Article Text

Abstract
The landscape in immuno-oncology (I-O) has undergone profound changes since its early beginnings up through the rapid advances happening today. The current drug development pipeline consists of thousands of potential I-O therapies and therapy combinations, many of which are being evaluated in clinical trials. The efficient and successful development of these assets requires the investment in and utilization of appropriate tools and technologies that can facilitate the rapid transitions from preclinical evaluation through clinical development. These tools include (i) appropriate preclinical models, (ii) biomarkers of pharmacodynamic, predictive and monitoring utility, and (iii) evolving clinical trial designs that allow rapid and efficient evaluation during the development process. This article provides an overview of how novel discoveries and insights into each of these three areas have the potential to further address the clinical management needs for patients with cancer.
- immunotherapy
- antigenic modulation
- biomarkers
- tumor
- immune tolerance
- clinical trials as topic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The principle(s) surrounding the potential applications of immuno-oncology (I-O) has been around for quite some time. Initial research to harness immunological mechanisms for treating cancers began more than a century ago in 1891, when the early pioneer William Coley attempted to treat osteosarcoma using heat-inactivated Streptococcus pyogenes and Serratia marcescens.1 2 The elucidation of cytotoxic T-lymphocyte antigen 4 (CTLA-4) by Allison and colleagues in the 1990s,3 and of the programmed cell-death protein 1 (PD-1) by Honjo and colleagues in the 2000s4 led to the development of a new class of anticancer agents, namely the immune checkpoint inhibitors (ICIs), which are today widely used to treat a variety of cancer types.
Over the years, the level at which I-O therapies have been embraced by the oncology community has fluctuated owing to the many breakthroughs as well as challenges and struggles to achieve clinical success.5–7 For instance, advances were seen with the application of the concept of tumor-infiltrating lymphocytes (TILs) and the generation of effector T cells expressing chimeric antigen receptors (CAR T).8 9 Promising preclinical data were sometimes followed by setbacks in later clinical trials, such as the phenomenon of hyperprogressive disease observed in a subset of patients treated with checkpoint blockade immunotherapies,10 and I-O combination therapy failures in many tumor types.11 Only in the last decade has this promising therapeutic approach become a robust clinical reality and, consequently, the field of I-O has rapidly gained momentum.12 13 I-O encompasses multiple mechanistic approaches, many with several approved therapies and a burgeoning pipeline of over 4700 I-O agents.14
For many difficult-to-treat malignancies, the rapid advances and practice-altering clinical successes within the I-O field have attracted significant investment from biopharmaceutical companies, as well as fuelled intensive research into new preclinical models and biomarkers with which to advance our understanding.15 The recent explosion of novel I-O programs has created the demand for better predictive preclinical models and translational strategies to understand combination immunotherapy, patient responses and resistance, and the identification of novel biomarkers and targets.16 Moreover, addressing current questions, controversies, and limitations in the completed and ongoing clinical trials of ICIs will help to continue bringing new I-O therapies to patients.
There are three principal elements which can make drug development more efficient: using the right preclinical models to infer the right dosing strategy, identifying the right biomarker(s) to select the right patient population and indicate engagement of the right target, and executing the right clinical development strategy.
Immuno-oncology models used in the preclinical setting
Several different types of preclinical oncology models exist (box 1): syngeneic mouse tumor models, genetically engineered mouse models (GEMMs), cell line-derived xenografts (CDX), patient-derived xenografts (PDX), humanized mouse models, and tumor organoids/spheroids, with each type having specific advantages and disadvantages (table 1). When choosing which model is most appropriate, researchers must consider the attributes of each model in the context of their specific research questions.
Glossary
Biomarker: a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.
Syngeneic mouse tumor model: derived following the implantation of a mouse cancer cell line into the same strain of mouse from which it was derived.
Genetically engineered mouse model: also known as a transgenic mouse, results from the use of genetic engineering technology to modify the genome of a mouse. In cancer models, these mice have been created to specifically express tumor promoting genes or tumor inhibitory genes.
