinguished by their affinity for the Fc fragment, cell distribution, and type of immune response they induce. Activating FcγRIIIa (CD16A) on natural killer cells plays a crucial role in mediating ADCC, and activating FcγRIIa (CD32A) and FcγRIIIa on macrophages are important for mediating ADCP. Polymorphisms in FcγRIIIa and FcγRIIa generate variants that bind to the Fc portion of antibodies with different affinities. This results in differential FcγR-mediated activities associated with differential therapeutic outcomes across multiple clinical settings, from early stage to metastatic disease, in patients with HER2+ breast cancer treated with the anti-HER2 mAb trastuzumab. Trastuzumab has, nonetheless, revolutionized HER2+ breast cancer treatment, and several HER2-directed mAbs have been developed using Fc glyco-engineering or Fc protein-engineering to enhance FcγR-mediated functions. An example of an approved anti-HER2 Fc-engineered chimeric mAb is margetuximab, which targets the same epitope as trastuzumab, but features five amino acid substitutions in the IgG 1 Fc domain that were deliberately introduced to increase binding to activating FcγRIIIa and decrease binding to inhibitory FcγRIIb (CD32B). Margetuximab enhances Fc-dependent ADCC in vitro more potently than the combination of pertuzumab (another approved mAb directed against an alternate HER2 epitope) and trastuzumab. Margetuximab administration also enhances HER2-specific B cell and T cell–mediated responses ex vivo in samples from patients treated with prior lines of HER2 antibody-based therapies. Stemming from these observations, a worthwhile future goal in the treatment of HER2+ breast cancer is to promote combinatorial approaches that better eradicate HER2+ cancer cells via enhanced immunological mechanisms.
- adaptive immunity
- immunity
- innate
- review
- breast neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Trastuzumab, a humanized human epidermal growth factor receptor 2 (HER2)-directed monoclonal antibody (mAb), increases disease-free survival (DFS) and overall survival (OS) in early stage HER2-overexpressing/amplified breast cancer and improves progression-free survival (PFS) and OS in metastatic HER2-positive (HER2+) disease.1 2 Trastuzumab was the first humanized mAb approved for cancer treatment and the first biologic agent approved for treatment of breast cancer. Since its initial regulatory approval in 1998, it is estimated that trastuzumab has been given to more than 2.5 million women worldwide and is on the WHO’s list of essential medicines. Trastuzumab has revolutionized therapy of HER2+ breast cancer.1 2 Both non-immune and immune-mediated mechanisms account for trastuzumab’s clinical activity. Non-immune-related mechanisms result directly from binding of antibody fragment antigen-binding (Fab) domains to HER2 receptors on the tumor cell surface, causing perturbation of HER2-signaling and resulting in antiproliferative effects.2 Immune-related mechanisms result from engagement of fragment crystallizable (Fc) domains of tumor cell-bound antibodies with Fc receptors (FcRs) expressed by immune cells. FcRs mediate cross talk between innate and adaptive immune responses and display polymorphic variants that exhibit different activation properties (figure 1).3–8

Mechanism of action of anti-HER2 mAbs: antiproliferative effects and immune activation. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; FcγR, fragment crystallizable gamma receptor; HER2, human epidermal growth factor receptor 2; mAb, monoclonal antibody; MHC II, major histocompatibility complex class II; NK, natural killer; TAA, tumor-associated antigen. The red X in the left panel indicates inhibition.
Fcγ receptors
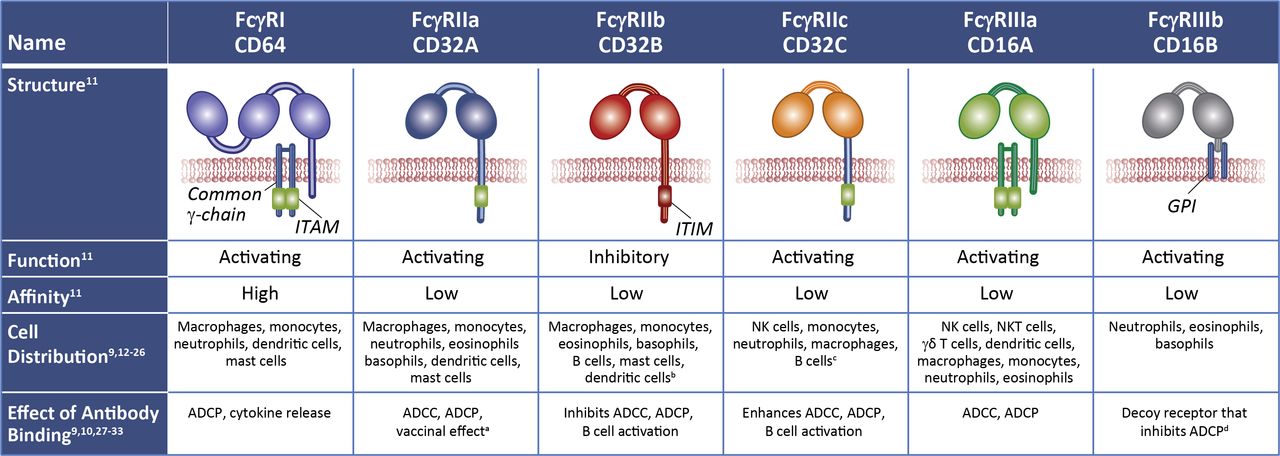
FcRs are expressed on immune cells and bind the Fc portion of immunoglobulin.9 10 Fcγ receptors (FcγRs), the largest group of FcRs, bind IgG and comprise several subtypes (figure 2).9–33 Low-affinity FcγRs, with binding affinities ranging from 30 nM to 1000 nM, are important mediators of antibody functions in vivo, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and induction of cytokines and chemokines.11 Under physiologic conditions, low-affinity FcγR binding is a function of avidity that occurs via multimerization in immune complexes or by cell opsonization.11 Low-affinity FcγRs include two activating receptors, FcγRIIIa (CD16A) and FcγRIIa (CD32A), as well as the sole inhibitory receptor, FcγRIIb (CD32B).34 Activating FcγRs signal via their immunoreceptor tyrosine-based activation motifs.9 34 The inhibitory FcγR contains an immunoreceptor tyrosine-based inhibition motif that counters cell activation when inhibitory and activating receptors become co-engaged.9 34

FcγRs differ in their function, cell distribution, immune response, signaling motifs, and affinity for IgG molecules. aDendritic cells internalize Ag:Ab immune complexes and present Ag to T cells. bCD32B is expressed on NK cells in ~3% of humans due to an FCGR2C-FCGR3B gene deletion that links the FCGR2C promoter to the FCGR2B coding sequence. cCD32C is expressed in ~20% of humans due to an unequal crossover of FCGR2A and FCGR2B genes. dSame ECD as CD16A but lacks intracellular signaling motifs. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; ECD, extracellular domain; FcγR, fragment crystallizable gamma receptor; GPI, glycophosphatidylinositol; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based inhibition motif; NK, natural killer; NKT, natural killer T cell.
