Article Text

Abstract
Background Adoptive transfer of CD19-specific chimeric antigen receptor (CD19CAR) T cells can induce dramatic disease regression in patients with B cell malignancies. CD19CAR T cell therapy may be limited by insufficient engraftment and persistence, resulting in tumor relapse. We previously demonstrated a proof of principle that cytomegalovirus (CMV)-specific T cells can be isolated and enriched prior to CD19CAR transduction to produce CMV-CD19CAR T cells, and that these CMV-CD19CAR T cells can be expanded in vivo through CMV vaccination, resulting in better tumor control in a murine model. Here we developed a clinical platform for generating CMV-CD19CAR T cells.
Methods Peripheral blood mononuclear cells (PBMCs) collected from CMV-seropositive healthy donors were stimulated with a good manufacturing practices-grade PepTivator overlapping CMVpp65 peptide pool and enriched for CMV-responsive interferon γ (IFNγ)+T cells using IFNγ Catchmatrix, within the CliniMACS Prodigy Cytokine Capture System (Miltenyi Biotec). Resulting CMV-specific T cells were transduced with a lentiviral vector encoding a second generation CD19R:CD28:ζ/EGFRt CAR and expanded with interleukin 2 (IL-2) and IL-15 for 15 days before characterization.
Results CMV-specific T cells were enriched from 0.8%±0.5 of input PBMC to 76.3%±11.6 in nine full-scale qualification runs (absolute yield of 4.2±3.3×106 IFNγ+T cells from an input of 1×109 PBMCs). Average CD19CAR transduction efficiency of CMV-specific T cells was 27.0%±14.2 in the final products, which underwent rapid expansion, resulting in a total cell dose of 6.2±0.9 × 106 CD19CAR-tranduced T cells with CMV specificity (ie, functionally bispecific). CMV-CD19CAR T cells were polyclonal, expressed memory markers but had low expression of exhaustion markers, responded to both CD19 and CMVpp65 stimulation with rapid proliferation and exhibited antigen-specific effector functions against both CD19-expressing tumors and CMVpp65 antigen. The final products passed release criteria for clinical use.
Conclusions We demonstrated the feasibility of our large-scale platform for generating CMV-CD19CAR T cells for clinical application. We plan to initiate a clinical trial at City of Hope using CMV-CD19CAR T cells for patients with intermediate/high-grade B cell non-Hodgkin’s lymphoma immediately after autologous hematopoietic cell transplantation followed by vaccination with a novel CMV vaccine based on Modified Vaccinia Ankara (Triplex) 28 days and 56 days post-T cell infusion.
- receptors
- chimeric antigen
- cell engineering
- hematologic neoplasms
- immunotherapy
- adoptive
- vaccination
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Adoptive transfer of virus-specific T cells can effectively prevent progressive viral infection, with long-term persistence of adoptively transferred cells.1–3 In the context of cytomegalovirus (CMV) disease, viremia refractory to antiviral chemotherapy, or both, the majority of patients treated with very low numbers (21×103 /kg) of T cells specific for the CMV peptide pp65 cleared their CMV infection or significantly reduced their viral burden.4 In contrast, adoptive T cell therapy for the treatment of cancer, including chimeric antigen receptor (CAR) T cell therapy, has been significantly more challenging and less effective than for viral diseases, primarily due to lack of persistence of the adoptively transferred tumor-specific T cells in patients.5 The mechanisms for differential persistence of adoptively transferred virus-specific T cells in hematopoietic cell transplantation (HCT) recipients versus tumor-reactive T cells in patients with cancer is not fully understood, but possibly reflects the environment into which the T cells are infused, qualitative attributes of the T cells that are isolated and expanded for adoptive transfer, and the fact that virus-specific T cells receive optimal co-stimulation after engagement of their native T cell receptors (TCRs).1–3 6 7
To improve persistance and efficacy of CAR T cells, several groups have generated T cells with dual specificity; T cells with virus-specific TCRs are first isolated and then transduced with a tumor-specific CAR construct. Thus, this strategy allows for expansion of tumor-specific CAR T cells following engagement of the virus-specific TCR. For example, isolated Epstein–Barr virus (EBV)–specific T cells modified to express GD2-targeting or CD30-targeting CARs recognize tumors of neural crest origin,8–10 while isolated influenza A matrix protein 1–specific T cells modified to express CD19-targeting CARs (CD19CAR) recognize B cell malignancies.11 These virus and CAR bispecific T cells demonstrate superior in vivo survival and antitumor activity compared with CAR T cells alone.8–11 Furthermore, clinical studies demonstrate that adoptively transferred EBV ×CD19 CAR or EBV ×CMV × CD19CAR bi (tri) specific T cells proliferate in patients as a result of viral reactivation;12 13 Lapteva et al reported that in patients with viral reactivation, up to 30,000-fold expansion of CD19CAR-virus specific T cells was observed, with depletion of CD19 +B cells.13
We previously developed CMV-specific CD19CAR (CMV-CD19CAR) T cells by transducing our CD19CAR construct into purified T cells with TCRs specific for CMVpp65. CMV-CD19CAR T cells proliferate vigorously after engagement through either the endogenous CMVpp65 TCR or the engineered CD19CAR and exhibit specific cytolytic activity and interferon γ (IFNγ) secretion against target cells.14 On adoptive transfer into immunodeficient mice bearing human CD19 +lymphomas, CMV-CD19CAR T cells exhibit antitumor activity, which we can enhance through delivery of a CMVpp65 peptide vaccine that promotes CMV vaccine-mediated CMV-CD19CAR T cell expansion.14
Dr Don Diamond’s laboratory at City of Hope has engineered CMV-Modified Vaccinia Ankara (MVA) Triplex vaccine, a multiantigen recombinant MVA virus vaccine with genes encoding three CMV immunogenic proteins, pp65, IE1, and IE2. In clincial trials of healthy donors, the Triplex vaccine induced robust expansion of pp65-specific, IE1-specific and IE2-specific CD8 and CD4 T cells in both CMV-seropositive and seronegative subjects without severe adverse events or dose-limiting toxicities at all dose levels.15 In a multicenter randomized, placebo-controlled phase II trial (NCT02506933), Triplex vaccine was well tolerated and highly immunogenic with clinical efficacy in recipients of allogeneic HCT.16
Based on our preclinical data, we anticipate that CMV-CD19CAR T cells will expand in patients following administration of the CMV-MVA Triplex vaccine. The in vivo expansion of CMV-CD19CAR T cells through the CMV-specific TCR is expected to improve antitumor activity and durable remission by improving CD19CAR T cell persistence. To translate this discovery into the clinic, we have established a large-scale platform under good manufacturing practices (GMP) guidelines to generate CMV-CD19CAR T cells and have characterized the final products for clinical application. Our data demonstrated the feasibility and consistency of generating CMV-CD19CAR T cells, which exhibited cytolytic activity against CD19 + tumors and responded to CMVpp65 stimulation with rapid proliferation.