Cell line-derived xenograft: derived following the implantation of a human cancer cell line into an immune-deficient mouse. These cell lines have been maintained and propagated through ex vivo cell culture conditions over time.
Patient-derived xenograft: derived following direct implantation of freshly acquired human patient tumor material into immune-deficient mice. These models have been maintained and propagated in vivo (in mice) over time and not exposed to cell culture conditions.
Tumor mutational burden: an emerging clinical biomarker that assesses the total number of somatic mutations found within the coding area of a tumor genome.
Immunostimulatory cytokine: a cytokine that causes stimulation of particular immune cell populations and/or results in general activation of the immune system.
Co-stimulatory agonist: a type of drug that stimulates proteins that drive activation of the immune system for therapeutic benefit.
Co-inhibitory antagonist: also known as immune checkpoint inhibitors, these are a type of drug that blocks inhibitory immune checkpoint proteins for therapeutic benefit.
Immunogenic cell death inducers: agents that induce the death of tumor cells in such a way that those cells then elicit a specific immune response.
Cytotoxic agents: agents that kill cells by interrupting their ability to proliferate.
Preclinical immuno-oncology models
Syngeneic mouse models have been most commonly used over the years in the I-O setting.17 These models are derived by transplanting established mouse cell lines or tumor tissues to strain-matched mouse hosts that retain an intact immune system. Several syngeneic mouse models are commercially available, spanning solid and hematological malignancies including some that have been modified to stably express the firefly luciferase gene to offer the ability to use in vivo small animal imaging.18 These models are logistically easy to use, are suitable for screening large numbers of drug candidates, and are useful for studying pharmacodynamics and mechanisms of action.19 Despite their widespread use and applicability to human cancers, syngeneic models are not without their drawbacks. For example, the tumor cells are murine derived and thus the mouse tumor histotypes may not always match directly with all aspects of human clinical disease. In addition, treatment responses in these models can be poorly predictive of clinical outcomes.20 However, their utilization to understand critical aspects around immune response to therapy outweighs many of these disadvantages.
Unlike syngeneic models, genetically modified, transgenic mouse models or GEMMs are shown to more faithfully mimic the stromal biology of human disease counterparts, including the tumor microenvironment, which plays a huge role in influencing tumor behavior.21 These models allow for a more natural progression of disease onset that, in many cases, is controlled via tissue-specific elements and driver mutations known to promote tumor formation in the tissue or organ of interest. These models have provided valuable insight into tumorigenesis and served as critical models for oncology drug development in the past.22 For example, Dow et al have shown that a transgenic model for colorectal cancer provided the validity to therapeutically target the Wnt signaling pathway in this cancer.23 In spite of these types of successes, the utilization of driver mutations in some transgenic models can limit broad neo-antigen formation making these models less well suited to studying I-O applications. However, a number of approaches have been undertaken more recently in an effort to circumvent some of the limitations described and Kersten et al provide a valuable review of this area.22
While syngeneic mouse cell line models and certain GEMMs provide value in the development of I-O drugs, human cancer CDX models also need to be considered. While they have been heavily used for more conventional oncology approaches, they have also become the industry standard for studying cell-based immunotherapies. CDX models are logistically easy to use with thousands of human cell lines commercially available, including many stably expressing the firefly luciferase gene again making these models suitable for in vivo small animal imaging.24 These studies must be performed in immune-deficient mouse strains which makes them less suitable to study the roles of the immune system in tumor development and therapy response.22 Moreover, due to the clonal nature of the cell lines and their continued growth on plastic in cell culture settings, these models have generally lost their heterogeneity and in some cases have shown irreversible changes in gene expression.25 26 Despite these challenges, researchers continue to evaluate approaches to improve the utilization of CDX models.27
PDX mouse models, based on transplantation and in vivo serial propagation of fresh human tumor biopsies in immunodeficient mice, have been developed to overcome some of the limitations of traditional CDX models.22 Unlike CDX, PDX tumors maintain the molecular, genetic, and histological heterogeneity observed in patients with cancer, even after serial passaging in mice, and thus have been shown to be better predictors of clinical outcome in humans. Furthermore, PDX tumors are often extensively characterized, thus enabling more complex studies of drug resistance and treatment response.28 However, a number of known challenges exist with these models. While hundreds of these lines exist, some tumor histotypes, such as prostate cancer and a range of hematological malignancies, are considerably more challenging to establish. Many of the PDX models have a slower growth rate than their CDX counterparts, which could be more indicative of the human setting but certainly results in considerably longer study durations. As with CDX models, PDX models also require immunodeficient mouse strains for utilization which hinders broad utilization for a range of I-O drug therapies.
