Article Text

Abstract
Background Toll-like receptors (TLRs) are pattern-recognition sensors mainly expressed in innate immune cells that directly recognize conserved pathogen structures (pathogen-associated molecular patterns-PAMPs). Natural killer (NK) cells have been described to express different endosomal TLRs triggered by RNA and DNA sequences derived from both viruses and bacteria. This study was addressed to establish which endosomal TLR could directly mediate NK activation and function after proper stimuli. It was also important to establish the most suitable TLR agonist to be used as adjuvant in tumor vaccines or in combined cancer immunotherapies.
Methods We assessed endosomal TLR expression in total NK cells by using RT-qPCR and western blotting technique. In some experiments, we purified CD56brightCD16− and CD56dimCD16+ cells subsets by using NK Cell Isolation Kit Activation marker, cytokine production, CD107a expression and cytotoxicity assay were evaluated by flow cytometry. Cytokine release was quantified by ELISA. NK cells obtained from ovarian ascites underwent the same analyses.
Results Although the four endosomal TLRs (TLR3, TLR7/8, and TLR9) were uniformly expressed on CD56brightCD16− and CD56dimCD16+ cell subsets, the TLR7/8 (R848), TLR3 (polyinosinic-polycytidylic acid, Poly I:C) and TLR9 (ODN2395) ligands promoted NK-cell function only in the presence of suboptimal doses of cytokines, including interleukin (IL)-2, IL-12, IL-15, and IL-18, produced in vivo by other environmental cells. We showed that R848 rather than TLR3 and TLR9 agonists primarily activated CD56brightCD16− NK cells by increasing their proliferation, cytokine production and cytotoxic activity. Moreover, we demonstrated that R848, which usually triggers TLR7 and TLR8 on dendritic cells, macrophages and neutrophils cells, activated CD56brightCD16− NK-cell subset only via TLR8. Indeed, specific TLR8 but not TLR7 agonists increased cytokine production and cytotoxic activity of CD56brightCD16− NK cells. Importantly, these activities were also observed in peritoneal NK cells from patients with metastatic ovarian carcinoma, prevalently belonging to the CD56brightCD16− subset.
Conclusion These data highlight the potential value of TLR8 in NK cells as a new target for immunotherapy in patients with cancer.
- adjuvants
- immunologic
- killer cells
- natural
- immunotherapy
- receptors
- immunologic
- tumor microenvironment
Data availability statement
No data are available. Deidentified subject data protocols.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Toll-like receptors (TLRs) are pattern-recognition sensors (PRR) predominantly expressed on cells of the innate immunity that directly recognize conserved pathogen structures (pathogen-associated molecular patterns,PAMPs), including peptidoglycans, lipopeptides, lipopolysaccharide (LPS), flagellin, double strand RNA (dsRNA), single strand RNA (ssRNA), and C-phosphate-G DNA (CpG DNA).1 Among the 10 human TLRs, TLR1, 2, 4, 5, 6, and 10 are expressed on the cell surface where they are primarily devoted to recognize extracellular macromolecular ligands from bacteria and fungi. By contrast, TLR3, 7, 8, and 9 are expressed inside the endosomal compartments of various cells where they recognize foreign nucleic acids from intracellular pathogens. PAMPs-TLR interaction triggers signaling pathways that, in turn, induce proinflammatory and anti-inflammatory gene transcription of cytokines and chemokines as well as costimulatory, adhesion and antimicrobial molecules.2 3
Natural killer (NK) cells are known for their ability to kill virus-infected and tumor cells by directly recognizing selective molecules on target cells through their inhibitory and activating receptors.4 NK cells have been described to express endosomal TLRs, but poor evidence exists on a direct activation of NK-cell functions through these TLRs. The response to their agonists depends on their activation state; therefore, only cytokine-activated NK cells can respond to Toll-like receptor ligands (TLR-Ls).5 6 TLR-activated NK cells, in turn, favor dendritic cells (DCs)maturation and cytokine secretion leading to the activation and regulation of both innate and adaptive immune responses.7 Whereas a critical cross-talk between DCs and NK cells has been well established,8 no data are available on which NK subset uses TLRs signals for pathogen recognition. Between the two distinct NK-cell subsets,9 the CD56dimCD16+ cells are predominant (>90%) in human peripheral blood and specialized for cytotoxicity, while CD56brightCD16− cells are non-cytolytic, mainly produce cytokines, and are less represented in periphery (<10%), while they are prevalent in tissues (T-cell areas of lymph nodes) and tumor microenvironment (TME).10 11
The immunotherapy based on the cross-talk between DCs and NK cells is becoming a promising approach to the treatment of infectious diseases and cancer, even though the clinical efficacy of systemically delivered TLR agonists has been limited by their toxicity or their ability to induce autoimmunity.12 13 In murine models, the local infusion of endosomal TLRs agonists enhanced both the recruitment and activation of immune cells into tumors and polarized antitumor immune response to a more effective type 1 profile, also improving the efficacy of checkpoint inhibitors.14 Thus, intratumor (IT) therapy with TLR agonists has been established as a novel potential strategy to treat solid tumors.15 Furthermore, dermal application of imiquimod (TLR7 agonist) or resiquimod (TLR7/8 agonist) to treat human skin tumors induced local immune activation, limiting systemic exposure and side effects.16
The purpose of the study was to evaluate the response of human NKcell subsets to different endosomal TLR-Ls in order to establish the most valid molecule to be employed as adjuvant in cancer vaccines or in combined tumor immunotherapy.
Using synthetic agonists of endosomal TLRs, we found that, besides the consistent expression of all four endosomal TLRs on both CD56brightCD16− and CD56dimCD16+ cell subsets, the TLR7/8 (R848) rather than TLR3 (Poly I:C) and TLR9 (ODN2395) ligands resulted as the most active molecule for NKcell function, only in combination with suboptimal doses of cytokines normally present in the microenvironment. Importantly, we demonstrated that the TLR7/8 agonist prevalently activated CD56brightCD16− NK cells by selectively increasing their proliferation, cytokine production and cytotoxic activity. We also showed that R848 was mostly active on the CD56brightCD16−NKcell subset essentially through TLR8. TLR8 agonists were capable of improving interferon gamma (IFN-γ) production and cytotoxic activity in CD56brightCD16− NK cells derived from healthy donors (HDs) or patients with metastatic ovarian carcinoma. These data highlight the potential value of TLR8 as a novel target for immunotherapy of cancer.
Materials and methods
Human samples and NKcell isolation
Peripheral blood was obtained from buffy-coat of healthy volunteers admitted to the blood transfusion service of Bambino Gesù Children’s Hospital, Rome, Italy. Peripheral blood mononuclear cells (PBMCs) were obtained from buffy coats by density gradient centrifugation with lympholyte cell separation media following the manufacturer’s protocol (Cederlane, Burlington, Canada). NKcell isolation was performed as previously described.17 In some experiments, CD56dimCD16+ and CD56brightCD16− NK cells were separated by using CD56+CD16− human NK Cell Isolation Kit (Miltenyi Biotech, Bergisch Gladbach, Germany). NK cells were also isolated from peritoneal fluids of nine patients with metastatic ovarian cancer admitted to the Agostino Gemelli Hospital, Rome, Italy, showing the following histotypes: three patients suffered from endometrial cancer stage IV, four from high-grade serous ovarian cancer stage III, and two from high-grade serous ovarian cancer stage IV. The mean age of patients was 59 years, ranging from 43 to 69.