Methods
Antibodies and flow cytometry
Human peripheral blood mononuclear cells (PBMCs) and T cells were analyzed by flow cytometry after staining with fluorochrome-conjugated monoclonal antibodies (mAbs) against CD3 (clone Leu-4), CD4 (clone SK3), CD8 (clone RPA-T8), CD28 (clone CD28.2), CD62L (clone SK11), CD27 (clone M-T271), CD127 (clone HIL-7R-M21), streptavidin (Catalog No. 554061), CD45RA (clone BHKG-2), IFNγ (clone B27), CD45RO (clone UCHL1), CD57 (clone NK-1) that were obtained from BD Biosciences. Antibodies against killer cell lectin-like receptor G1 (KLRG1) (clone 2F1), Tim3 (clone F38-2E2) and epidermal growth factor receptor (EGFR) (clone AY13) were purchased from BioLegend. The anti-LAG3 (Catalog No. LS-C130398) and anti-PD1 (MIH4) antibodies were from Lifespan Biosciences and eBiosciences, respectively. Anti-IFNγ (45–15, Miltenyi) was used to check the purity of IFNγ+cells postenrichment. Biotinylated cetuximab (Erbitux) was generated from Erbitux purchased from the City of Hope pharmacy. Briefly, 200 mg Erbitux was buffer exchanged to phosphate buffered saline (PBS) (D-PBS, pH 7.5±0.1) using a MidGee Hoop Cartridge (UFP-30-E-H42LA). The material (2 mg/mL) was modified with Sulfo-NHS-LC-biotin (30:1) in a 2-hour reaction on ice and diafiltered to remove excess biotin. The biotinylated Erbitux was then buffer exchanged to PBS and frozen. Product purity was confirmed on NuPAGE Novex Bis-Tris gels with or without sodium dodecyl sulphate (SDS) reduction. For cell-surface phenotyping experiments, we stained PBMCs with optimized antibody panels for 20 min at 4°C followed by two washes with PBS. The IOTestBeta Mark TCR Vβ Repertoire Kit (representing ~70% of normal TCR Vβ repertoire) was obtained from Beckman Coulter and analysis was performed according to the kit instructions. Isotype-matched mAbs served as controls. Data acquisition was performed on MACSQuant Analyzer 10 (Miltenyi Biotech) and analyzed using FCS Express V.3-V.7 Software (De Novo Software).
Cell lines
EBV-transformed lymphoblastoid cell lines (LCLs) were made from PBMCs as previously described.17 To generate muromonab-CD3 (OKT3)-expressing LCLs, LCLs were resuspended in nucleofection solution using the Amaxa Nucleofector kit T, OKT3-2A-Hygromycin_pEK plasmid was added to 5 µg/107 cells, the cells were electroporated using the Amaxa Nucleofector I, and the resulting cells were grown in Roswell Park Memorial Institute (RPMI)-1640 with 10% fetal calf serum (FCS) containing 0.4 mg/mL hygromycin.14 To generate enhanced green fluoresent protein (eGFP) +LCLs, LCL cells were transduced with lentiviral vector encoding eGFP and eGFP +cells were purified by FACS sorting and expanded for use in these experiments. KG1a cells were purchased from American Type Culture Collection (ATCC) and transduced with lentiviral vector encoding eGFP. Banks of all cell lines were authenticated for the desired antigen/marker expression by flow cytometry prior to cryopreservation and thawed cells were cultured for less than 6 weeks prior to use in assays. To generate cells capable of stimulating the CMV-CD19CAR T cells via the CMV-specific TCR, autologous PBMCs were loaded with human cytomegalovirus (HCMV) PepMix (pp65pepmix) (>90%) (JPT peptide Technologies GmbH) at 1 µg/mL for 2 hours at 37C°. Cells were washed in PBS twice before use.