Monocytes, macrophages, and dendritic cells express both low-affinity activating FcγRs and the inhibitory FcγR, with antibody-mediated activation of these innate immune cells influenced by the FcγR activating/inhibitory ratio.10 Monocytes, macrophages, and dendritic cells also express FcγRI, a high-affinity receptor that binds monomeric uncomplexed IgG molecules.11 FcγRI saturation by endogenous circulating IgGs in vivo may attenuate its role in mediating antibody function.35 Other circulating blood cells generally express more limited repertoires of FcγRs.11 36 Natural killer (NK) cells mainly express FcγRIIIa.11 36 NK cells and monocytes may also express FcγRIIc (CD32C); however, the gene for this receptor is polymorphic at a site that determines a premature stop codon in one of its exons; hence, translation yields a functional activating FcγR in only a minority (~20%) of humans.17 22 An even smaller subset (~3%) of humans have NK cells that also express FcγRIIb, the inhibitory receptor, due to a chromosomal deletion that juxtaposes the promoter of the gene for FcγRIIc with the coding sequences for FcγRIIb.16 37 Neutrophils express high levels of FcγRIIIb (CD16B), a glycosylphosphatidyl-inositol–linked receptor that lacks intracellular signaling motifs and likely serves as a decoy receptor.32 Neutrophils also express FcγRI, FcγRIIa, and low levels of FcγRIIIa.30 Platelets express FcγRIIa.38 B cells mainly express inhibitory receptor FcγRIIb,11 where it counters activation mediated by the B cell receptor. B cells may also express FcγRIIc, but only in a minority (~20%) of humans.27 36 While naïve T cells do not express FcγRs, subsets of activated CD4+ T cells may express FcγRIIIa, FcγRIIa, and FcγRIIb,39 40 and subsets of activated memory CD8+ T cells may express FcγRIIb.41 CD56+ (neural cell adhesion molecule) subsets of CD3+ T cells express FcγRs and are capable of mediating ADCC, such as NK T cells expressing FcγRIIIa and γδ T cells expressing FcγRIIa and FcγRIIIa.24 These small subsets of FcγR-expressing T cells potentially could influence the linkage between innate and adaptive immunity. Inhibitory receptor FcγRIIb is also expressed at high levels in non-immune cells, such as endothelial and some stromal cells, where it functions as a scavenger receptor and mediates clearance of small immune complexes.42
FcγR polymorphisms
Numerous polymorphisms and allelic variations have been identified for FcγRs.10 43 FcγRIIa and FcγRIIIa allelic polymorphisms generate variants that bind to IgG Fc with different affinities correlating with relative ADCC potency. FcγRIIa 131H (histidine) and FcγRIIIa 158V (valine) have a higher affinity for IgG Fc, compared with FcγRIIa 131R (arginine) and FcγRIIIa 158F (phenylalanine), respectively. FcγRIIb allelic polymorphism generates variants with altered signaling function. The FcγRIIb 232T (threonine) signaling variant is unable to associate with lipid rafts, resulting in impaired negative regulatory activity compared with FcγRIIb 232I (isoleucine). Prevalence of these polymorphisms in the general population, including healthy subjects and patients with cancer, is provided in table 1 and online supplemental table 1.44–73 FcγRIIIa-158V homozygotes represent ~12% of the population, across race.44–64 FcγRIIa-131H homozygotes represent ~27% of Caucasians and Blacks44 45 49 50 54–56 58 65–67 versus ~60% of Asians.48 59 60 62 FcγRIIb-232T homozygotes represent ~2% of Caucasians54 56 58 68–70 and ~6% of Asians or Blacks.48 68 70–73
Supplemental material
Prevalence of polymorphic variants of FcγRIIIa, FcγRIIa, and FcγRIIb, based on available literature
ADCC mediated by therapeutic mAbs
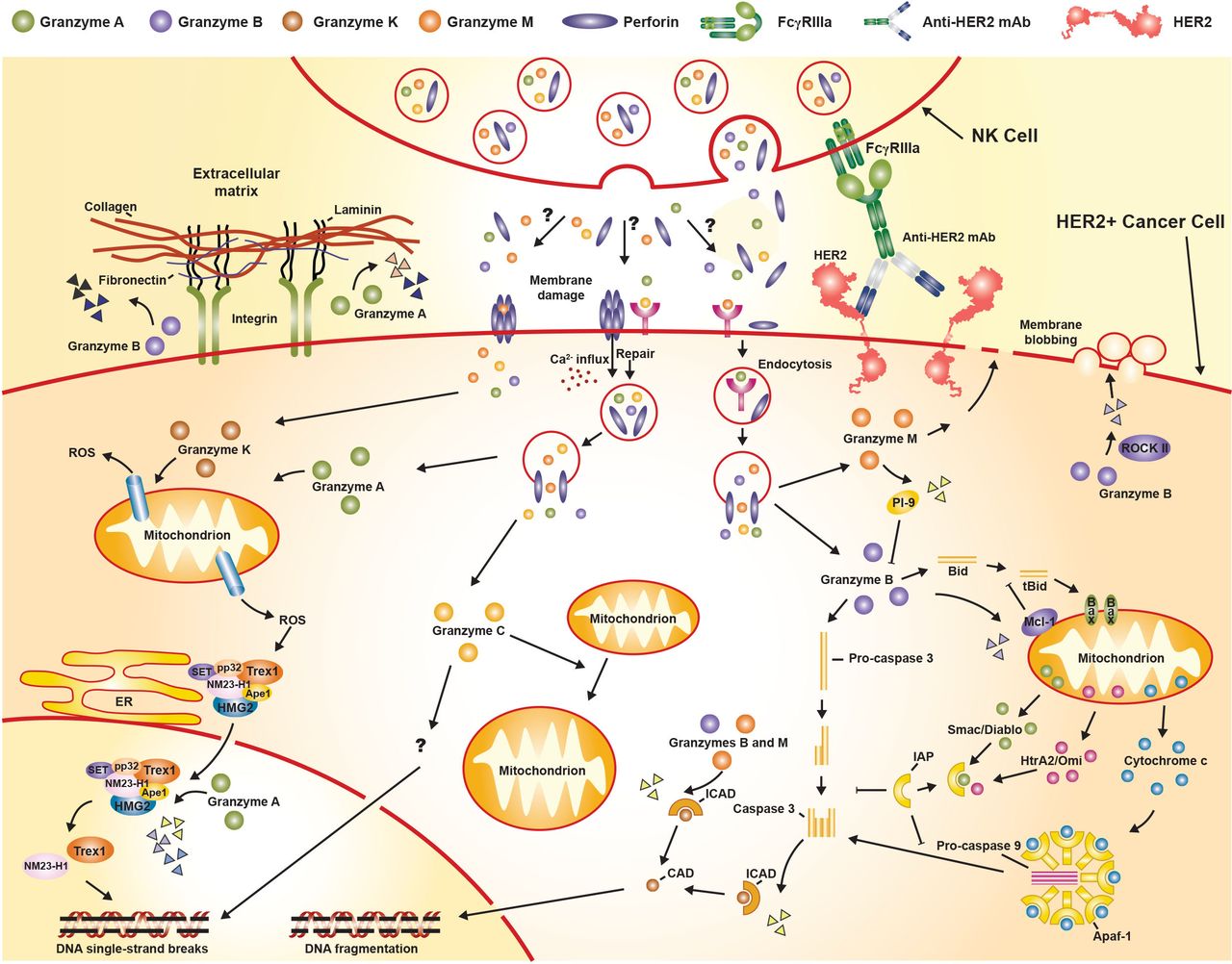
ADCC is an Fc-dependent mechanism mediated by innate immune cells.11 36 ADCC by NK cells is mediated through its FcγRIIIa receptors,28 74 which bind Fc regions of tumor-bound antibodies to form immunological synapses, triggering secretion of perforins and granzymes that induce tumor cell death (figure 3).75 76 Five different granzymes have been described in humans: A, B, H, K, and M. Granzyme B induces caspase-dependent apoptosis, whereas granzymes A, H, K, and M induce caspase-independent cell death.75 76