Cell cultures, cytokines and TLR-L
Purified NK cells were cultured in Roswell Park Memorial Institute (RPMI-1640) (Euroclone, Milan, Italy) medium supplemented with 2 mM L-glutamine (Euroclone), 1% penicillin and streptomycin (Euroclone) and 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, USA). NK cells were cocultured with the following cytokines at suboptimal doses: interleukin (IL)-2 (20 U/mL) (Novartis, Basel, Switzerland), IL-12 (1 ng/mL), IL-15 (1 ng/mL) (Miltenyi Biotech), IL-18 (20 ng/mL) (R&D Systems, Bio-Techne, Minneapolis, Minnesota, USA). Daudi (human Burkitt’s lymphoma) and HEK293T (human embryonic kidney) cell lines were purchased from American Type Culture Collection (Manassas, Virginia, USA). The TLRs agonists and the doses used are R848 (6 µM), Poly-I:C (10 ng/mL), ODN 2395 (1 µM), IMIQUI (10 µM), TL8-506 (TL8) (100 nM) and loxoribine (1 µM); these reagents were all purchased from Invivogen (San Diego, California, USA).
Antibodies and flow cytometry
To perform flow cytometry experiments, cells were stained in phosphate buffered saline (PBS) with 5% FBS for 20 min at 4°C with the following antibodies: CD45-Alexa-Flour700, CD16-BV510, CD19-PE/Dazzle, CD56-PECy7, CD57-PB, CD226 (DNAM-1)-PE, CD336 (NKp44)-APC purchased from BioLegend (San Diego, California, USA); anti-CD3-FITC, CD25-PE, CD69-FITC, CD158e (KIR3DL1)-FITC from Miltenyi Biotech; CD14-ECD, NKG2A-APC, CD337 (NKp30)-PE, CD158e1/e2-PE from Beckman Coulter (Brea, California, USA); anti-CD335 (NKp46)-EF450 from Thermo Fisher Scientific; and anti-CD314 (NKG2D)-BV605 from Becton-Dickinson (Franklin Lakes, New Jersey, USA).
CD107a expression on NK cells was performed as follows: 2×105 NK cells were stimulated with 2×105 Daudi used as target cells, in 200 µL of medium. The effector:target (E:T) ratio was 1:1, and the cell density was the same for all conditions. Monensin (1:500) and anti-CD107a-APC antibody (Miltenyi Biotech) were added to the medium during stimulation. For intracellular staining, monensin and brefeldin-A (1:500) were added during the last 4 hours of treatment.
To detect intracellular cytokine production, the cells were fixed, permeabilized, and stained with the FoxP3 Staining Buffer Set (Miltenyi Biotech) according to the manufacturer’s instructions. Anti-IFN-γ-PE (Miltenyi Biotech), antitumor necrosis factor alpha (TNF-α)-EF450 (Thermo Fisher Scientific), anti-Granulocyte-macrophage colony-stimulating factor (GM-CSF)-PE, anti-granzyme A-PE, anti-granzyme B-BV421 (Becton-Dickinson), and antiperforin-FITC (Ancell, Hamburg, Germany) antibodies were used.
Data about cells were acquired at Cytoflex LX or Cytoflex S and analyzed with Cytexpert software V.2.4 (Beckman Coulter).
Immunofluorescence and confocal microscopy
Purified NK cells were fixed with cold paraformaldehyde 4% (Sigma) and then permeabilized with 0.1% Triton X-100 (Sigma Aldrich) for 5 and 8 min at room temperature, respectively. After blocking in PBS-bovine serum albumin (BSA) 5% goat serum 5% for 30 min at room temperature, cells were incubated with the following antibodies overnight at 4°C in 1% PBS:BSA: α-TLR7 1:340 (Novus Biologicals, LLC, Littleton, Colorado, USA), α-TLR8 1:25 (Abcam, Cambridge, UK), α-Rab7 0.6 µg/mL (Abcam), or α-Rab7 1:100 (Cell Signaling Technologies, Danvers, Massachusetts, USA). After washes in PBS, cells were incubated with goat-α-mouse Alexa Fluor 488 1:500 (Thermo Fisher, #A11001) or goat α-rabbit Alexa Fluor 555 conjugated secondary antibody 1:800 (Thermo Fisher, #A21430). For double staining, cells were incubated with both primary antibodies and subsequently with both secondary antibodies at the same time as specified previously. After washes in PBS, nuclei were stained with Hoechst 33342 1:10,000 (Life Technologies) for 5 min at room temperature. Slides were mounted with PBS:glycerol 1:1. In order to verify specific staining, HEK293T cells were used as negative control. Confocal microscopy was performed as described in Urciuoli et al.18 Images are processed using Adobe Photoshop V.CS4 software (Adobe Systems Inc).
Detection of cytokines
Cytokine levels in the supernatants from in vitro NKcell cultures were measured by DuoSet ELISA kits (R&D Systems) according to the manufacturer’s instructions. The cut-off limits of the assays were 18.76 pg/mL for IFN-γ (DY285b), 15.6 pg/mL for TNF-α (DY210), and 15.6 pg/mL for GM-CSF (DY215).
NK-cell cytotoxicity assay
NKcell cytotoxic activity was evaluated by a flow cytometric detection as previously described,19 and Daudi cells were used as targets at different E:T ratios. Percentage (%) of cell lysis was calculated as follows:

Chemokine profile
Supernatants obtained from freshly purified NK cells stimulated with TLR agonists for 20 hours, in the presence of IL-2 and IL-12, were tested to measure chemokine release.
Chemokines were quantified using a customized MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel assay (Millipore, Darmstadt, Germany), and the plate was read by a MAGPIX with xPONENT software, following the manufacturer’s instructions.
Migration assay
Purified NK cells were stimulated for 20 hours with TL8 (+IL-2 and IL-12), then washed, and cultured for a further 20 hours to collect conditioned media (CM). PBMC migration assays were performed using transwell inserts containing filters (pore size: 8 µm) (Corning, Tewksbury, USA) in a 24-well plate.
For the migration assay, 900,000 PBMCs per well (in 200 µL medium) were used. Lower chambers contained NK CM (1:2 dilution in complete RPMI 1640 medium). Cells were allowed to migrate for 3 hours at 37°C and 5% CO2, and transwell inserts were removed. Migrated cells were then stained using the following antibodies: CD56-PE, CD4-BV650, CD8-APC (Beckton-Dickinson), CD3-FITC, CD15-vioBlue (Miltenyi Biotech), CD16-BVB510 (BioLegend), and CD19-APCeFluor780 (Thermo-Fisher), and were analyzed by flow cytometry.
Results were plotted as fold change of the absolute number of migrated cells (for each subset) relative to NT samples.
Transfections
For TLR8 silencing experiments, freshly isolated NK cells were electroporated with Silencer Select TLR8 siRNA (assay ID: s27921) or negative control #2 siRNA using Neon Transfection system (Thermo Fisher Scientific), as previously described with minor changes.19 At day 0, for each experimental point, 2×106 cells were electroporated with 330 nM of siRNAs and then seeded in 0.75 mL of RPMI medium (Euroclone) supplemented with 20% FBS (Thermo Fisher Scientific), 2 mM L-glutamine (Euroclone), IL-2 600 u/mL (Miltenyi Biotech), and without antibiotics. At day 1, transfected cells were harvested and a part was used for evaluation of viability by trypan blue assay and TLR8 expression by qRT-PCR. The remaining part of transfected cells was seeded and treated with TL8-L in the presence of IL-2 and IL-12, as we described previously. We collected the supernatants after 20 hours of treatment and the IFN-γ release was measured by ELISA assay.