CliniMACS Prodigy immunomagnetic CMV-specific T cell enrichment after CMVpp65 stimulation
Leukapheresis products were obtained from healthy donors using protocols approved by the City of Hope Institutional Review Board or purchased from StemCell Technologies. PBMCs were isolated by density gradient centrifugation over Ficoll-Paque (Pharmacia Biotech). PBMCs (1×109) were loaded on the closed and automated CliniMACS Prodigy system (Miltenyi Biotec) and unused PBMCs were frozen for later use. CMV-specific T cells were isolated on the CliniMACS Prodigy and cytokine capture system (CCS) system according to the manufacturer’s instruction. Briefly, after PBMCs (109) were added to the application bag connected to the tubing set, the CliniMACS Prodigy device automatically performed successive processes, including sample washing, antigen stimulation with PepTivator CMVpp65, Catchmatrix labeling, anti-IFN-γ microbead labeling, magnetic enrichment and elution. CMV-specific cells and non-CMV-specific cells were eluted in separate bags following magnetic enrichment.
Generation of CMV-CD19CAR T cells
After overnight incubation in RPMI medium containing 10% human AB serum (Gemini Bio Products), IL-2 (50 U/mL) and IL-15 (1 ng/mL), recovered IFNγ+cells (~1 × 106) were transduced at multiplicity of infection (MOI) 3 with current good manufacturing practice (cGMP) grade CD19CAR lentivirus that is used in current clinical trials (NCT02146924 and NCT02153580). The CD19R:CD28:ζ/EGFRt-epHIV7 contains: (1) VH and VL gene segments of the CD19-specific FMC63 mAb, IgG4 hinge with mutations at two sites (L235E; N297Q), CD28 costimulatory molecule, and CD3ζ chain;18 (2) The ribosomal skip T2A and truncated human EGFR (EGFRt) sequence.19 Fresh culture medium and cytokines were added every other day for ~15 days at 37°C and 5% (v/v) CO2 before phenotypic and functional characterization. Where indicated, PBMCs from the same donor were activated with anti-CD3/CD28 beads (Fisher Scientific) and transduced with CD19CAR to generate conventional CD19CAR T cells. CD19CAR and CMV-CD19CAR T cells were expanded in parallel prior to use.
Phenotypic characterization of CMV-CD19CAR T cells
After 14 days ex vivo expansion in the presence of IL-2 and IL-15, CMV-CD19CAR T cell products were analyzed with multicolor flow cytometry for the expression of immune receptors representing memory, activation and exhaustion. To analyze the clonal type of the products, we performed flow cytometry-based Vβ repertoire analysis using The IOTestBeta Mark TCR Vβ Repertoire Kit (representing ~70% of normal TCR Vβ repertoire) per the manufacture’s instruction. To simultaneously analyze protein and gene expression by flow cytometry, the PrimeFlow assay (Thermo Fisher) was performed per the manufacturer’s suggested protocol.
Cytolytic activity assay and intracellular IFNγ staining
Effector CMV-CD19CAR T cells (2.5×105) and eGFP +LCL target cells were co-cultured at various E:T ratios (0:1, 0.06:1, 0.3:1 and 0.6:1) for 3 days. Co-cultures with OKT3 LCL, and KG1a were used as positive and negative controls, respectively. The percentage of viable eGFP +CD3- tumor cells was measured using multicolor flow cytometry. Cytotoxicity induced by CMV-CD19CAR T cells was normalized to the number of tumor cells remaining in the 0:1 E:T ratio control well. To assess INFγ release, effector CMV-CD19CAR-T cells (1×105) were stimulated overnight with LCL, OKT3 LCL, KG1a cells or CMVpp65pepmix-pulsed autologous PBMCs (1×105) in 96-well tissue culture plates in the presence of Brefeldin A (BD Biosciences). The cell mixture was stained with either anti-CD8 and anti-EGFR antibodies or biotinylated cetuximab and streptavidin to analyze surface expression of CD8 and CAR, respectively. Cells were then fixed and permeabilized using the BD Cytofix/Cytoperm kit (BD Biosciences). After fixation, the cells were stained with an anti-IFNγ antibody and analyzed using multicolor flow cytometry on MACSQuant (Miltenyi Biotec).
Proliferation assays
CMV-CD19CAR T cells (2.5×105) were labeled with 0.5 µM CellTrace Violet dye (CTV) and co-cultured with 8000 centigray (cGy)-irradiated stimulator cells LCL-OKT3, KG1a, or 3700 cGy-irradiated autologous CMVpp65-peptide pulsed PBMCs at 1:1 ratio for 7 days. Proliferation of the CD3 +population was determined using multicolor flow cytometry.
Cytokine production assays
CMV-CD19CAR T cells (1×105) were co-cultured overnight in 96-well tissue culture plates with 1×105 OKT3 LCL, LCL, KG1a, or autologous CMVpp65-pepmix-pulsed PBMCs. Supernatants were analyzed with the Luminex IS100 bead array technology with kits purchased from Life Technologies (Invitrogen) for Human Cytokine 30-Plex according to the manufacturer’s instructions.