{kind=link}
{kind=link}
{kind=link}
Classical granzyme/perforin-mediated apoptosis pathway (adapted from Bots and Medema76). Bid, BH3 interacting domain death agonist; CAD, caspase-activated DNase; ER, endoplasmic reticulum; FcγR, fragment crystallizable gamma receptor; HER2, human epidermal growth factor receptor 2; HMG2,high-mobility group protein 2; ICAD, inhibitor of caspase-activated DNase; mAb, monoclonal antibody; Mcl-1, myeloid-cell leukemia 1; NK, natural killer; PI-9, proteinase inhibitor 9; ROS, reactive oxygen species; tBid, truncated Bid.
Diverse therapeutic mAbs, targeting CD20, HER2, and epidermal growth factor receptor, are capable of mediating ADCC to eliminate cancer cells.77 Correspondingly, rituximab, ofatumumab, veltuzumab, ocrelizumab, trastuzumab, and cetuximab have demonstrated ADCC activity mediated by human peripheral blood mononuclear cells (PBMCs) or NK cells in vitro.3 77–79 ADCC stimulation by trastuzumab against HER2+ breast cancer cells was demonstrated in ex vivo assays using human PBMCs or NK cells from patients with HER2+ breast cancer treated with trastuzumab.80 Subsequently, FcγR knockout mouse models demonstrated that activating FcγRs mediate in vivo antitumor activity of therapeutic antibodies such as trastuzumab or rituximab, whereas FcγRIIb inhibits in vivo antitumor activity.77 Trastuzumab Fc domain mutants engineered to disrupt FcγR binding also attenuate antitumor effects in vivo.77 Interestingly, in vitro ADCC assays confirm that the HER2-directed antibody drug conjugate ado-trastuzumab emtansine (T-DM1) maintains ADCC activity of trastuzumab, which potentially may supplement the antitumor activity mediated by its cytotoxic payload.81
Pertuzumab binds independently (ie, without competing with trastuzumab) to a distinct epitope on HER2, which overlaps the dimerization domain; however, it carries the same wild-type IgG1 Fc domain as trastuzumab.82 83 Pertuzumab is more effective than trastuzumab in inhibiting HER2 dimer formation with other HER family members, such as HER3 or HER1.83 Both pertuzumab and trastuzumab can trigger ADCC.83 It is postulated that simultaneous binding of trastuzumab and pertuzumab to HER2 enhances the density of FcγR binding sites on HER2+ tumor cells, increasing the possibility for NK cell–mediated ADCC and macrophage-mediated ADCP antitumor responses.74 When the antibodies were combined, a modest additive effect on ADCC was reported.83 However, the combination of trastuzumab and pertuzumab exhibited strongly enhanced antitumor activity in nude mice bearing HER2-overexpressing human KPL-4 breast cancer xenografts, which appears to be solely attributed to Fc-dependent effects because KPL-4 cells are resistant to the direct, antiproliferative effects of trastuzumab or pertuzumab.83 In the metastatic (CLEOPATRA)84 and neoadjuvant (NeoSphere)85 settings of HER2+ breast cancer, significantly improved responses were observed when trastuzumab and pertuzumab were combined. In CLEOPATRA, the end-of-study result for median OS was 57.1 months (95% CI 50 to 72) for trastuzumab + pertuzumab + docetaxel compared with 40.8 months (95% CI 36 to 48) for trastuzumab + docetaxel (HR 0.69; 95% CI 0.58 to 0.82).84 In NeoSphere, the 5-year DFS result was 84% (95% CI 72% to 91%) for trastuzumab + pertuzumab + docetaxel compared with 81% (95% CI 72% to 88%) for pertuzumab + docetaxel (HR 0.60; 95% CI 0.28 to 1.27).85
Improved clinical responses are observed when trastuzumab or trastuzumab + pertuzumab therapy is combined with taxane-based chemotherapy.2 85 Taxanes are microtubule assembly inhibitors that disrupt cell division, but they can also exert effects on the immune system by inducing immunogenic cell death and triggering immunostimulatory stress responses.86 For example, trastuzumab + taxane treatment of patients with HER2+ breast cancer has been shown to increase NKG2D expression on circulating NK cells and enhance trastuzumab-mediated ADCC measured ex vivo.87 NKG2D is a key receptor for NK cell activation that recognizes ligands (MHC class I chain-related A or B (MICA or MICB) proteins or UL-16 binding proteins), which can be induced on breast tumor cells after taxane treatment.