RNA extraction, cDNA synthesis and PCR analysis
Total RNA extraction from NK cells was performed with RNeasy Plus mini kit (starting material ≥1×106 cells) or micro kit (starting material <1×106 cells), following the manufacturer’s protocols (Qiagen GmbH, Hilden, Germany). RNA concentration and purity were evaluated by spectrophotometric analysis (Nanodrop 2000, Thermo Fisher Scientific). For gene expression analysis, total RNA was reverse transcribed with oligo-dT primers using Super Script IV first-strand synthesis system following the manufacturer’s instructions (Thermo Fisher Scientific). Real-time PCR for TLR7 and TLR8 expression were carried out in 96 wells in triplicate with PowerUp Sybr Green reagent (Applied Biosystems, Foster City, California, USA). GAPDH was used as endogenous control using the ΔΔCt method for comparing relative fold expression differences. The following primers were used: TLR3 fw: 5′-TCCCTGATGAAATGTCTGGA-3′, TLR3 rev: 5′-ATGCACACAGCATCCCAAAG-3′, TLR7 fw: 5′-CTGCTCTCTTCAACCAGACCTC-3′, TLR7 rev: 5′-CATCTAGCCCCAAGGAGTTTGG-3′, TLR9 fw: 5′-GAAGGGACCTCGAGTGTGAA-3′, TLR9 rev: 5′-TGCACCAGGAGAGACAGC-3′, TLR8 (BIO-RAD Laboratories, UniqueAssayID qHsaCED0036379), GAPDH fw: 5′-TCTTTTGCGTCGCCAGCCGA-3′, and GAPDH rev: 5′- ACCAGGCGCCCAATACGACC-3′.
PCR array for CCL3, CCL4, CXCL8, and CXCL11 in NK cells was performed in 384-well TaqMan array microfluidic cards with a custom configuration for the detection of selected genes implicated in NKcell biology (Thermo Fisher Scientific). Briefly, 600 ng of cDNAs for each sample was mixed with an equal volume of TaqMan Advanced Master Mix 2× and loaded in duplicate in the array cards (150 ng/channel). Reactions were carried out on a QuantStudio 7 Flex instrument using thermal PCR cycling conditions suggested by the manufacturer (Applied Biosystems). Data analysis was performed on Thermo Fisher Cloud with Design and Analysis New qPCR application (Thermo Fisher Scientific). PCR array data were analyzed with relative threshold algorithm and normalized using the mean of β-actin expression, used as reference gene (TaqMan assay ID Hs99999903_m1, Applied Biosystems and Thermo Fisher Scientific).
Amplified products of real-time PCR using TLR3, TLR7, TLR8, TLR9, and GAPDH primers (shown in figure 1A and online supplemental figure 2) were loaded and resolved in 2% agarose gel (TAE1X). DNA staining was performed with GelRed fluorescent dye (Biotium, Freemont, California, USA). 1 Kb Plus DNA ladder (Thermo Fisher Scientific) was used as marker.
Supplemental material

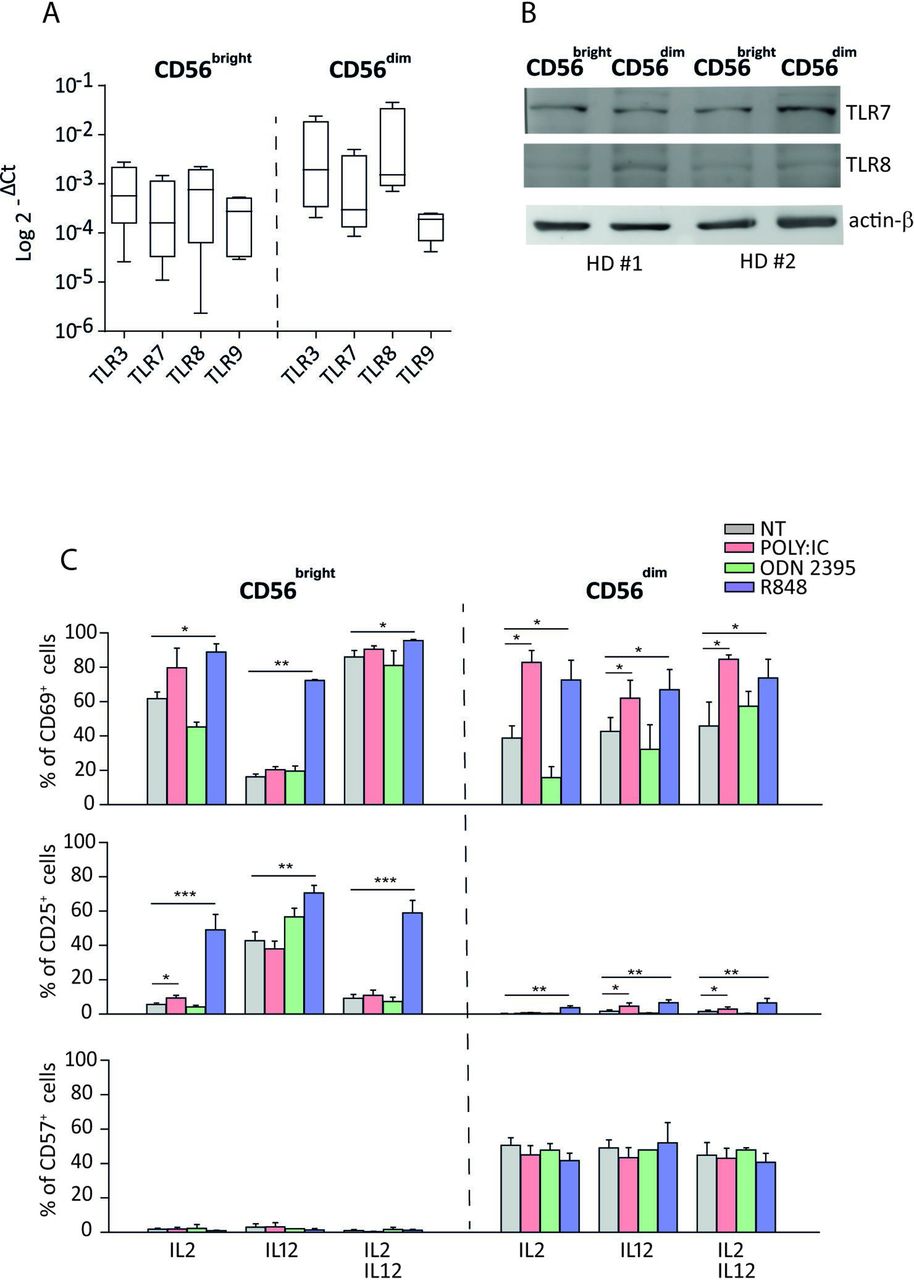
Endosomal TLR expression on CD56bright and CD56dim NK-cell subsets and their involvement in NK-cell activation. Expression of endosomal TLRs on freshly isolated NK cells evaluated by real-time PCR (n=4) (A) and TLR7 and TLR8 protein expression on freshly isolated NK cells assessed by western blotting analysis (n=2) (B). Analysis of CD69, CD25, and CD57 expressions on NK-cell surface after 20 hours of culture with indicated TLR-L plus either IL-2, IL-12, or their combination (n=4) (C). *p<0.05; **p<0.01; ***p<0.001. HD, healthy donor; IL, interleukin; NK, natural killer; NT, not treated; TLR, toll-like receptor.