The lack of human targets as well as inherent interspecies immunological differences constitute major limitations of syngeneic and GEMMs. Accordingly, considerable efforts have been invested in humanizing the murine immune system.29 While a variety of approaches to humanize mouse models exist, and a full summary is beyond the scope of this review, one of the more common is the immunoavatar model in which immunodeficient mice were reconstituted with human peripheral blood mononuclear cells (PBMCs), thus allowing human cancer cell lines or primary patient-derived tumors to be studied in an autologous or heterologous immunologic context. Despite omitting the need of sophisticated purification techniques, prominent human xenograft-versus-host disease usually develops a few weeks following the engraftment of human PBMCs and is thought to be because of the major histocompatibility complex (MHC) mismatch between mouse and human T lymphocytes. Alternatively, engrafting the immunocompromised-NOD SCID gamma (or NSG) mice with human hematopoietic stem and progenitor cells has been demonstrated to ultimately give rise to multiple lineages of human hematopoietic and immune cells. These so-called HuNSG mice develop partially functional human immune systems. The development of these humanized mouse models has made them approaches that can be readily used to characterize human CAR T cell function in vivo and facilitate the development of novel CAR therapies, making it an active area of current research. Despite this, each humanized mouse approach has its own unique advantages and disadvantages that should be investigated prior to use.29
A novel cell-based approach to study tumor immunobiology has been recently developed with the use of organoid technology that is rapidly being adapted to the I-O setting.30 Organoids are three-dimensional in vitro cultures of normal and/or tumor tissues with multiple cell lineages, including stem cells and differentiated cells, and tissue architecture. Their close resemblance to the original tumors and the ability to culture organoids from every individual patient holds promise that organoids will become a more clinically relevant preclinical model system compared with cell lines for drug discovery and precision medicine.30 A number of other in vitro or ex vivo systems are also being used in the discovery and development of oncology and I-O therapies. An insightful review article on these approaches is provided by Rodrigues et al and highlights the importance of understanding the tumor microenvironment and why it may be critical to consider therapeutics targeting these components for successful treatment of cancer.31
In summary, the explosion of I-O clinical studies has spurred additional research into mouse models with competent immune systems, and hence the number of tumor histotypes represented in syngeneic models is expanding rapidly. Demand is also growing for syngeneic models with tumors grown in orthotopic settings. This provides a means to surgically implant tumor cells directly into the organ and track and monitor growth and treatment response. Utilization of luciferase expressing cell lines can allow for use of longitudinal, non-invasive small animal bioluminescence imaging in these instances which greatly reduces the number of mice on study and can provide a semi-quantitative analysis of tumor burden. Thus, this approach can obviate some of the downsides of genetically modified or subcutaneous models yet harness many of their advantages, including the tumor microenvironment and an intact immune system without the problems associated with some of the humanized mouse models.32
Syngeneic mouse models can help identify biomarkers of treatment response