Immunoglobulin Fc region glycosylation can significantly affect receptor binding and ADCC activity. Hence, robust quality management systems are important to ensure process and product consistency when manufacturing such mAbs and their biosimilars.88 High levels of fucosylated glycans in the Fc region of some trastuzumab lots were found to be associated with reduced binding of trastuzumab’s Fc region to FcγRIIIa, which led to decreased ADCC activity.88 These commercial reference lots of originator trastuzumab were used in the control arm of the phase III neoadjuvant trial of the Samsung trastuzumab biosimilar SB3 in patients with HER2+ early breast cancer.89 Higher event-free survival rates in the SB3 versus control arm were attributed to altered lots of trastuzumab used in the trial, underscoring the importance of Fc-dependent mechanisms, including ADCC, in clinical outcome.89 High levels of Fc region mannose glycans observed in other lots of trastuzumab were found to cause increased binding of trastuzumab Fc to FcγRIIIa, which led to an increase in ADCC activity.88
Fc-mediated activities of the antibody-drug conjugates (ADC) T-DM1, DS-8201a (or fam-trastuzumab deruxtecan-nxki), and SYD985 (or vic-trastuzumab duocarmazine), do not appear to be altered compared with the unconjugated parental antibody, since in vitro ADCC mediated by these ADCs was comparable to unconjugated trastuzumab.81 90 91
Strategies to enhance ADCC activity of therapeutic mAbs
Diverse mAbs targeting various antigens have been developed using Fc glyco-engineering or Fc protein-engineering.92 Antibody Fc glyco-engineering specifically improves FcγRIIIa binding affinity, which can enhance ADCC.93 94 As an example, obinutuzumab is a CD20-directed mAb that binds to a distinct but overlapping epitope compared with rituximab and has an afucosylated Fc domain that allows ADCC to be mediated more effectively in vitro than rituximab;93 obinutuzumab is approved for treatment of patients with chronic lymphocytic leukemia or follicular lymphoma. Compared with trastuzumab, afucosylated trastuzumab enhanced ADCC mediated by human NK cells in vitro, delayed tumor progression in xenograft models of HER2-amplified breast cancer in immune-deficient mice transgenic for human FcγRIIIa-158F (the weaker binding variant), and improved antitumor responses in patients with solid tumors.95 96
Fc protein-engineering represents another strategy to enhance ADCC activity. For example, margetuximab, approved for HER2+ metastatic breast cancer (MBC), is an Fc-engineered anti-HER2 mAb that targets the same epitope as trastuzumab.97 Five amino acid substitutions in the IgG1 Fc domain (L235V/F243L/R292P/Y300L/P396L) led to increased binding to activating FcγRIIIa, but also decreased binding to inhibitory FcγRIIb.97 98 Margetuximab mediated enhanced ADCC in vitro compared with trastuzumab. In a xenograft model of HER2-amplified breast cancer in immune-deficient mice transgenic for human FcγRIIIa-158F, margetuximab exhibited greater antitumor activity than an otherwise identical variant with a wild-type IgG1 Fc domain.97 98
Editing of the glycocalyx, a thick coat of proteins and carbohydrates on the outer surface of tumor cells, with an antibody-enzyme conjugate that selectively removes sialic acids also improves ADCC.99 A trastuzumab-sialidase conjugate desialylated tumor cells in an HER2-dependent manner and this led to enhanced ADCC in vitro.99 Another approach to enhance ADCC is combination therapy; for example, treatment with trastuzumab and lapatinib increased ADCC in vitro by stabilizing the display of cell surface HER2.55 100 101 Lastly, a bispecific tribody ([HER2]2×CD16) that comprises two HER2-specific single chain fragment variable domains fused to a Fab specific for the extracellular domain of FcγRIIIa was shown to mediate in vitro ADCC of HER2-expressing tumor cells more efficiently than trastuzumab.102
ADCP mediated by therapeutic mAbs
ADCP, which is mediated by phagocytic cells such as macrophages, monocytes, or neutrophils, is another important Fc-mediated mechanism of action of antibodies that target HER2+ tumors. Macrophages express all classes of FcγRs. Tumor-associated macrophages (TAM) from primary human breast tumors have been shown to promote tumor progression, and increased TAM infiltration often correlates with poor progression.103 Nevertheless, TAMs express elevated levels of activating receptors FcγRIIa and FcγRIIIa and retain the ability to phagocytose tumor cells in an antibody-dependent manner.104 In vitro studies of trastuzumab-mediated ADCP of HER2-overexpressing tumor cells demonstrate that FcγRIIIA has greater influence than FcγRIIa.105 Breast cancer xenograft studies in mice, in which macrophages are depleted by treatment with clodronate liposomes, demonstrate that the antitumor activity of trastuzumab depends on macrophage recruitment in the tumor tissues.21 Combination of trastuzumab and pertuzumab, presumably owing to increased avidity of binding to macrophage FcγRs, elevates ADCP potency in vitro compared with the individual antibodies.105
Strategies to enhance ADCP
The glyco-engineered Fc version of trastuzumab with enhanced FcγRIIIa binding affinity mediates ADCP in vitro better than wild-type trastuzumab.106 Variants with Fc domains engineered for increased affinity for activating FcγRIIa have enhanced ability to mediate ADCP but surprisingly, variants with altered binding affinity to inhibitory FcγRIIb have little effect on ADCP.107 CD47 is an antiphagocytic ‘don’t eat me’ signal that is highly expressed in many cancers, including breast cancer, which functions to suppress phagocytosis through binding to and triggering signaling of macrophage SIRPα.108 Combination of trastuzumab and a CD47-blocking antibody (MIAP410) enhanced ADCP in vitro, and in immunocompetent mice bearing HER2+ tumors, the combination improved antitumor responses and prolonged survival due to expansion and activation of TAMs and emergence of a hyperphagocytic macrophage population.109 In another study, combination of trastuzumab with an anti-CD47 mAb (magrolimab: Hu5F9-G4) enhanced ADCP in vitro and improved inhibition of HER2+ xenograft growth in vivo, such that inhibition of tumor growth persisted even after treatment discontinuation. Significantly increased susceptibility to ADCP was also observed in vitro against HER2+ breast cancer cell lines selected for tolerance to trastuzumab-mediated ADCC (yet retained cell surface HER2 expression levels).110 It will be interesting to discover whether margetuximab may further enforce ADCP mechanisms with magrolimab, as compared with trastuzumab. Future clinical translation of combinatorial therapeutic approaches targeting HER2 and CD47 are warranted. Other promising approaches to improve ADCP against HER2-overexpressing tumor cells include combining trastuzumab with B7-H4 blockade111 or combining trastuzumab with a histone deacetylase inhibitor, such as vorinostat or valproic acid.112
FcγR polymorphisms are associated with the clinical outcome of HER2+ breast cancer after trastuzumab in the neoadjuvant, adjuvant, and metastatic settings
FcγRIIIa and FcγRIIa polymorphism effects on breast cancer clinical outcomes have been studied in multiple settings (table 2).