Western blotting
NK whole-cell extracts were quantified by a bicinchoninic acid assay (BCA) (Thermo Fisher Scientific), resolved on 8% SDS-PAGE, and transferred onto nitrocellulose membrane. Filters were probed with primary antibodies overnight at 4°C, followed by biotinylated anti-mouse IgG (H+) (1:1000) or biotinylated anti-rabbit IgG (H+) (1:7500) (Vector Laboratories, Burlingame, California, USA) for 30 min at room temperature. Finally, filters were incubated for 30 min at room temperature with streptavidin-horseradish peroxidase (HRP) conjugate diluted 1:1000 in Tris-NaCl-Tyramide Signal Amplification (TNB-TSA) blocking buffer (0.1 M Tris–HCl, pH 7.5; 0.15M NaCl; 0.5% TSA blocking reagent) (PerkinElmer, Waltham, Massachusetts, USA). The following primary monoclonal antibodies were used: TLR3 (TLR3.7) (Thermo Fisher Scientific); TLR7 (4G6) (Bio-Techne, Abingdon, UK); TLR8 (D3Z6J) and TLR9 (D9M9H) XP rabbit monoclonal antibodies (Cell Signaling Technologies). β-actin (C4,Santa Cruz Biotechnology, Dallas, Texas, USA) was used as housekeeping. To detect β-actin, filter was further incubated with anti-mouse HRP-conjugated IgG (Cell Signaling Technologies).
Statistical analysis
For all the experiments, statistical significance was calculated using unpaired Student’s t-test. P values ≤0.05 were considered statistically significant. Significant p values were reported as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Graphs and statistical analysis were generated with GraphPad Prism V.9.1 software.
Results
R848 activates CD56brightCD16− NK cells in the presence of suboptimal doses of cytokines
Previous studies have demonstrated that resting NKcells express activation markers after endosomal TLRs triggering in the presence of suboptimal doses of cytokines.20–22 We first evaluated the expression of CD69 and CD25 activation markers, by culturing freshly isolated NK cells for 20 hours with TLRs-L, namely, R848 (ligand of TLR7/8), Poly I:C (ligand of TLR3), or ODN2395 (ligand of TLR9). In the absence of additional cytokines, no changes in the proportion of CD69+ and CD25+ NK cells or cytokines producing NK cells were observed (data not shown and online supplemental figure 1A). Conversely, and in agreement with the majority of previous reports,20–22 when NK cells were cultured in the presence of suboptimal doses of IL-2, IL-12, or both, R848 and Poly I:C increased significantly the proportion of CD69+ and CD25+ NK cells compared with untreated controls, while ODN2395 did not induce any significant increase of expression of such activation markers (online supplemental figure 1B). Moreover, the presence of each cytokine or their combination did not seem to strongly increase TLR7 and TLR8 protein levels on NK cells (online supplemental figure 1C).
Supplemental material
To assess whether specific NKcell subsets were more responsive to endosomal TLR stimulation, we evaluated the expression of endosomal TLR3, TLR7, TLR8, and TLR9 in freshly isolated CD56brightCD16− and CD56dimCD16+ NK-cell subsets. Notably, the four TLRs are consistently expressed in NK cells as shown at the RNA and, for TLR7 and TLR8 specifically, also at the protein level in freshly isolated and purified CD56brightCD16− and CD56dimCD16+ NK-cell subsets. However, previous reports show a high variability of TLR7 and TLR8 expression among individuals (figure 1A,B and online supplemental figure 2). Notably, TLR7 and TLR8 have also been also visualized by confocal microscopy, confirming their expression in NK cells (online supplemental figure 3).
Supplemental material
When CD56brightCD16− and CD56dimCD16+ NKcell subsets were specifically analyzed in the same samples, we confirmed that ODN2395 did not change the proportion of CD69+ cells in both NKcell subsets, whereas Poly I:C significantly upregulated CD69 expression on CD56dimCD16+ NK cells in the three experimental conditions analyzed (figure 1C). More importantly, R848 increased the expression of CD69 on both CD56dimCD16+ and CD56brightCD16− NK cells. On the other hand, CD25 expression was significantly upregulated only on CD56brightCD16− NK-cell subset on the three conditions with R848, whereas it was only slightly upregulated in these cells with Poly I:C plus IL-2. In agreement with the results on bulk NK cells, ODN2395 did not change the CD25 expression on both NK subsets (figure 1C). Importantly, the exhaustion marker CD57 was undetectable on CD56brightCD16− NK subsets with TLR agonists in all culture conditions (figure 1C).
Similar results were obtained by culturing cells with low doses of other growing cytokines such as IL-15, IL-18 alone, or in combination with IL-2 (online supplemental figure 4)
Supplemental material
Notably, on the previous experimental conditions, no changes in the expression of inhibitory (KIR3DL1 and NKG2A) and activating (NKG2D, DNAM-1, KIR2DL1/S1, NKp30, NKp40, and NKp46) receptors were observed on NKcell subsets (online supplemental figure 5A).
Supplemental material
These results demonstrate that R848 is the strongest activating stimulus of endosomal TLRs in NK cells and that it is the unique signal able to trigger the CD56brightCD16− NK subset in the presence of suboptimal doses of several environmental cytokines.
R848 increases the functional activity of CD56brightCD16− NK subset
In order to assess the impact of TLRs-Ls on the function of NKcell subsets, freshly purified NK cells were cultured for 20 hours with R848, Poly I:C, or ODN2395 in the presence of IL-2, IL-12, or both, and were analyzed for cytokine production, proliferative response, and cytotoxic activity.
Significant increased levels of IFN-γ,TNF-α, and GM-CSF were detected in the supernatants of R848-stimulated NK cells (cultured in the presence of cytokines), while Poly I:C and ODN2395 had poor or no effect (figure 2A). These results obtained with R848 stimulation were also confirmed by real-time PCR assay (figure 2B). When CD56brightCD16− and CD56dimCD16+ NK subsets were analyzed on the same samples, R848 dramatically increased the proportions of IFN-γ-producing cells in CD56brightCD16− and only slightly in CD56dimCD16+ cells (figure 2C). Similar results were obtained for CD56brightCD16− TNF-α-secreting cells. Unexpectedly, the number of GM-CSF-producing cells did not change on R848 (figure 2C). The discrepancy of this result with the previous data observed in the supernatants of R848-driven NK cells could suggest that GM-CSF is produced earlier (first 16 hours) in culture, likely declining before the flow cytometric detection, as suggested by some authors.23 However, this is not true for IFN-γ; indeed, by performing a time-course experiment, we found that such cytokine was upregulated on CD56brightCD16− NK subset by R848 only after 20 hours of culture, thus excluding an earlier release (online supplemental figure 5B).

R848 increases cytokine production and proliferation of CD56brightCD16− NK-cell subset. (A) IFN-γ, TNF-α and GM-CSF levels detected by ELISA in culture supernatants of NK cells treated 20 hours with TLR-L in the presence either of IL-2 or IL-12 or both (n=3). (B) Analysis of IFN-γ, TNF-α, and GM-CSF gene expression level by real-time PCR represented as fold change (2-ΔCt treated/2-ΔCt untreated) in the presence of suboptimal doses of IL-2 plus IL-12 (n=4). (C) Percentages of cytokines (IFN-γ, TNF-α, and GM-CSF) producing CD56brightCD16− and CD56dimCD16+ NK cells after 20 hours of stimulation with TLR-L and suboptimal doses of cytokines (n=3). (D) Representative CFSE proliferation profile of CD56brightCD16− (upper panels) and CD56dimCD16+ (bottom panels) NK-cell subsets after 2, 4, and 7 days of treatment with TLR agonists (n=4). Proliferation profile data are summarized in the right panel representing the percentage of NK cells divided at the referred time point. (E) Time course expression of CD25 on CD56brightCD16− (left panels) and CD56dimCD16+ (right panels) NK-cell subsets after TLR-L stimulation. Mean values±SEM are reported. Statistical significance has been determined by Mann-Whitney test. IFN-γ, interferon gamma; IL, interleukin NK, natural killer; NT, not treated; TLR, Toll-like receptor; TNF-α, tumor necrosis factor alpha, *p<0.05, **p<0.01, ***p<0.001
Importantly and in agreement with data on total NK cells, Poly I:C or ODN2395 did not induce any increase on the proportion of NKcell subsets producing the three tested cytokines (figure 2C). Similar results on IFN-γ and TNF-α secretion by NK subsets were obtained by culturing NK cells with R848 or Poly I:C plus suboptimal doses of IL-15, IL-18, or their combinations with IL-2 (online supplemental figure 5C).