Syngeneic mouse models have been reported to respond differently to ICIs, such as PD-1, programmed death-ligand 1 (PD-L1), and CTLA-4 antibodies, and this responsiveness is well characterized for many of the commonly used models.33 In addition to treatment response variations, other differences in these models include type of immune cell infiltration, gene expression profiles, and overall tumor microenvironment components.33 These differences have resulted in many of the models being classified as either ‘cold’ or ‘warm/hot’ such that cold models display a more refractory phenotype with less cytotoxic T cells and a more immunosuppressive environment while warm/hot models tend to have more cytotoxic T cell infiltration and a less immunosuppressive tumor microenvironment.34 Knowing, and understanding these models’ baseline response to these agents is useful for studying drug combinations and identifying phenotypic markers of response: (1) to characterize the baseline tumor by (a) infiltrating leukocyte population, (b) other molecular characteristics (RNASeq, whole exome sequencing, etc) of the models, and (c) baseline treatment response; all of which enable selection of the most appropriate model for preclinical studies, and then (2) to use the most appropriate models to determine antitumor activity or proof-of-concept for novel therapeutics either as monotherapy or in combination with other treatments like novel ICIs, chemotherapy, focal radiation, etc. As an example, in an immunologically ‘cold’ mouse mammary carcinoma model, 4T1-luciferase, the primary tumor is known to spontaneously metastasize to the thoracic region, including lung and axillary lymph nodes.35 Adding focal radiation therapy to systemic anti-CTLA-4 antibody therapy only slightly reduces the size of the primary tumor when compared with either anti-CTLA-4 or focal radiation therapy alone in this model; however, it greatly delays the initial onset of metastatic disease.35 36 These treatment responses can be visualized, quantified, and compared using bioluminescence imaging techniques.35 37 Furthermore, flow cytometry and serial blood sampling can be used to identify and quantify the phenotypic markers, such as immune cells and cytokines, which underlie these responses.37
Biomarkers can also be used to identify the most appropriate model for studying a particular I-O agent, or to identify changes in gene expression following treatment. For example, microarray data show that the immunologically cold 4T1-luciferase mouse model and immunologically warm CT26 mouse colon model have very different patterns of gene expression following treatment with immuno-modulatory agents, and this information can help identify genetic determinants of treatment response.19
Predictive biomarkers in immuno-oncology: drug development and clinical uses
As mentioned previously, biomarkers are useful for assessing various facets of treatment response in preclinical models, but they are also being more widely used in the clinic for patient management. The US Food and Drug Administration (FDA), defines biomarkers as: ‘a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions’.38 Many types of biomarkers are used to inform treatment decisions to maximize patients’ responses to various treatments, including diagnostic, prognostic, and predictive biomarkers.39 Diagnostic biomarkers are used to detect or confirm the presence of a disease or condition of interest, whereas prognostic biomarkers are used to identify the likelihood of a clinical outcome such as disease recurrence or progression.39 Prognostic biomarkers are typically used in clinical trials to set trial entry and exclusion criteria to identify higher-risk populations.39 Predictive biomarkers are defined by the change in biomarker that predicts an individual or group of individuals more likely to experience a favorable or unfavorable effect from exposure to a given therapeutic agent; they are especially useful in the design and conduct of clinical trials.39 Diagnostic biomarkers (table 2) may become more useful to redefine the classification of some diseases. Many predictive biomarkers are used in diagnostics, and others are used to inform treatment decisions to maximize patients’ responses to various treatments.40 Predictive biomarkers for immuno-oncology agents include PD-1/PD-L1 expression,41 mutations in mismatch repair genes and microsatellite instability,42 tumor mutational burden (TMB), and various signatures.40 43 For example, expression of