Clinical outcome of HER2-positive breast cancer by FcγR polymorphisms
The neoadjuvant randomized phase II CHER-LOB trial evaluated preoperative chemotherapy plus trastuzumab (Arm A) or lapatinib (Arm B) or trastuzumab and lapatinib (Arm C) in 121 patients with operable HER2+ breast cancer.55 Combined chemotherapy plus trastuzumab and lapatinib (Arm C) provided a statistically significant improvement in pathologic complete response (pCR) rate in the whole-study population.55 Efficacy analysis by FcγRIIIa-158 genotype in 73 patients from the CHER-LOB study showed that pCR rate improvement on chemotherapy plus trastuzumab and lapatinib was restricted to FcγRIIIa-158V carriers (pCR rate in Arm C vs A: 67% vs 27%, p=0.043; Arm C vs B: 67% vs 22%, p=0.012). By contrast, FcγRIIIa-158F homozygotes had no significant improvement in pCR rate on chemotherapy plus trastuzumab and lapatinib (pCR rate in Arm C vs A: 42% vs 25%, p=0.642; Arm C vs B: 42% vs 50%, p=0.737).55 Separately, a small prospective study with 15 patients with early stage HER2+ breast cancer showed that FcγRIIa-131H homozygotes had higher pCR on trastuzumab-based neoadjuvant chemotherapy (71% (5/7)), compared with FcγRIIa-131R carriers (0% (0/8); p=0.015).59 Another small prospective study with 26 patients with HER2+ ductal breast cancer treated with trastuzumab-based neoadjuvant chemotherapy, found that among the 12 patients who achieved pCR, half were FcγRIIa-131R homozygotes (50% (6/12)), while the other half were either FcγRIIa-131H homozygotes (25% (3/12)) or FcγRIIa-131H/R heterozygotes (25% (3/12)), showing a statistically significant association between FcγRIIa-131R homozygous genotype and pCR (p=0.012).113 No association was detected for the FcγRIIIa-158 polymorphism (p=0.590).113
The adjuvant randomized phase III NSABP B-31 trial demonstrated that addition of trastuzumab to postoperative chemotherapy improves outcomes after surgically resected HER2+ breast cancer.114 Retrospective analysis of 1156 patients from this study found that adjuvant chemotherapy plus trastuzumab provided greater DFS benefit in FcγRIIIa-158V carriers (HR 0.31; 95% CI 0.22 to 0.43; p<0.001), compared with FcγRIIIa-158F homozygotes (HR 0.71; 95% CI 0.51 to 1.01; p=0.05).45 Of note, large retrospective analyses of over 1000 patients enrolled in the phase III BCIRG-006 trial or the phase III NCCTG-N9831 study found no correlation between FcγRIIIa or FcγRIIa polymorphisms and outcome (DFS) with adjuvant trastuzumab in early breast cancer.46 56 The analysis on patients from the NCCTG-N9831 trial found a statistically significant treatment interaction between the FcγRIIb polymorphism (I/I vs T carriers) and treatment arms (p=0.03), with FcγRIIb-232I homozygotes treated with adjuvant trastuzumab having a better DFS than those treated with chemotherapy alone (p<0.0001).56 Both studies had sampling biases that may have reduced power to detect genotype-treatment interaction. In BCIRG-006, the subset of genotyped patients did not show significant benefit from trastuzumab, unlike the effect seen in the overall BCIRG-006 population. Also, FcγRIIIa-158 genotype frequencies significantly deviated from Hardy-Weinberg equilibrium. By contrast, genotyped patients in the NCCTG-N9831 study had substantially better DFS than the entire NCCTG-N9831 population. A subgroup analysis conducted on 132 patients enrolled in the phase III UNICANCER-PACS04 trial treated with adjuvant chemotherapy followed by trastuzumab found that FcγRIIa-131H carriers had a significantly higher 5-year event-free survival rate (90%), compared with FcγRIIa-131H homozygotes (70%; p=0.0278); whereas, clinical outcome was not associated with FcγRIIIa-158 polymorphism.115
A retrospective analysis of 54 patients with HER2+ MBC receiving trastuzumab plus taxane revealed that FcγRIIIa-158V homozygotes experienced an improvement in PFS, objective response rate (ORR), and ex vivo ADCC activity, compared with FcγRIIIa-158F carriers (median PFS: V/V—not reached, F carriers—12.9 months (p=0.0035), F/V—15.0 months (p=0.008), F/F—11.1 months (p=0.005); ORR: V/V—82%, F carriers—40%, F/V—42%, F/F—35%, p=0.03; normalized ADCC: V/V vs F carrier, p=0.04).54 Finally, the randomized phase III SOPHIA trial (NCT02492711) investigated margetuximab versus trastuzumab, each combined with physician’s choice chemotherapy in 536 patients with MBC after at least two prior anti-HER2 therapies. In SOPHIA, treatment with margetuximab and chemotherapy improved independently assessed PFS over control in the whole-study population.58 Efficacy analysis by FcγRIIIa-158 allele expression in 506 genotyped patients from the SOPHIA study showed that PFS benefit of margetuximab over trastuzumab was increased in FcγRIIIa-158F carriers (median PFS: 6.9 months vs 5.1 months; HR 0.68; 95% CI 0.52 to 0.90; p=0.005).58 Conversely, no margetuximab benefit over trastuzumab was seen in FcγRIIIa-158V homozygotes (HR 1.78; 95% CI 0.87 to 3.62; p=0.110).58 In the SOPHIA trial, no association of FcγRIIa-131 genotypes with benefit was observed for margetuximab, whose engineering did not increase binding to FcγRIIa.58 Of note, there also was no association between margetuximab benefit and FcγRIIb-232 genotypes; however, this signaling polymorphism does not affect IgG1 Fc binding, with margetuximab demonstrating reduced binding to either variant.58 Importantly, Fc domain engineering to enhance immune effector function was shown to be clinically feasible in SOPHIA. Based on a recent press release, the SOPHIA final OS analysis for the intent-to-treat population did not demonstrate a statistically significant advantage in the margetuximab group compared with the trastuzumab group, while a numerical OS advantage was observed in the subgroup of patients homozygous for the FcγRIIIa-158F low-affinity allele. In this trial, similar safety profiles between the margetuximab and trastuzumab treatment groups were observed, with infusion-related reactions more common in the margetuximab group.58 Finally, in the SOPHIA safety database, the adverse event ‘left ventricular cardiac dysfunction’ (all instances of which were asymptomatic and reversible) occurred in seven patients (3%) in each treatment group.58