Thereafter, the proliferative response of NKcell subsets cultured with TLRs-L stimuli was assessed in time-course experiments. According to previous results, R848 plus IL-2, IL-12, or both induced the proliferation of CD56brightCD16−, but not that of CD56dimCD16+ NK cells. We observed a peak of the proliferative response at 96 hours. Importantly, on the same culture conditions, Poly I:C induced a lower and delayed proliferation of CD56bright CD16− NK cells (figure 2D). In agreement, we observed that in the same time-course experiments, R848, but not the other TLRs agonists, was able to increase CD25 expression mostly on CD56brightCD16− NK cells in all culture conditions, peaking between 24 and 72 hours (figure 2E).
Lastly, we assessed the cytotoxic activity of NK cells under TLRs-L stimuli. NK cells, previously activated with R848 plus cytokines, were able to exert significantly higher cytotoxic activity than untreated NK cells, against Daudi cells (as targets), at different E:T ratios. By contrast, Poly I:C-stimulated and ODN2395-stimulated NK cells did not exert any change of cytotoxicity (figure 3A). On these conditions, CD56brightCD16−, but not CD56dimCD16+, NK cells expressed significantly higher levels of perforin (in the presence of IL-2) and granzymes A and B as well as CD107a expression when activated with R848, but not with the other ligands (figure 3B,C).

R848 increases the cytotoxic activity of CD56brightCD16− NK-cell subset. (A) The cytotoxic activity of bulk NK cells unstimulated (NT) or stimulated for 20 hours with R848 and Poly I:C (plus IL-2, IL-12, or both) was assayed against Daudi cells (as targets) at different E:T ratios by flow cytometry (n=4). (B) Intracellular expression of granzyme A, granzyme B and perforin on CD56brightCD16− and CD56dimCD16+ NK-cell subsets cultured for 20 hours with R848 and Poly I:C. Relative mean fluorescence intensity (rMFI) espression levels are reported (n=7). (C) Degranulated cells, measured by the expression of CD107a, on unstimulated (NT) and R848-stimulated or Poly I:C-stimulated CD56brightCD16− and CD56dimCD16+ NK cells (n=4). Mean values±SEM are reported. E:T, effector:target; IL, interleukin; NK, natural killer; NT, not treated; rMFI, Relative mean fluorescence intensity, *p<0.05, **p<0.01, ***p<0.001
These data confirmed that, among the TLRs agonists, the major activity on NK-cell function is exerted by R848, acting predominantly on the CD56brightCD16− subset.
R848 stimulates CD56brightCD16− NK cells via TLR8
Since R848 triggers TLR7 and TLR8 on DCs and both receptors are constitutively expressed on CD56brightCD16− and CD56dimCD16+ NK cells, subsequent experiments have been devoted to evaluate which of the two receptors is functionally active on these cells.
Thus, we assessed the function of freshly isolated NK cells cocultured for 20 hours with selective TLR7 (IMIQUI) or TLR8 (TL8-506) agonists and R848, as controls, in the presence of IL-2 and IL-12 or both. When we checked the proportion of CD69+ and CD25+ cells on purified NK cells cocultured with selective agonists or R848 (plus cytokines), we found that IMIQUI did not modify the expression of activation markers on both NK-cell subsets. By contrast, TL8-506 and R848 significantly increased the proportion of CD69+ and CD25+ cells on CD56brightCD16−, but not on CD56dimCD16+, NK cells (figure 4A and data not shown). In agreement, by evaluating the cytokine levels in the culture supernatants on the previous experimental conditions, we found that IFN-γ, TNF-α, and GM-CSF were significantly increased on TL8-506 and R848, but not IMIQUI, treatment (figure 4B). Similarly to the previous experiments, IFN-γ-producing cells, but not TNF-α-producing