WPRE qPCR assay
To analyze the T cell products produced during qualification run for vector copy number, genomic DNA (gDNA) was extracted from 2×106 CMV-CD19CAR T cells for each product using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s protocol. The gDNA concentrations were measured on the NanoDrop One Spectrophotometer (Invitrogen) and then tested for the detection of Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE) DNA by quantitative PCR (qPCR). The release criteria for CMV-CD19CAR T cell products was set to ≤5 WPRE copies per cell.20
VSV-G qPCR assay
Detection of overt replication competent lentivirus (RCL) was measured by qPCR evaluation of VSV-G copy number as an indicator of virion production (ie, RCL). gDNA was extracted from 2×106 transduced T cells for each product using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s protocol. Standards were made for VSV-G using pCMV-G plasmid at the concentrations 1×105, 1×104, 1×103, 1×102, and 10 copies/µL. To calculate the VSV-G copy number/50 ng gDNA, the average number of copies detected in the 10 sample wells was taken. The release criteria for CMV-CD19CAR T cell products was set to ≤2.5 VSV-G copies per 50 ng gDNA.20
PrimeFlow RNA assay
The PrimeFlow RNA Assay is an in situ hybridization assay that combines the power of branched-DNA technology with the single-cell resolution of flow cytometry, enabling the simultaneous detection of up to four RNA targets in combination with immunophenotyping for cell surface and intracellular proteins using fluorochrome-conjugated antibodies. Genes related to T cell persistence (telomerase Tert, IL-2), memory (KLF2, TCF1, Lef1), homing (CCR7, ITAG5, ITAG3) and activation/exhaustion (PDCD1) were included following instructions from the manufacturer (Thermo Fisher Scientific). The gene/protein expression of stimulated cells that were double positive for surface EGFR and intracellular IFNγ by flow cytometry (10,000 cells were acquired) were analyzed.
Statistical analysis
Mean±SD of replicate samples are presented. We used unpaired T tests for statistical analysis of cytokine production and cytolytic activity assays.
Results
Clinical scale CliniMACS prodigy immunomagnetic selection of CMV-specific T cells after stimulation with cGMP grade CMVpp65 peptide pool
To demonstrate the consistency and clinical feasibility of isolating CMV-specific T cells (figure 1A), we performed nine selection process runs using PBMCs from eight different healthy CMV-seropositive donors. IFNγ+T cells were consistently enriched from pre-enrichment levels of 0.8±0.5% to postcapture levels of 76.3%±11.6% (figure 1B and table 1). On average, we recovered a total of 4.2±3.3×106 IFNγ+T cells from an input of 1×109 PBMCs, which is the maximum loading capacity of the CliniMACS Prodigy according to the manufacturer. The freshly isolated IFNγ+, CMV-specific T cells consisted of polyclonal CD8+ (44.0%±21.0%) and CD4 +T cells (49.8%±21.2%). Phenotypic characterization of the purified CMV-specific IFNγ+T cells showed the presence of several T cell subpopulations including CD45RA+CD27+T stem cell memory (Tscm), CD45RA-CD27+T central memory (Tcm), and CD27-CD45RA+effector memory cells T cells re-expressing CD45RA (TEMRA) (figure 1C).