PD-L1 in tumors is predictive of response to immunotherapies and correlates with improved overall survival. Therefore, immunohistochemistry (IHC) staining is recommended before initiating several such therapies, including pembrolizumab, which requires the PD-L1 IHC assay 22C3 pharmaDx developed by Dako,44 atezolizumab, which uses the PD-L1 SP142 IHC assay developed by Ventana Medical Systems,45 and TMB testing,46 which shows promise as a new tool for predicting the response to various I-O agents, including ICIs.47 48 Studies have shown that progression-free survival and duration of response to either nivolumab, ipilimumab, or pembrolizumab is higher in those with high TMBs compared with those with lower mutational burdens.47
Biomarkers in drug development
Determining cut-off levels at which tumor mutation is considered high,49 as well as developing useful diagnostic assays and methods of interpretation, is an active area of research.50 Such is the importance of predictive biomarkers for informing clinical decisions, that many cancer drugs are now being approved not based on where in the body they first originated, but based on the tumor’s key associated biomarkers.51 TMB promotes the production of neoantigens expressed on MHC which in turn boost the capability of the immunosurveillance system.52 Exome sequencing of two preclinical syngeneic tumor models revealed heightened somatic mutations and TMB in the more ICIs-responsive colorectal cancer (CT26.WT) compared with the non-responsive TNBC (4T1) model.52 BRCA1-mutated TNBC tumors with high TMB are more responsive to ICIs primed with chemotherapy suggesting that assessing BRCA1 status clinically could serve as a predictive biomarker.53 Intriguingly, analyzing the genomic and clinical data of 1662 patients with advanced cancer treated with FDA-approved ICIs revealed that higher TMB (calculated by normalizing the total number of somatic non-synonymous mutations to the total number of sequenced megabases—assessed using MSK-IMPACT assay) is associated with increased overall survival.54 For the patient, this means that along with concentrating efforts on finding exactly where their tumors originated, it is also important to focus on tumor-agnostic biological pathways that are driving cancer growth, when making decisions about definitive treatment. Until recently, biomarkers have been largely studied independently of one another, however, current research is shifting toward studying how combinations of biomarkers that address features of the tumor as well as the tumor microenvironment, including genetic and gene expression ‘signatures’ are being applied to treatment responses and patient outcomes.55 56
There is also increasing interest in biomarkers that reflect the immune response to immuno-oncology therapies, measured either in the blood or in the tumor/tumor microenvironment.57 A better understanding of the immune response can be helpful in the translation of studies from the preclinical setting into clinical trials, but may also assist in the reverse translation to drive and test new hypothesis in preclinical models, either for new agents or in combination therapies.58 Such approaches may improve the success rate in clinical trials for novel therapies or novel combination therapies.59 Gene expression profiling of the tumor and tumor microenvironment has been a valuable approach to assess this important interaction to characterize patient response as well as to facilitate reverse translational approaches. Multiple gene expression signatures have been developed and include tumor inflammation signatures, signatures related to tumor infiltrating CD8-positive cells, and those that incorporate gene expression of immune response genes along with other genomic biomarkers.60 61
In 2016, the FDA and National Institutes of Health published the ‘Biomarkers, EndpointS and other Tools’ framework, which described seven types of biomarkers.62 In addition to the diagnostic, prognostic, and predictive biomarkers discussed above, this framework also defined monitoring, susceptibility/risk, safety, and pharmacodynamics biomarkers.