and GM-CSF-producing cells, were significantly increased in CD56brightCD16− NK cells stimulated with TL8-506 and R848 but not with IMIQUI (figure 4C), suggesting that the last two cytokines could be produced earlier in the culture. To definitively confirm that TLR8 is responsible of the functionally activation of NK cells when triggered by TLR8 agonist, we performed the TLR8 genetic silencing of resting NK cells by siRNA transfection. The IFN-γ production of silenced NK cells was drastically reduced compared with the same NK cells transfected with control siRNA (figure 4D).
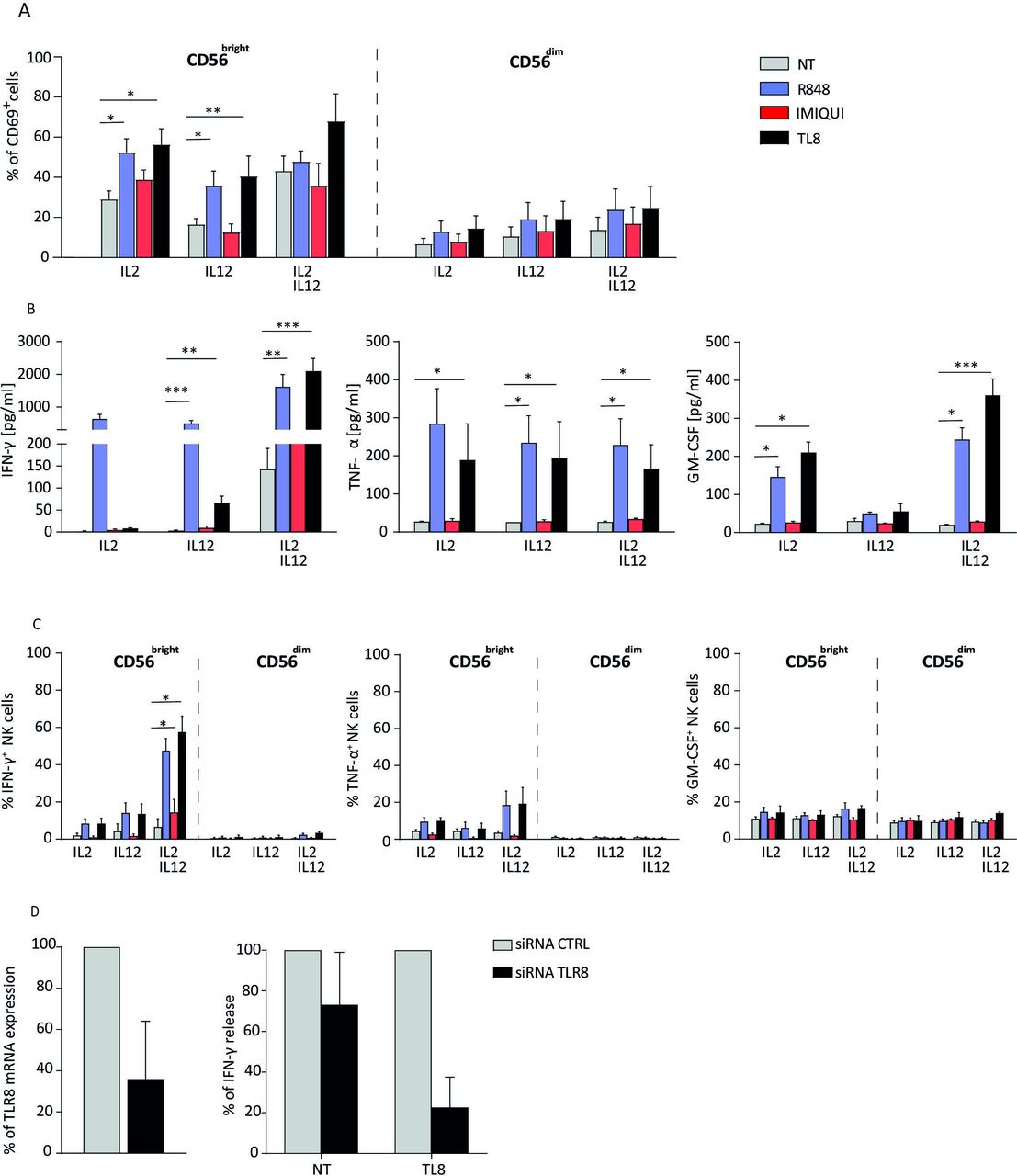
TLR8 triggering mediates activation of CD56brightCD16− NK-cell subset. (A) CD69 expression on CD56brightCD16− and CD56dimCD16+ NK cells stimulated for 20 hours with TLR7 (IMIQUI), TLR8 (TL8-506), or TLR7/8 (R848) agonists in the presence of IL-2 or IL-12 or both (n=4). (B) IFN-γ, TNF-α, and GM-CSF levels detected by ELISA in the supernatants of bulk NK cells stimulated with TLR7 or TLR8 agonists (n=4). (C) IFN-γ-producing, TNF-α-producing and GM-CSF-producing cells in CD56brightCD16− and CD56dimCD16+ NK-cell subsets precultured with TLR7 or TLR8 agonists plus suboptimal doses of cytokines (n=4). (D) Percentage of TLR8 mRNA expression in NK cells transfected with TLR8 siRNA versus NK cells transfected with a CTRL siRNA (n=4) (left panel) and relative analysis of IFN-γ released by transfected NK cells (n=3) (right panel). CTRL, control; IFN-γ, interferon gamma; IL, interleukin; NK, natural killer; NT, not treated; TLR, Toll-like receptor; TNF-α, tumor necrosis factor alpha, *p<0.05, **p<0.01, ***p<0.001
TLR8 agonist induces chemokine production, increases cytotoxicity, and synergizes with other TLR agonists on NK cells
To evaluate additional function of TLR8-driven NK cells, we assayed the chemokine production. Results showed that TLR8 agonists upregulate genes encoding for some chemokines as CCL3, CCL4, CXCL8, and CXCL10, on NK cells from HDs more than IMIQUI (figure 5A). In addition, we confirmed that TLR8-506 and R848 (rather than other endosomal TLR agonists) were able to induce the protein production of the same chemokines, thus eliciting a broad chemotactic activity from NK cells (figure 5B). In agreement, we showed that the culture supernatants of TLR8-triggered NK cells were able to promote migration of several types of PBMC (figure 5C).
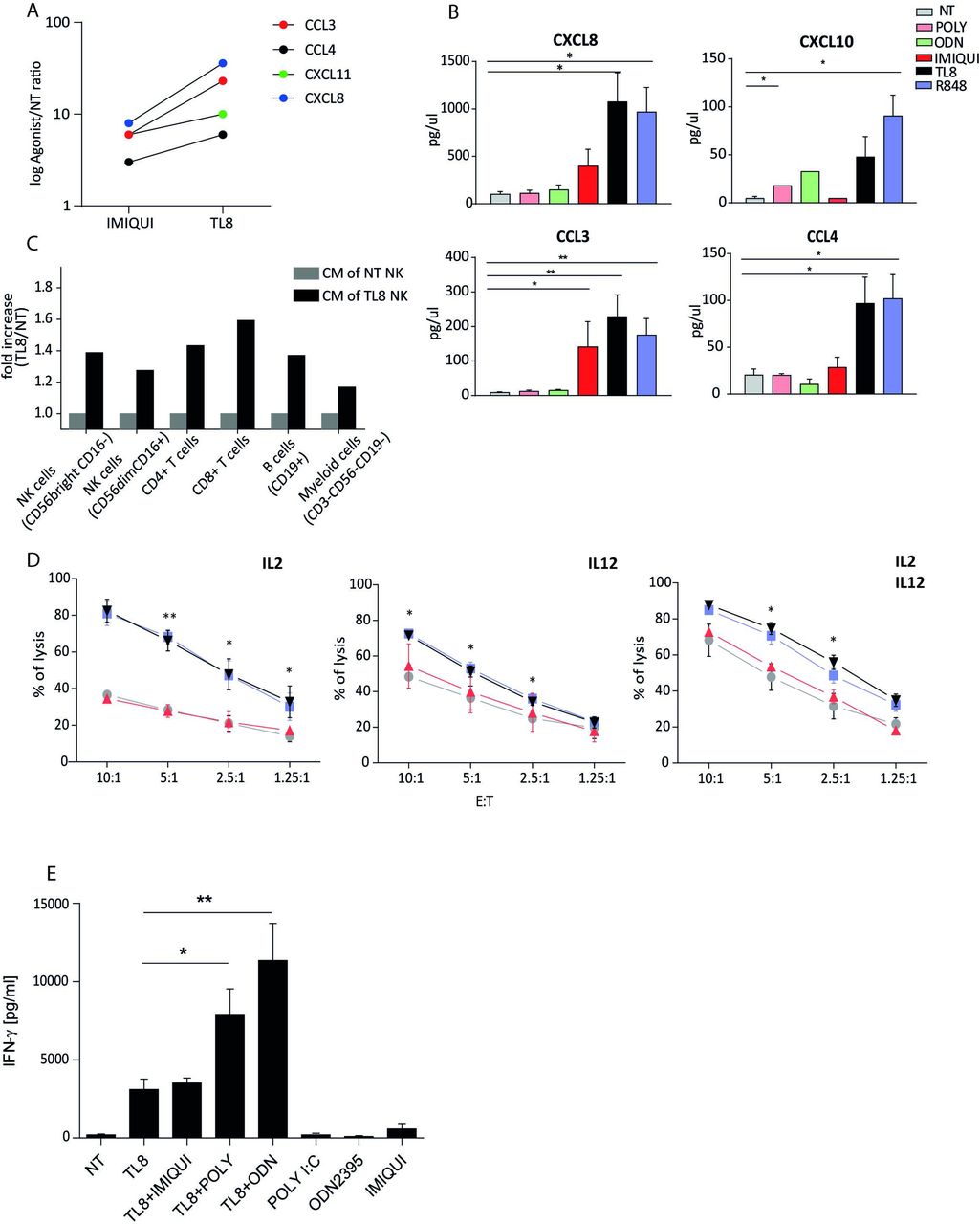
TLR8 agonist modulates NK-cell function. (A) Chemokine mRNA expression by NK cells on IMIQUI or TL8 stimulation, indicated as log 2-ΔCt agonist/2ΔCt untreated cells (NT) (n=3). (B) Chemokine proteins release analysis under indicated TLR agonist treatment (n=4). (C) Migration assay of PBMC under conditioning with CM of NK cells treated with TL8 or untreated (NT) (n=2). Migrated cells were harvested from the bottom chamber and stained for immune cell surface markers. NK cells were gated as CD56+CD3− and further gated into CD56brightCD16− and CD56dimCD16+ subsets. T cells were identified as CD56−CD3+ and further divided into CD8+ and CD4+. B cells were gated as CD56−CD3−CD19+, while within the CD56−CD3−CD19− gate, myeloid monocytic cells were considered as CD15− and CD16 either + or −. (D) Cytotoxic activity of unstimulated and stimulated bulk NK cells against Daudi cells (as targets) at different E:T ratios assessed by flow cytometry (n=3). (E) Evaluation of IFN-γ release by NK cells treated with TL8 agonists in the presence or absence of other TLRs-L (n=4). Mean values±SEM are reported. *p<0.05, **p<0.01. CM, culture medium; E:T, effector:target; IFN-γ, interferon gamma; IL, interleukin; IMIQUI, imiquimod; NK, natural killer; NT, not treated; TLR, toll-like receptor.