Clinical scale CliniMACS Prodigy immunomagnetic selection of cytomegalovirus (CMV)-specific T cells. (A) Development of clinically feasible platform for derivation of CMV-CD19CAR T cells. (B) Leukapheresis products were obtained from CMV-immune healthy donors and peripheral blood mononuclear cells (PBMCs) were isolated and purified by density gradient centrifugation over Ficoll-Paque. PBMCs (1×109) were loaded on the closed and automated CliniMACS Prodigy cytokine capture system (CCS) system (Miltenyi Biotec) and CMV-specific T cells were isolated. The percentage of live, purified interferon γ (IFNγ) positive cells (black) over pre-enrichment (red) from three representative donors are depicted. (C) Phenotypic characterization of IFNγ-positive gated cells are presented. Data from three representative donors are presented. APC, allophycocyanin; FITC, fluorescein; PE, phycoerythrin.
Large-scale isolation and enrichment of cytomegalovirus (CMV)-specific T cells
Lentiviral vector transduction and expansion of CMV-CD19CAR T cells
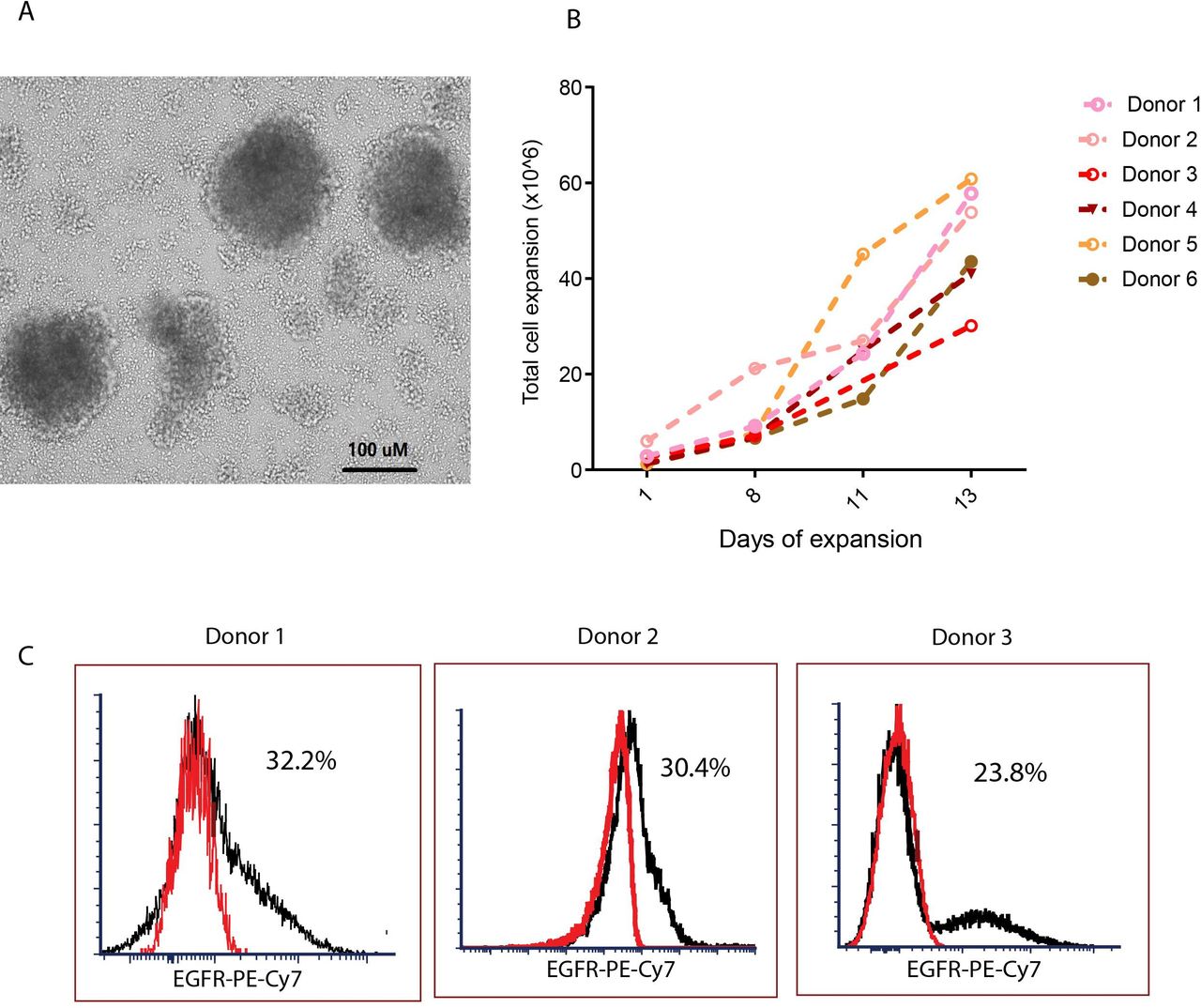
One day after isolation, we transduced CMV-specific T cells with a lentivirus encoding our second generation CD19CAR.14 Because the CMV-specific, IFNγ+T cells were stimulated with the pp65 protein prior to selection and were thus already activated, we transduced these cells without further CD3/CD28 stimulation. The transduced CMV-CD19 CAR T cells formed clusters and grew rapidly within the first 10 days (figure 2A). Cell density was maintained at approximately 0.7×106 cells/mL; growth kinetics of the CMV-CD19CAR T cell products indicated that exponential growth occurred over 7–14 days after transduction. After 14 days expansion, we consistently observed a 10-fold to 40-fold expansion of CMV-CD19CAR T cells (figure 2B), with an average of 27.0%±14.2% CAR +cells (figure 2C and online supplemental table 1).
Supplemental material
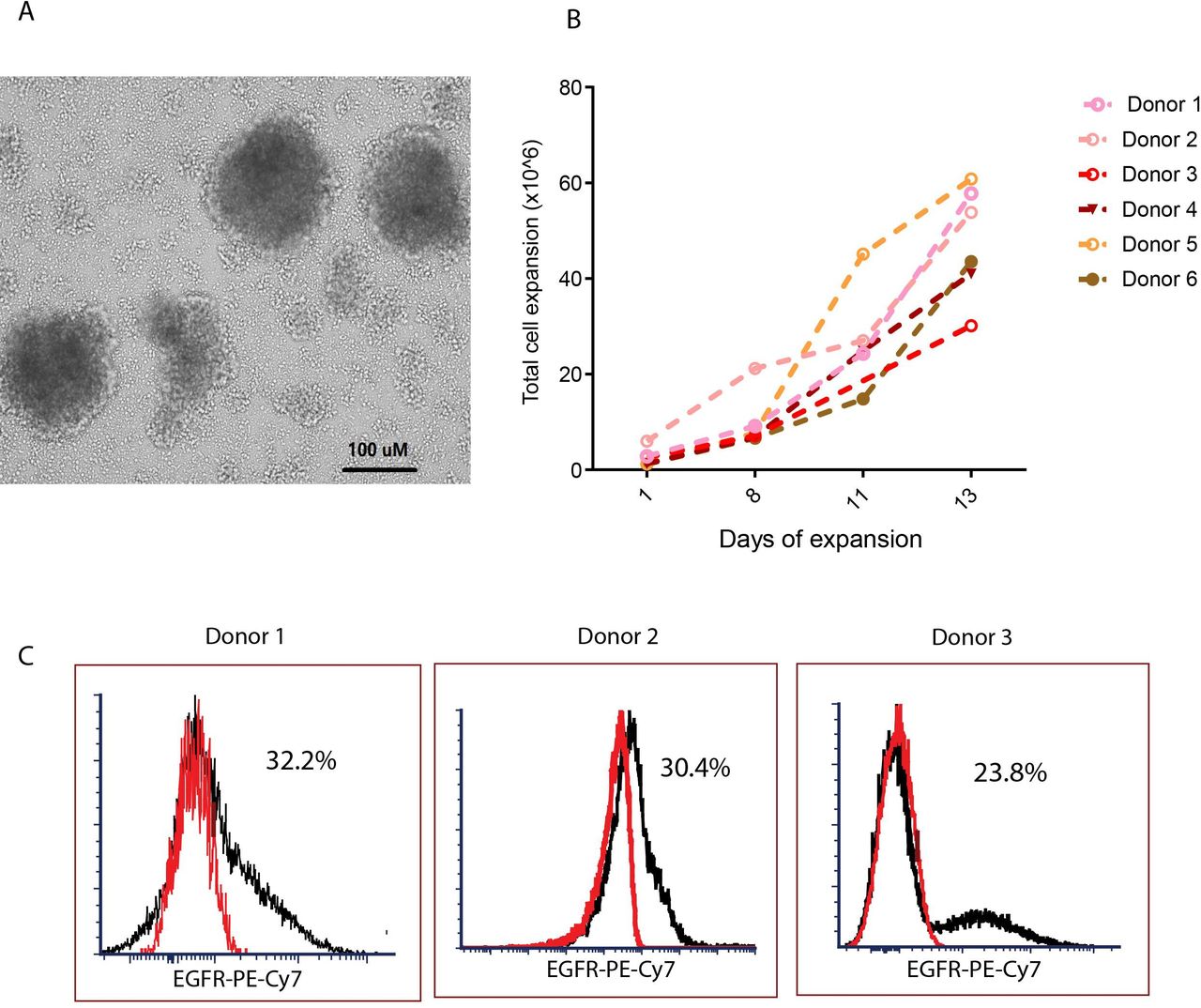
Generation of cytomegalovirus (CMV)-specific CD19 chimeric antigen receptor (CAR) T cells. (A) One day after isolation, CMV-specific T cells were transduced with a second-generation CD19CAR/CD28/EGFRt lentiviral vector with IgG4 Fc hinge region mutations (L235E; N297Q) at MOI 3 in the presence of 50 U/mL IL-2, 1 ng/mL IL-15, and 100 µg/mL protamine sulfate. The transduced CMV-CD19CAR T cells formed clusters as seen under light microscopy in 10 × magnification. (B) Total viable cell count was performed every other day for 14 days to assess in vitro expansion. (C) The proportion of CAR +cells in the products after 14 days of in vitro expansion was analyzed with flow cytometry. EGFR +cells after subtraction of isotype control (red) are presented. Representative data from multiple donors are presented.