Immuno-oncology is changing the paradigm for clinical trials
Over the past decades, there has been a shift in the general approach to systemic anticancer therapy. Cytotoxic chemotherapy consisted of chemicals that targeted any dividing cell. Next came targeted agents that preferentially acted on cancer cells that displayed specific molecular aberrations, exemplified by imatinib for chronic myeloid leukemia and trastuzumab for HER2-positive breast cancer.63 More recently developed immunotherapeutic agents act against various aspects of the cancer cell and its interaction with the tumor microenvironment and immune system, and may be broadly classified into agents that act directly on cancer cells (‘passive’ I-O) and those that stimulate immune cells to exert antineoplastic effects (‘active’ I-O). I-O agents (see figure 1) include antitumor antibodies, adoptive cells, immunostimulatory cytokines, co-inhibitory antagonists or ICIs, co-stimulatory agonists, immunological cell death inducers, and therapeutic cancer vaccines.64 65

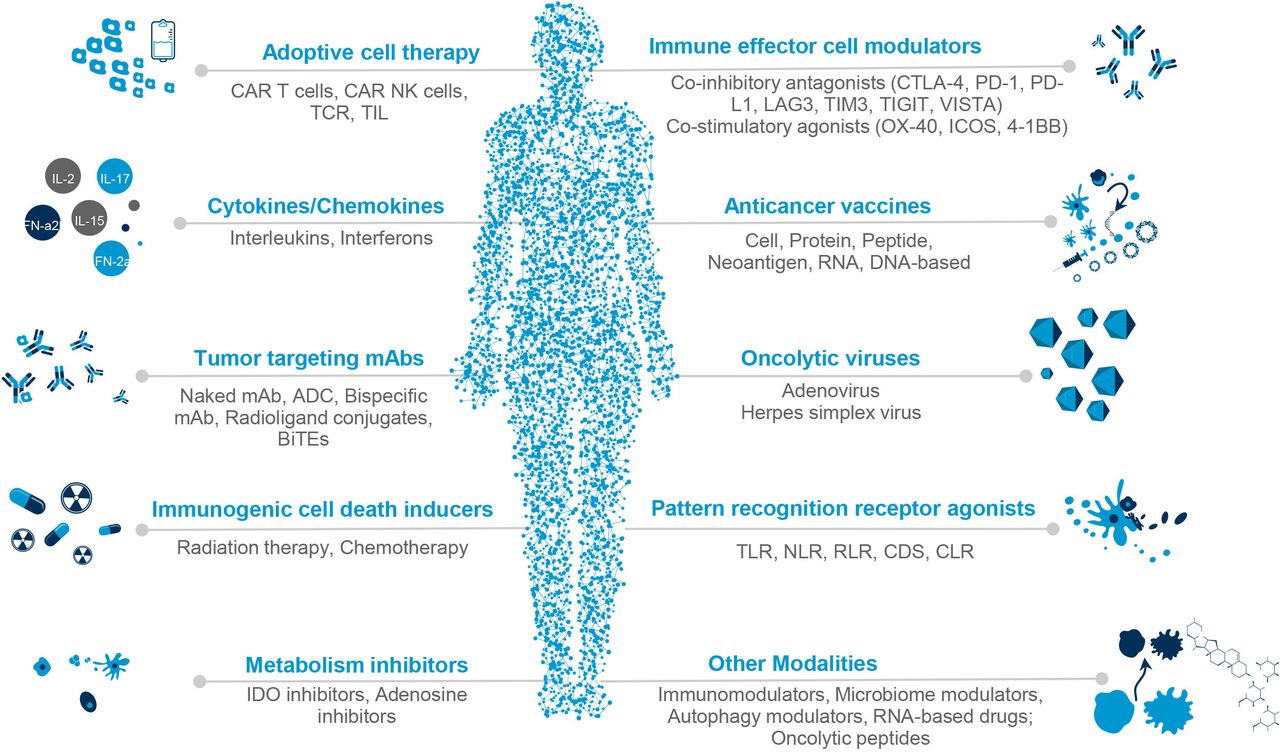{kind=link}
Classification of immuno-oncology agents used in cancer treatment. ADC, antibody-drug conjugate; BiTEs, bispecific T cell engager; CAR T, chimeric antigen receptor; CDS, cytosolic DNA sensors; CLR, C-type lectin receptors; CTLA-4, cytotoxic T-lymphocyte associated protein 4; ICOS, inducible costimulatory receptor; IDO, indoleamine 2,3-dioxygenase; LAG3, lymphocyte-activation gene 3; mAb, monoclonal antibody; NK, natural killer cell; NLR, NOD-like receptors; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; RLR, RIG-I-like receptors; TCR, T cell receptor; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIL, tumor infiltrating lymphocyte; TIM3, T-cell immunoglobulin domain and mucin domain 3; TLR, toll-like receptor; VISTA, V-domain immunoglobulin suppressor of T cell activation.