FcγRIIa gene polymorphisms show more limited influence on outcomes, relative to FcγRIIIa. In a small prospective study of 35 patients with HER2+ MBC treated with trastuzumab, FcγRIIa-131H homozygotes experienced higher ORR and longer PFS compared with FcγRIIa-131R carriers (ORR: H/H—40%, R carriers—10%, p=0.043, Fisher’s exact test; median PFS: 9.2 months vs 3.5 months, p=0.034).59 Similarly, FcγRIIa-131H homozygotes had superior PFS compared with FcγRIIa-131R carriers (HR 0.36; 95% CI 0.16 to 0.82; p=0.02) in a retrospective analysis conducted on 42 patients with metastatic gastric cancer (GC) treated with trastuzumab and chemotherapy.62
FcγRIIIa polymorphism effects on clinical outcomes are also seen in other mAb-treated cancers. Rituximab and cetuximab have been assessed across studies in lymphoma and chronic lymphocytic leukemia (rituximab), colorectal cancer, and head and neck cancers (cetuximab).116 Analyses of rituximab studies did not show statistically different PFS based on FcγR genotype, and analyses of cetuximab studies were inconsistent.116
Association of FcγR genotypes with the clinical activity of immunomodulatory antibodies that target molecules expressed by immune cells also has been investigated, although studies in patients with breast cancer have not been reported in the literature yet. Antibodies targeting programmed cell death protein 1 (PD-1) are most commonly IgG4 isotype or IgG1 isotype engineered for debilitated FcγR binding; these Fc domains are selected for their minimal interaction with FcγRs to focus the antibody effects on blocking PD-1 binding to its ligands, programmed cell death ligand 1 (PD-L1) and PD-L2, and avoiding Fc-mediated deletion of tumor-reactive PD-1-expressing T cells.117 Indeed, anti-PD-1 mAbs with IgG1 isotype are substantially less effective than those with IgG4 isotype.118 Thus, FcγR genotypes are irrelevant for anti-PD-1 mAbs, which is supported by the lack of association between FcγRIIIa polymorphism in patients with advanced melanoma treated with pembrolizumab or nivolumab, both of which are IgG4 isotype.119 By contrast, the activity of anti-PD-L1 mAbs may be enhanced by the IgG1 isotype. Studies in mouse models demonstrated that antitumor activity mediated by PD-L1 mAbs was enhanced by engagement of activating FcγRs and that this effect correlated with elimination of monocytes and modulation of myeloid cells within the tumor microenvironment.118 However, analysis of FcγRIIa and FcγRIIIa polymorphisms showed no impact on PFS in the recent JAVELIN study of avelumab, an anti-PD-L1 antibody of IgG1 isotype, in patients with renal cell carcinoma.120 Fc-mediated effects also contribute to the activity of mAbs targeting cytotoxic T lymphocyte–associated antigen-4. Studies in mouse models demonstrate that the antitumor activity of ipilimumab (IgG1 isotype) is associated with depletion of intratumoral regulatory T (Treg) cells and increases in the CD8+ to Treg cell ratio.119 A meta-analysis of patients with advanced melanoma treated with ipilimumab revealed significantly higher response rates among FcγRIIIa-158V carriers with high insertion-deletion mutations (p=0.016) or high neoantigen burden (p=0.043) compared with FcγRIIIa-158F homozygotes.119 Significantly longer OS was also found in FcγRIIIa-158V carriers with high neoantigen burden (p=0.014) compared with FcγRIIIa-158F homozygotes.119 The same meta-analysis did not find a correlation between FcγRIIa-H131R polymorphisms and response rates or OS.
Promotion of adaptive immunity by FcγRs
FcγRs can play a role in adaptive immune responses.36 121 Innate immune activation is known to contribute to T cell–mediated adaptive antitumor responses.36 ADCC by activated NK cells or ADCP by macrophages or other immune cells cause tumor cell lysis, releasing tumor antigens that can be taken up and displayed on antigen-presenting cells to prime adaptive, T cell–mediated antitumor responses.9 121 Activated NK cell cytokines facilitate macrophage and dendritic cell activation that in turn stimulates cytotoxic T cell migration to the intratumoral space.74 121 ADCC and ADCP induced by treatment with antitumor mAbs generates antibody:tumor antigen immune complexes.29 FcγR-dependent mechanisms contribute to the uptake and processing of these immune complexes by antigen-presenting cells, thus facilitating tumor antigen presentation to T cells, resulting in T cell memory responses and long-term antitumor vaccinal effects.29 Trastuzumab was found to increase HER2 antigen uptake by dendritic cells via an FcγR-mediated mechanism.4 Specifically, increased HER2 antigen uptake resulted in cross-presentation of the E75 peptide, the immunodominant epitope derived from the HER2 protein, by dendritic cells, and this triggered priming of an antitumor immune response with increased antigen-specific cytotoxic T cell generation.4
Innate and adaptive immune systems cooperate in patients with breast cancer after trastuzumab therapy.6 7 122 In a phase II study of neoadjuvant chemotherapy plus trastuzumab in patients with HER2+ breast cancer, those achieving a pCR had increased activated NK cell percentages and multiepitopic, polyfunctional (including HER2-specific) antitumor T cell responses.122 Patients in the N9831 clinical trial who received adjuvant chemotherapy plus trastuzumab had higher post-treatment endogenous polyclonal anti-HER2 antibodies relative to those who received adjuvant chemotherapy alone.7 These data support that trastuzumab therapy could promote an adaptive immune response that in turn generates additional patient-derived anti-HER2 antibodies. Importantly, higher post-treatment anti-HER2 antibodies were associated with improved DFS.7 Similarly, analysis of patients with HER2+ MBC from two phase II trials (N0337 and N983252) revealed that trastuzumab-containing therapy led to generation of anti-HER2 antibodies that associated with improved PFS.6