Phenotypic characterization of CMV-CD19CAR T cells and vector copy number
We characterized the final CMV-CD19-CAR T cell products by flow cytometry. We also assessed surface expression of the CD19CAR using an anti-EGFR antibody or biotinylated cetuximab followed by streptavidin. The T cell products possess memory features, but did not show features of exhaustion such as high levels of PD1, LAG3 and Tim3 (figure 3A). The propagated CMV-CD19CAR T cells have broad Vβ usage with no skewed Vβ repertoire observed (online supplemental figure 1), supporting that neither CMV-specific TCR isolation nor CD19CAR engineering would lead to clonal expansion.19 21 To investigate if stimulation through the TCR impacted phenotype of CMV-CD19CAR T cells, we conducted phenotypic characterization of CMV-CD19CAR T cells following a 7-day stimulation with pp65pepmix loaded autologous PBMC (CMV stimulator) or LCL (CD19 stimulator). Across multiple donors, we showed that stimulation via TCR had no significant phenotypic impact on CMV-CD19CAR T cells when compared with unstimulated cells (figure 3B).

Phenotypic characterization of cytomegalovirus (CMV)-specific CD19CAR T cells. (A) The final products were stained with antibodies against indicated immune receptors. Per cent positive cells (means±SD) under chimeric antigen receptor (CAR) +population from three different T cell products are presented. (B) CMV-CD19CAR T cells were labeled with CellTrace Violet dye (CTV) and co-cultured with 8000-cGy-irradiated lymphoblastoid cell line (LCL) cells and 3700 cGy-irradiated, autologous CMVpp65-pepmix-pulsed peripheral blood mononuclear cells (PBMCs) at 1:1 ratio for 7 days. Dilution of CTV was analyzed by flow cytometry. Per cent positive cells (means±SD) under CAR +population from three different donor-derived T cell products are depicted. Replicates were conducted for each product. (C) To simultaneously quantify specific mRNA levels and both cell surface and intracellular proteins, CMV-CD19CAR T cells were incubated overnight with either pp65pepmix-loaded autologous PBMC to stimulate the CMV-specific T cell receptor (TCR) (pp65 stim) or LCL cells to stimulate the CD19CAR (CAR stim). Cells were subsequently stained with Erbitux and anti-CD3 antibodies to detect CAR +and CD3+cells. After fixation, cells were permeabilized with PrimeFlow RNA permeabilization buffer in the presence of RNAase inhibitors followed by intracellular interferon γ (IFNγ) staining. Next day, the cells were labeled with RNA probes including genes that are related T cell persistence (telomerase Tert, IL-2), memory (KLF2, TCF7, Lef1), homing (CCR7, ITAG5, ITAG3), and activation/exhaustion (PDCD1). Gene expression on gated EGFR (CAR+) and intracellular IFNγ double positive cells were analyzed with flow cytometry (10,000 cells were acquired for analysis).