With the increasing availability and decreasing cost of high-throughput approaches such as next-generation sequencing, genomic features of various cancers are getting better defined. As a result, patients with cancer are becoming fragmented into smaller subpopulations and biomarker-positive populations are now being recruited into I-O clinical trials based on the specific molecular characteristics of their tumors.66 67
This has resulted in I-O trials enrolling more molecularly homogenous but smaller patient populations67; one study found that the average number of patients per oncology trial reduced significantly from 429 in 2014 to 129 in 2019.68
The total time required for clinical development of new anticancer agents has also significantly reduced over the past few decades. For cytotoxic agents,69 the drug development process typically consisted of a sequence of trials: phase I to determine dose and safety, phase II to confirm dose, document side effects, and determine efficacy, and phase III to confirm efficacy and compare the new treatment against the existing standard; with regulatory approval granted after successful phase III results.70 During the era of targeted therapies, promising phase II results were sometimes sufficient to gain accelerated approval, on the condition that future confirmatory phase III studies would follow.71 In the current immunotherapy era, development often begins with a small phase I study, with the progressive addition of different expansion cohorts to the same study, sometimes including randomized arms. A single such ‘seamless’ trial can be sufficient to secure accelerated approval from regulatory agencies, as was demonstrated by the first-in-human pembrolizumab trial KEYNOTE-001 that recruited 1260 patients and led to the approval of this ICI for melanoma and non-small cell lung cancer (NSCLC).72
Furthermore, this paradigm shift has shortened development timelines from around 10 years to <5 years,71 with median drug development times being 112.7, 87.1, and 64.6 months for cytotoxic, targeted non-precision and precision strategies, respectively.73
The use of ‘master’ protocols allows the application of one treatment to several diseases (‘basket’ study), or several treatments to one disease (‘umbrella study’).74 The development of adaptive trials that allow modification of the design and statistical plan of ongoing studies based on analysis of data accumulating while the trial is still ongoing has allowed greater flexibility and efficiency into the drug development process.75
One interesting phenomenon that has been noted with I-O therapies is that the Kaplan-Meier curves (I-O vs standard chemotherapy) tend to separate late, but then lead to a sustained separation (sometimes evocatively described as ‘lifting the tail of the curve’), implying durable benefit observed in a proportion of patients.76 77 This poses methodological challenges from the statistical point of view, and therefore apart from progression-free survival, other newer approaches such as milestone survival, landmark analysis, and restricted mean survival time are also being investigated in the I-O era.78
Newer challenges have emerged in the medical monitoring of I-O trials, given that many immunotherapies lead to late, severe, or unusual toxicity, which may initially present beyond the protocol-specified dose-limiting toxicity period.48 79 80 Additionally, atypical patterns of radiological response, such as pseudoprogression or hyperprogression, may be observed in a minority of patients treated with I-O agents; hence criteria for assessment of tumor response have been modified for the I-O era, examples include RECIST 1.1 for immune-based therapeutics (iRECIST) and response criteria for intratumoral immunotherapy in solid tumors (itRECIST).81 82
Simultaneous with the continued growth of I-O compounds, there has been a significant shift toward precision medicine in the field of oncology, using information about an individual’s genes, proteins, and other cancer features to diagnose or treat their particular disease. These rapid advances and new frontiers necessitate accelerated research into preclinical models and assays and when appropriate translation into later stage clinical trials; particularly as more biomarkers and drug targets are identified, to support this burgeoning therapy area and underpin the value of early research into tomorrow’s therapies.