Furthermore, NK cells stimulated with TLR8-506 and R848, but not with IMIQUI, were able to significantly increase the cytotoxic activity towards Daudi cells at different E:T ratios (figure 5D). The role of TLR8 on NK cells was highlighted by the finding that TLR3 or TLR9 agonists (which were not effective on NK cells when singularly used) induced the cytokine production when stimulated in combination with TLR8-L, even potentiating its activity (figure 5E). On the whole, these data confirm that TLR8 is functionally active in NK cells and is responsible of inducing/potentiating different functions.
TLR8 agonists improve peritoneal natural killer (pNK)-cell function in metastatic ovarian cancer
Since the majority of NK cells detectable in TME belongs mainly to the CD56brightCD16− cell subset, we checked the activity of TLR8-L on tumor-associated NK cells. pNK cells were purified from ascites fluids of nine patients with metastatic ovarian cancer and assayed for phenotype and function. In the purified pNK-cell samples the number of contaminating cells were consistently less than 1%, and the proportion of CD56brightCD16− NK cells ranged from 70% to 100% of total NK cells, irrespective of histotypes. Virtually all these cells expressed CD69. However, when pNK cells were cultured for 20 hours with R848, plus IL-2, IL-12, or combination, the proportion of CD25+ cells was significantly increased in comparison with controls (figure 6A). Furthermore, IFN-γ and TNF-α levels in the supernatants of TLR8 (but not TLR7-) agonist-cultured NK cells were significantly increased compared with controls (figure 6B). Finally, the cytotoxic activity against Daudi cells of pNK preactivated with TLR7/8 agonists was significantly improved in comparison with those untreated (plus suboptimal doses of cytokines) (figure 6C). Interestingly, preliminary data indicated that the TLR8 agonist is able to induce the production of IFN-γ by pNK cells also in the context of autologous (highly suppressive) mononuclear plus tumor cells, as surrogate assay to establish which patient can potentially benefit from adoptive therapy with in vitro conditioned NK cells (online supplemental figure 6). Therefore, our conclusion is that TLR8 agonists may trigger NK cells in the TME, increasing their functional activity.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TLR8 agonist improves function of pNK cells from patients with metastatic ovarian cancer. (A) CD25 expression of pNK cells stimulated with TLR7/8-agonists for 20 hours in the presence of IL-2, IL-12, or both (n=4). (B) IFN-γ and TNF-α levels detected by ELISA in the supernatants of pNK cells stimulated with TLR7 or TLR8 agonists (n=3). (C) Cytotoxic activity of unstimulated and stimulated pNK cells against Daudi cells (E:T ratio=1:1) assayed by flow cytometry (n=4 for IL-2, n=1 for IL-12, and n=3 for IL-2+IL-12 costimulation). Mean values±SEMs are reported. *p<0.05. E:T, effector:target; IFN-γ, interferon gamma; IL, interleukin, NT, not treated; pNK, peritoneal natural killer; TLR, toll-like receptor
Discussion
This study has been addressed to evaluate the response of human NK cells and their subsets to the stimulation of endosomal TLRs agonists, with the specific aim to establish the most suitable molecules to be used as adjuvant in cancer vaccines or in combined immunotherapy of cancer. We show that the R848 agonist delivers a potent and selective stimulus to TLR8. In addition, the most responsive NK subset is the CD56brightCD16−, ready to prevalent cytokine production.
The endosomal TLRs and some cytosolic sensors of RIG-I family recognize specifically motifs present on DNA or RNA of several pathogens, and their triggering on antigen presenting cells (APCs) contributes to induce type I/III interferon, proinflammatory cytokines and chemokines, or costimulatory molecules. Controversial observations have been reported on the expression of endosomal TLRs in human NK cells at mRNA and protein levels,21 24–26 likely explained with the use of different primer sets and detecting antibodies. Other causes for discrepancy may be the (1) activation degree of NK cells expressing TLRs in different donors; (2) DC–NK interaction mediated by different surface and soluble signals which, in turn, may favor or impair the expression and triggering of endosomal TLRs on NK cells; (3) cross-talk among such receptors after triggering of one of them; and (4) interference of cytosolic miRNAs with TLR7 or TLR8 agonists for the receptors.22 26–29 Despite several reports having been published on the expression and function of TLRs on NK cells, the activity of each agonist on endosomal TLRs has been poorly examined.
Once the TLR detection was optimized, we initially demonstrated that the four endosomal TLRs were consistently expressed on freshly isolated CD56brightCD16− and CD56dimCD16+ NK subsets both at the mRNA level and, in particular, TLR7 and TLR8 have also been checked for protein expression and endosomal localization. TLR8, but not TLR7, mostly localizes at late endosome level. Since it is known that the endosomal pH is important for the functional activation of TLRs,30 this may explain the functional differences between TLR7 and TLR8 in NK cells. However, this finding does not mean that all TLRs are functionally active.
Then, we optimized the experimental conditions (timing, doses, etc), allowing the TLRs triggering of NK cells by using different ligands. Taking advantage of synthetic agonists of human TLRs, we confirmed that resting NK cells cannot be activated by endosomal TLRs-L (as R848, Poly I:C, and ODN2395) in the absence of costimuli as suboptimal doses of growing cytokines. Even though some reports indicate that NK cells can be directly activated by some TLRs-L,21 24 the majority agrees that the cytokines produced by DCs and other innate cells may play a major role in such indirect activation of NK cells.20 22 26 27 29
Coculturing with suboptimal doses of IL-2, IL-12, IL-15, and IL-18 (alone or in combination with IL-2) allowed us to demonstrate the different effects of TLRs-L on NK-cell function. The first relevant result of the study is that R848, rather than Poly I:C and ODN2395, gives the strongest signal upregulating NK-cell function. Indeed, we found that the activation markers CD69 and CD25 were significantly increased by both R848 and Poly I:C on NK cells, but the cytotoxic activity was only improved by the former. Also, the levels of IFN-γ, TNF-α, and GM-CSF in the supernatants of NK cells cultured with R848 were much higher than those detected with the other two agonists (Poly I:C and ODN2395). These data clearly demonstrate that, when the different stimuli were simultaneously compared, R848 consistently gave the strongest signal to NK cells.
Moreover, R848 is the unique agonist able to activate the CD56brightCD16− NK subset. Indeed, while Poly I:C stimulates only CD56dimCD16+ NK cells, R848 activates both subsets and even at the higher extent the CD56brightCD16− on suboptimal doses of cytokines. Importantly, R848 did not induce CD57 expression on the CD56brightCD16− subset, thus suggesting no detectable effects on their maturation.
In agreement with these data, R848, but not the other TLRs agonists, carried out its activity almost prevalently on CD56brightCD16− NK cells by increasing several functional activities of such cell subset as proliferation, cytotoxicity, and cytokine production. R848 induced selective proliferation of CD56brightCD16− NK cells, which peaked at 4 days and was preceded by an increase of CD25 expression, while it did not exert any proliferation and a slight increase of CD25 expression on CD56dimCD16+ NK cells. Poly I:C also induced a proliferative response of CD56brightCD16+ NK cells, which, however, was much lower and delayed (7 days) in comparison to R848.