To further characterize and compare the consequences of T cell fate following CMV and CD19 stimulation, we stimulated CMV-CD19CAR T cells from a product with 26.3% CAR + as above overnight. Simultaneous quantification of specific mRNA as well as cell surface and intracellular proteins was performed using PrimeFlow technology as a more sensitive approach to understand the influence of TCR stimulation on CMV-CD19CAR T cells. In one representative analysis, CMV-CD19CAR T cells tended to have higher expression (>2 folds) of genes associated with persistence, memory (KLF2, TCF7, Lef1) and homing (ITAG5, ITAG3) following TCR stimulation with pp65 compared with CAR stimulation with CD19 +tumor (figure 3C). To ensure that the CMV-CD19CAR T cells could pass FDA-requested release criteria for lentivirus transduced CAR T cells, we evaluated the lentivirus vector integration copy number and VSV-G lentiviral envelope genes by qPCR specific for the vector’s WPRE sequence and lentiviral envelope pCMV-G, respectively. The analysis of three CMV-CD19CAR T cell products passed the release criteria as ≤2.5 copies/50 ng gDNA VSV-G and ≤5 copies/cell WPRE (online supplemental figure 2).
Dual functionality of CMV-CD19CAR T cells against CD19 and CMVpp65 antigens
To demonstrate the bifunctionality of CMV-CD19CAR T cells, we incubated the propagated CMV-CD19CAR T cells with either CD19 +tumor cells (LCL) to assess signaling through the CD19CAR or pp65-pulsed PBMCs to assess signaling through the CMV-specific TCR. We used OKT3-LCL cells as a positive control. The CMV-CD19CAR T cells consistently secreted IFNγ on stimulation through both the endogenous CMV-specific TCR and the CD19CAR (figure 4A), supporting their capability to respond to CMV vaccine and CD19-positive tumor.

Dual functionality of cytomegalovirus (CMV)--specific CD19CAR T cells against CD19 and CMV pp65 antigens. (A) Propagated CMV-CD19CAR T cells were stimulated overnight with CD19 +tumor (lymphoblastoid cell line (LCL)) and CMVpp65pepmix-pulsed PBMC. OKT3 LCL and medium were used as positive and negative controls, respectively. The frequency of intracellular interferon γ (IFNγ) in CD3 +T cells from three representative CMV-CD19CAR T cell products are depicted. (B) The composition of T cell subsets in the final CMV-CD19CAR T cell products within the CD3 +population and (C) the relative proportion of IFNγ+cells within the chimeric antigen receptor (CAR) + populations from three different donors are presented.
Although we enriched CMV-specific T cells with high purity prior to lentiviral transduction as indicated by IFNγ-positivity (figure 1 and table 1), the T cell populations in the final CMV-CD19CAR T cell product may still be heterogeneous and may include CMV-specific T cells, CD19CAR +T cells, T cells that respond to both CMVpp65 and CD19, and a subset of T cells that are neither CMV-specific nor CD19CAR+. We therefore characterized the relative proportions of true CMV-CD19CAR T cells in the final products. To that end, we stimulated large-scale manufactured CMV-CD19CAR T cells overnight with pp65pepmix-loaded autologous PBMCs and analyzed IFNγ-positivity by intracellular flow cytometry staining. The functional CMV-CD19CAR T cell subset was defined as cells that were EGFR and IFNγ double positive on CMVpp65 stimulation. The final products contained CD4 +and CD8+CAR T cells, the proportion of which varied from donor to donor (figure 4B). We observed that 50%–60% (6.2±0.9×106 cells; online supplemental figure 3) of the CAR +T cells secreted IFNγ+ on stimulation with CMVpp65 (figure 4C), representing the CAR population that can be expanded by CMV vaccine.