Concluding remarks and future perspectives
With the burgeoning pipeline of I-O candidates, the need for appropriate preclinical models as well as prognostic and predictive biomarkers has never been more important. The explosion of clinical studies of I-O therapies has spurred additional research into mouse models with competent immune systems.83 Consequently, more detailed preclinical experiments are needed that begin to address underlying immune cell involvement and biomarker discovery that can be further investigated during later stages of drug development. Moreover, the methodology and the design of cancer clinical trials as well as the methods of their implementation are undergoing significant change, with more focused effort to find the right patients for each trial. In order to accomplish this, biomarkers are now an integral part of clinical trials that enable the refinement of the patient population.78 The example of the approval of pembrolizumab based on a phase I expansion study, with biomarker-driven patient populations, points the way to this new methodology of clinical development, in which fewer patients are required and the time to approval is shortened considerably. New technologies, like liquid biopsies84 are now being used to screen and monitor patients and have been recently approved as companion diagnostics in the area of NSCLC and breast cancer; this new technology will make it easier for patients to further participate in new I-O trials. The general trend toward smaller precision trials defined by biomarkers is shortening drug development timelines and paving the way for more flexible clinical trial models. Indeed, the FDA approval for histology-agnostic cancer indications based purely on patient biomarkers in recent years offers promise for the future,48 but newer and more flexible models still need to be fully evaluated. While preclinical models have provided an amount of knowledge to support the clinical development of ICIs, further optimization of these models is urged to better recapitulate human cancer. In the era of precision medicine state-of-the-art NGS and tumor-targeted delivery technologies are all expected to provide an objective roadmap to dissect the molecular basis beyond the ‘therapeutic as well as off-target toxic’ responses to ICIs, identify predictive/prognostic biomarkers and devise durable and tolerable combination regimens for ICI-resistant patients with cancer.
Highlights
With an increasing focus on immuno-oncology-based therapies, such as ICIs to treat a variety of cancers, there must be stronger linkages established between preclinical and clinical development.
Biomarkers that can assist in the prediction of response or help understand the nature of the immune response are critical elements of making the linkage between preclinical and clinical development.
Translational and/or reverse translational approaches will allow for improvements in trial design and execution and thereby improve the potential success rates of novel therapies and combination therapies for immuno-oncology.
Outstanding questions
Can preclinical models be successfully used to evaluate and predict efficacy of new combinations of chemotherapy, targeted agent and immuno-oncology agents?
Are there biomarkers that predict which patients will respond to novel I-O agents or combinations in which one of the therapies is an I-O agent?
Are we closing the loop on research from the bench to the clinic and from the clinic back to the bench, with the results from clinical trials helping inform preclinical models and which new agent combinations might be useful for future drug development?
Can preclinical experiments and biomarker studies address the underlying immune cell involvement for I-O drugs?
Are the evolving clinical trial formats increasing our understanding of the interplay between the tumor and the immune system to help design clinical trials of the future?
Are clinical trials using I-O agents now able to identify the right drug, or drugs, for the right patient at the right time?
Ethics statements
Patient consent for publication
Ethics approval
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Acknowledgments
Medical writing and editorial assistance in the preparation of this manuscript was provided by Klara Belzar, PhD, and Prescript Communications, Hertfordshire, UK. Financial support for this assistance was funded by Labcorp Drug Development, Princeton, New Jersey, USA in compliance with international guidelines for good publication practice.
References
Footnotes
Twitter @KSainiMD
Contributors MRF, SP, KSS, and SA conceptualized the manuscript, all authors wrote the manuscript, all authors reviewed, edited, and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests KSS reports consulting fees from the European Commission, and stock and/or other ownership interests in Labcorp and Quantum Health Analytics SPRL, outside the submitted work. GC reports personal fees for consulting, advisory role and speakers’ bureau from Roche/Genentech, Novartis, Pfizer, Lilly, Foundation Medicine, Samsung, and Daichii-Sankyo; honoraria from Ellipses Pharma; fees for travel and accommodation from Roche/Genentech, and Pfizer outside the submitted work. The other authors do not report any conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.