The upregulation of CD107a, perforin, granzymes A and B in CD56brightCD16−, but not in CD56dimCD16+ NK cells cultured with R848, clearly indicates that the increase of cytotoxic activity observed on total NK cells was mainly related to this subset. Notably, in our conditions, Poly I:C did not modify CD107a and granzyme A and B expression on both NK subsets. This is in odds with previous results reporting that TLR3 or TLR9 agonists increase several NK functions.20 27 29 The discordant results are likely due to some differences in culture conditions (such as the timing of cultures, the stimulant dose, and the type and amounts of costimulatory cytokines), the type of TLRs-L (especially of TLR9), or the high variability of TLRs expression and signaling on NK cells, as previously described.20 27 29
Finally, R848, but not the other TLRs agonists, was able to induce the production of IFN-γ by both CD56brightCD16− and CD56dimCD16+ NK cells, even though the proportion of IFN-γ-secreting cells was significantly much higher in the former subset. Regarding the other two cytokines (TNF-α and GM-CSF), a discrepancy exists between the increased levels detectable in the supernatants of R848-driven NK cells and the proportion of NK cells producing the same cytokines, which is unchanged on R848 stimulation. Since at the mRNA level both cytokines were highly increased on R848, the previous result could suggest that TNF-α and GM-CSF are produced earlier in culture, likely declining before the flow cytometric detection, as previously reported.23 24 However, this is not true for IFN-γ, since, in a time-course experiment, we confirmed that such cytokine was upregulated in the CD56brightCD16− NK subset by R848 with a peak at 20 hours of culture, thus excluding an earlier release. Overall, these findings confirm that R848 exerts the strongest signal on NK cells since it is the only TLR-L able to stimulate both NK subsets and primarily the CD56brightCD16− subset.
Another relevant finding of this study is that R848 triggers NK cells only through TLR8, whereas R848 is well known to activate both TLR7 and TLR8 in DCs. Indeed, the TLR8-agonist TL8-506 upregulates significantly the activation markers, the cytotoxic activity, and the cytokine production by the CD56brightCD16− NK-cell subset, whereas the TLR7 agonist IMIQUI or other TLR7 agonists (as loxoribin) failed to modify NK-cell function when assayed in the same experimental conditions (online supplemental figure 7). The selective activation of TLR8 by TL8-506 was confirmed by the decrease of IFN-γ production by NK cells silenced for TLR8 and stimulated with its specific agonist. Some previous reports underscored the ability of different TLR7/8 agonists (including R848) to stimulate total NK cells. However, they did not report the selective functional activity of TLR8 alone on these cells,22 25 26 whereas our data show that TLR8 is also able to induce a massive IFN-γ release when combined with TLR3 or TLR9, but not TLR7, agonists which are ineffective if used alone. This suggests that TLR8 can interact with other TLRs, likely upregulating their expression. Notably, similar to NK cells, TLR8, but not TLR7, has recently been shown to be functionally active in other innate cells as neutrophils.31 32 The presence of a functional TLR8 in NK cells is a relative new notion since it confirms (1) the prevalence of TLR8 signaling on human innate cells in comparison to murine counterparts which largely prefer TLR7 expression33; (2) TLR8, but not TLR7, is expressed in a dimeric (active) form in a ligand-independent way, perhaps favored by DC-driven cytokines34; (3) the IFN-γ produced in NK cells by several stimuli is a strong inducer (in an autocrine way) of expression of TLR8, much more than TLR735; and (4) the miRNAs contained in exosomes derived from NK cells, as well as tumor and tumor environmental cells, bind almost exclusively to TLR8, impacting on NK-cell function.36 37
Supplemental material
As a whole, these findings highlight the potential role of TLR8 targeting infiltrating TME-NK cells as novel immunotherapy of tumors. The predominant activity of TLR8 agonists on CD56brightCD16− NK cells, a subset largely detectable in TME, makes the local infusion of NK cells stimulated and largely amplified by TLR8 agonists, or the combination of TL8 with others TLRs agonists, more than a potential strategy for cancer therapy. Indeed, we showed that TLR8 targeting was able to improve function (IFN-γ production and cytotoxic activity) of purified NK cells from the ascites of patients with metastatic ovarian carcinoma which are almost exclusively represented by CD56brightCD16− cells. Moreover, the latter cells are able to produce IFN-γ on TL8 stimulation also in an autologous setting where pNK cells undergo to highly suppressive environment. In these tumors, in vitro activated NK cells which, in turn, may produce cytokines and chemokines devoted to further amplify NK cells themselves by both proliferation and recruitment, enroll and modulate the functional profile of other cells favoring antitumor response. Indeed, our data indicate that TL8 induces NK cells to produce CXCL8, CXCL10, CCL3, and CCL4 chemokines which can promote the recruitment of many cells of innate and adaptive immunity expressing CXCR1/2, CXCR3, CCR1, and CCR5 receptors respectively, namely, neutrophils and their precursors, monocytes/macrophages, T and NK cells. A feasible strategy is to design a rapid and easy assay to check which patient is more susceptible to the modulatory activity of the TLR8 agonist on NK cells. A promising approach suggested by our preliminary data is to directly stimulate all mononuclear cells from ascites (which include all types of suppressive cells of TME) with this molecule and to test for cytokine production and cytotoxicity.
Indeed, in vitro and in vivo data indicated that TLR8 agonists may (1) reverse the suppressive functions of human tumor-derived CD4+ T cells, CD8+ T cells, and γδ T cells12 13 38; (2) induce a switch from M2 to M1 profile of intra tumor (IT) macrophages39; (3) enhance apoptosis of myeloid-derived suppressor cells40; and (4) prevent T-cell senescence/exhaustion.41 Moreover, TLR8 agonists were able to induce in vivo the metabolic control of CD4+ T-regulatory cells in ovarian cancer microenvironment.42 Some data on murine models indicated that locally infused TLR7/8 agonists delayed the tumor growth and, in some reports, induced tumor regression as in an acute myeloid leukemia model.43 44 Lastly, in humans, TLR8 agonists have been locally assayed in clinical phase Ib–II trials on patients with different metastatic solid tumors14 45 46; indeed, there are few data about the possibility that such a soluble agonist may display detrimental effects on a number of TME cell types bearing the active receptor.
Our data could suggest that the complex effects referred to TLR8 agonists are mediated, at least in part, by NK cells activated by the same molecules, as shown in some in vivo models.47 48 Next research efforts must be devoted to establish the interactions among NK cells activated by TLR8 agonists and other cells in TME, concentrating on drug safety, efficiency, and specificity.49 Collectively, if confirmed on a larger number of patients, these data highlight the potential value of locally infused TLR8 agonists as adjuvants of tumor vaccines or combined immunotherapy of cancer.
Data availability statement
No data are available. Deidentified subject data protocols.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human subjects and was approved by Bambino Gesù Paediatric Hospital (AIRC5X1000 #21147) and was conducted in accordance with the tenets of the Declaration of Helsinki. Subjects and donors gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
IV and CA contributed equally.
Correction notice This article has been corrected since it was first published. An error with figure 2 and the supplemental material has been amended.
Contributors IV, CA, and EM designed the project and wrote the manuscript. IV, CA, AP, BR, NL, and VDO performed the experiments. IV and CA analyzed the data. GS and AF provided the ovarian cancer samples. EM and LM discussed the results, provided critical comments and are responsible for the overall content.
Funding This work was supported by grants from the Ministero della Salute (grant number RC-2020 OPBG to LM and EM) and from Associazione Italiana per la Ricerca sul Cancro (project number 5x1000 2018 Id 21147 and IG 2017 Id 19920 to LM). IV was supported by FIRC-AIRC fellowship for Italy; CA was a recipient of a grant awarded by Fondazione Umberto Veronesi; NL received funding from European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant (grant agreement number 800924).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.