Fc-engineered margetuximab was also associated with enhanced HER2-specific adaptive immune responses in patients with HER2+ breast, gastric, or other cancers treated with prior lines of HER2 antibody therapy.123–125 In these patients, post-treatment blood samples exhibited increased T cell clonality together with greatly increased frequencies of HER2-specific T cells and increased levels of HER2-specific antibodies compared with pretreatment samples.124 125 Similarly, increased frequencies of HER2-specific T cells were observed in blood samples of patients with HER2+ GC in response to treatment with margetuximab and pembrolizumab.123
Mechanism of action of margetuximab, an Fc-engineered anti-HER2 mAb
The Fab portion of margetuximab shares HER2 specificity of trastuzumab, whereas the Fc portion is engineered.97 98 While trastuzumab is humanized, margetuximab is chimeric, comprising variable domains from the murine trastuzumab precursor and human IgG1 constant (Fc) domains.97 98 Direct (Fc-independent) properties of margetuximab are similar to those of trastuzumab; consequently, margetuximab and trastuzumab have similar binding affinity to HER2 protein and HER2+ cells, and antiproliferative activities of margetuximab and trastuzumab towards HER2+ tumor cells are similar (figure 1).82 Moreover, direct activity of both drugs can be improved by combination with pertuzumab, which binds to a different HER2 epitope.82 Fc-dependent properties of margetuximab, however, are enhanced compared with those of trastuzumab: margetuximab has higher binding affinity for both stronger-binding 158V and weaker-binding 158F allotypes of activating FcγRIIIa and decreased binding affinity for the inhibitory FcγRIIb.97 98 Notably, margetuximab binds FcγRIIIa-158F with higher affinity than trastuzumab binds FcγRIIIa-158V.82 97 98
Margetuximab mediates ADCC more potently than trastuzumab in vitro97 98 and ex vivo126 across all FcγRIIIa genotypes.82 Correspondingly, margetuximab promotes greater NK cell activation and expansion/proliferation in vitro than does trastuzumab.82 Margetuximab also mediates ADCC in vitro with greater potency than pertuzumab.82 Moreover, margetuximab, with or without pertuzumab, mediates ADCC in vitro with greater potency than trastuzumab with pertuzumab.82 127
There are no direct comparisons of adaptive immune responses associated with margetuximab versus those associated with trastuzumab and pertuzumab. Based on comparisons across different independent studies that used comparable assay methods, increases in circulating HER2-specific antibody levels (mediated by B cells) were found in 42%–69% of trastuzumab-treated patients6 7 and in 94% of margetuximab-treated patients.125 Increases in T cell–mediated responses were found in 50%–78% of trastuzumab-treated patients8 and in 98% of margetuximab-treated patients.125
Combinations of anti-HER2 antibodies with checkpoint inhibitors or co-stimulators
Breast cancer has been traditionally considered poorly immunogenic, being characterized by relatively low tumor mutation burden. Nevertheless, recent evidence has revealed high tumor-infiltrating lymphocytes and PD-L1 expression in tumor-infiltrating lymphocytes and breast cancer cells in a considerable proportion of patients with HER2+ breast cancer.128 Moreover, trastuzumab has been shown to upregulate expression of PD-L1 in HER2+ breast cancer cells in the presence of immune effector cells,129 which may limit the extent of trastuzumab-mediated antitumor activity, since upregulation of the PD-1/PD-L1 pathway leads to immune evasion. In the NeoSphere trial testing neoadjuvant HER2-directed therapies in patients with early stage HER2+ breast cancer, higher mRNA expression of PD-1/PD-L1 in tumor tissue samples was associated with lower probability of pCR in the pertuzumab + trastuzumab + docetaxel arm.101
Based on these findings, numerous clinical trials evaluating combinations of anti-HER2 mAbs with checkpoint inhibitors are underway, with the goal of leveraging engagement of both innate and adaptive immunity. In the phase II portion of the PANACEA study, trastuzumab plus pembrolizumab (anti-PD-1, IgG4 isotype) showed 15% ORR in patients with PD-L1-positive, trastuzumab-resistant, advanced HER2+ breast cancer, but there were no objective responses among the PD-L1-negative patients.130 In the Canadian Cancer Trials Group IND.229 phase Ib trial, trastuzumab plus durvalumab (anti-PD-1, IgG1 isotype engineered for reduced FcγR binding) failed to demonstrate any responses in patients with HER2+, PD-L1-negative MBC.131 The combination of anti-HER2 therapy with checkpoint inhibitors has proven successful in patients with advanced HER2+ gastric or gastroesophageal junction adenocarcinoma (GEA).123 132 Based on the first interim results of the KEYNOTE-811 study, showing a 74% ORR in the pembrolizumab arm versus 52% in the placebo arm, the US Food and Drug Administration granted accelerated approval of pembrolizumab in combination with trastuzumab and fluoropyrimidine and platinum-based chemotherapy as first-line therapy for patients with HER2+ advanced GEA.132 A phase II study of margetuximab plus pembrolizumab in previously treated patients with advanced or metastatic HER2+ GEA showed encouraging antitumor activity, particularly in the subgroup of HER2 immunohistochemistry 3+ and PD-L1-positive patients, with 44% ORR and 72% disease control rate.123 Furthermore, preliminary results of margetuximab plus tebotelimab (a bispecific anti-PD-1 × anti-lymphocyte-activating gene-3 dual-affinity re-targeting molecule) in a phase I study in patients with relapsed or refractory HER2+ tumors show an encouraging 21% ORR.133 134
A phase II/III study (MAHOGANY) is underway to investigate margetuximab plus checkpoint inhibitors with or without chemotherapy in the first-line setting for patients with HER2+ GEA.135 An ongoing neoadjuvant investigator-sponsored phase II trial (MARGOT) is comparing margetuximab plus pertuzumab plus chemotherapy to trastuzumab plus pertuzumab plus chemotherapy in FcγRIIIa-158F carriers with stage II/III HER2+ breast cancer. In this setting, the hypothesis of a superior efficacy in the margetuximab arm may be tested by using clinical end points (eg, pCR) as well as molecular and immune end points (eg, tumor-infiltrating lymphocyte rate, immune phenotype, natural anti-HER2 antibodies, or immune gene expression profiling).
FcγRIIb expressed by immune effector cells serves as a checkpoint molecule based on its strong inhibitory effect on tumor targeting antibodies, which was unambiguously demonstrated in studies comparing the antitumor activity of trastuzumab in HER2+ tumor-bearing mice that were wild-type or genetically deleted for FcγRIIb.77 Adding to the complexity, there is preclinical evidence that for some agonistic anticancer antibodies, such as anti-DR5, anti-CD40, anti-CD137, and anti-OX40, cross-linking by FcγRIIb is necessary to successfully elicit antitumor responses.36 117 In addition, FcγRIIb expressed on tumor cells can contribute to resistance to tumor-targeting antibodies by facilitating internalization of tumor antigens.136 Targeted blockade of FcγRIIb may help overcome resistance and boost activity of clinically validated and emerging antibodies in cancer immunotherapy.36 Antibodies specific to human FcγRIIb (which do not react with the FcγRIIa) have been isolated137 138 and shown to be capable of blocking the inhibitory effect of FcγRIIb.137 Early stage clinical trials are ongoing to evaluate FcγRIIb-blocking antibody BI-1206 as a single agent and combined with rituximab or pembrolizumab in B cell malignancy.36
CD137 (4-1BB) is an activation-induced costimulatory molecule that is expressed on activated T cells, NK cells, dendritic cells, eosinophils, mast cells, endothelial cells, and some tumor cells.139 Ligation of CD137 by agonistic antibodies provides a costimulatory signal in multiple immune cell subsets, including enhancement of ADCC and ADCP.140 For example, an anti-CD137 mAb enhances trastuzumab-induced, NK-mediated ADCC against pancreatic cancer cell lines, even with relatively low amounts of HER2 expression.141 A phase I clinical study (NCT01307267) tested the CD137 agonist mAb utomilumab in combination with rituximab in patients with relapsed/refractory follicular lymphoma and other CD20+ non-Hodgkin’s lymphomas.142 The study demonstrated anecdotal clinical activity and a favorable safety profile.142 Although the utomilumab sponsor recently deprioritized its further development in solid tumors, an investigator-initiated phase IB/II clinical trial of utomilumab plus either trastuzumab or T-DM1 in refractory HER2+ MBC is ongoing and has not yet reported results (NCT03364348). Whether or not this trial, or future investigation of margetuximab combined with a CD137 agonist, could renew interest in this approach remains to be explored. Another approach, consisting of a bispecific trivalent HER2×CD137×CD137 construct, could be useful to further assess the validity of the combinatorial strategy of CD137 agonism plus HER2 blockade.143
Future directions
The anti-HER2 mAb landscape continues to evolve, with several approved trastuzumab biosimilars, a recently approved novel anti-HER2 ADC (DS-8201a, trastuzumab deruxtecan),144 and another (SYD985)145 in late-stage clinical trials. Equally notable are recent approvals of anti-HER2 tyrosine kinase inhibitors tucatinib and neratinib. In addition, there are HER2-bispecific mAbs in phase II clinical trials. Zanidatamab (or ZW25; binding the two distinct HER2 epitopes targeted by trastuzumab and pertuzumab)146 is being tested in phase II studies (in HER2+/HR+ advanced breast cancer, in HER2+ advanced GEA, and in advanced HER2+ biliary tract cancers). Also, zenocutuzumab (or MCLA-128; targeting both HER2 and HER3)147 is being tested in a phase II study in patients with MBC with either HER2+ tumors or with estrogen receptor+/low HER2 expression.
Novel chimeric antigen receptor-T cells have also been developed to engage the Fc domain of tumor-specific mAbs. In particular, an innovative construct (antibody-coupled T cell receptor) has been designed with FcγRIIIa-158V extracellular domain, CD8 hinge and transmembrane domains, and 4-1BB and CD3ζ intracellular signaling domains.148 When these engineered T cells are transferred back into patients, they can be directed against HER2+ tumors by co-administering anti-HER2 mAbs with functional Fc domains, such as trastuzumab.
Additional bispecific anti-HER2 mAbs are in preclinical development. HER2(Per)-S-Fab, developed by linking the pertuzumab Fab to an anti-FcγRIIIa single-domain antibody, showed potent cytotoxicity against HER2+ tumor cells in vitro and tumor growth inhibition in vivo.149 HER2-BsAb, designed for bivalent binding to HER2 (same specificity as trastuzumab) and monovalent binding to CD3, includes a silenced Fc domain to reduce risk of cytokine release syndrome.150 HER2-BsAb is able to redirect T cells against established tumors and has exhibited increased antitumor activity versus trastuzumab both in vitro and in vivo.150
Conclusions
We have reviewed that FcγR-dependent activity is an important contributor to the mechanism of action of antitumor therapeutic mAbs, as demonstrated by (1) Differential antitumor responses based on patterns of Fc domain glycosylation (whether deliberate or accidental) that impact on FcγR binding, (2) Influence of FcγR genotypes on clinical response to trastuzumab in HER2+ breast cancer, and (3) Fc-domain engineering to enhance binding to activating FcγRIIIa and attenuate binding to inhibitory FcγRIIb. Fc domain engineering has been shown to be clinically feasible and active in the case of margetuximab,58 leading to US Food and Drug Administration approval (the first for an Fc-engineered antibody) in patients with HER2+ MBC who have received two or more prior anti-HER2 regimens, with at least one for metastatic disease.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank Emily Cullinan, PhD, CMPP, and Francesca Balordi, PhD, CMPP, of The Lockwood Group (Stamford, Connecticut, USA), for the professional editorial support provided by them in accordance with Good Publication Practice (GPP3) guidelines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MDP provided direction and guidance throughout the preparation of this manuscript. All authors reviewed and edited the manuscript. All authors approved the final manuscript. MDP is responsible for the overall content as the guarantor.
Funding MacroGenics, Inc. (Rockville, Maryland, USA) provided funding.
Competing interests AM reports grants from Roche and Eisai; personal fees from MacroGenics, Roche, Eisai, Novartis, and Lilly; participation in advisory boards from MacroGenics, Roche, Eisai, Novartis, Lilly. WJG has nothing to disclose. HSR reports personal fees for short-term consulting from Puma and Samsung; institutional grants for clinical research study activities from MacroGenics, Roche, Pfizer, Novartis, Lilly, Merck, Seattle Genetics, Odonate Therapeutics, Eisai, Sermonix, and Immunomedics, Daiichi Sankyo. MDP reports personal consulting fees from MacroGenics, AstraZeneca/Daiichi Sankyo, Pfizer, and Roche/Genentech, and grant support on this topic from the Parker Institute for Cancer Immunotherapy and the Mary Kay Foundation. JLN is an employee of MacroGenics. EPR was an employee of MacroGenics and is now an employee of Partner Therapeutics. FA was an employee of MacroGenics and is now an employee of AstraZeneca.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.