Cytolytic potency and effector cytokine secretion by CMV-CD19CAR T cells
To test the functionality of the CD19CAR portion of the CMV-CD19CAR T cells, we analyzed the direct cytolytic activity against CD19 + target cells (LCL). Although CMV-CD19CAR T cells did not kill CD19- KG1a cells, we observed consistent, specific killing of CD19 + tumor cells in a dose-dependent manner, even at the very low effector:target (E:T) ratio of 0.06:1 (figure 5A).
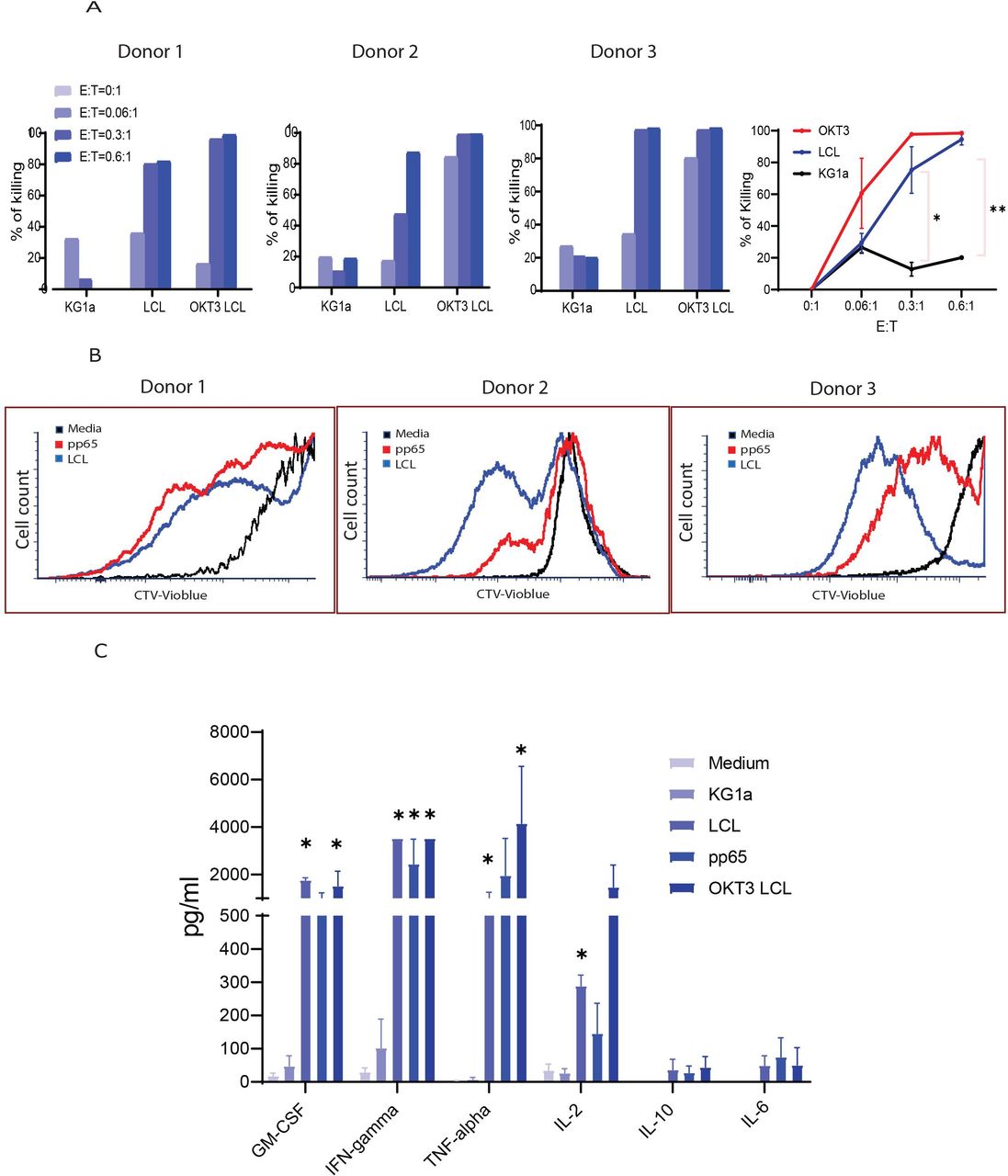
Cytolytic potency and effector cytokine secretion by cytomegalovirus (CMV)-specific CD19CAR T cells. (A) CMV-CD19CAR T cells were co-cultured with GFP+ target cells (lymphoblastoid cell line (LCL)) at different effector:target ratios for 3 days and remaining eGFP+ tumor cells were analyzed by flow cytometry. KG1a (CD19-) and OKT3 LCL were used as negative and positive controls, respectively. Cytotoxicity was normalized to the tumor only cultures. Data from three different donor-derived CMV-CD19CAR T cells are presented. (B) To test the proliferative capacity against CD19+ (LCL) and CMVpp65 targets, CMV-CD19CAR T cells were labeled with CellTrace Violet dye (CTV) and co-cultured with 8000-cGy-irradiated LCL cells and 3700 cGy-irradiated autologous CMVpp65-pepmix pulsed peripheral blood mononuclear cells (PBMCs) at 1:1 ratio for 7 days. Dilution of CTV was analyzed by flow cytometry. Data from two representative CMV-CD19CAR T cell products are depicted. (C) CMV-CD19CAR T cells (1×105) were cocultured overnight in 96-well tissue culture plates with 1×105 LCL, OKT3 LCL, KG1a, or autologous CMVpp65-pepmix pulsed PBMCs. Supernatants were collected and analyzed with the Luminex IS100 bead array technology for Human Cytokine 30-Plex according to the manufacturer’s instructions. Mean±SD from three different donors are presented. #: out of range above. ***p<0.001. **p<0.01, *p<0.05.
Our ultimate goal is to expand the CMV-CD19CAR T cells in patients with our CMV vaccine. To validate the capability of the CMV-CD19CAR T cells to proliferate following stimulation through the CMV-specific TCR, we performed a proliferation assay by labeling the CMV-CD19CAR T cells with CTV prior to stimulating with either CD19+LCL or CMVpp65-pepmix-pulsed PBMCs. Across three donors, we observed vigorous proliferation of CMV-CD19CAR T cells against both antigens as indicated by dilution of CTV dye following LCL or CMVpp65-pepmix stimulation, but minimal proliferation of unstimulated cells (figure 5B). To examine the broader cytokine profile of CMV-CD19CAR T cells against CD19 and CMVpp65 antigens, we compared levels of cytokines released by unstimulated cells or following target stimulation using a 30-plex bead array cytokine detection assay. Compared with unstimulated cells, CMV-CD19CAR T cells secreted significantly higher levels of Th1 cytokines, including tumor necrosis factor α (TNFα) and IFNγ, following CD19+ LCL or CMVpp65 target stimulation, which was comparable to that following OKT3 LCL stimulation (figure 5C). Increased granulocyte-macrophage colony-stimulating factor (GM-CSF), TNFα, and IFNγ following target stimulation suggested that the T cell products are highly Th1 polarized. Interestingly, autocrine IL-2, the major determinant of T cell memory and persistence,22 was greatly elevated in the CMV-CD19CAR T cells on both CD19 and CMVpp65 antigen engagement. However, production of the T cell immunosuppressive Th2 cytokine IL-10 and the inflammatory cytokine IL-6 that is related to the development of cytokine release syndrome (CRS) in the clinic were not elevated after stimulation (figure 5C).
CMV-CD19CAR T cells possess stronger effector function compared with CD19CAR T cells
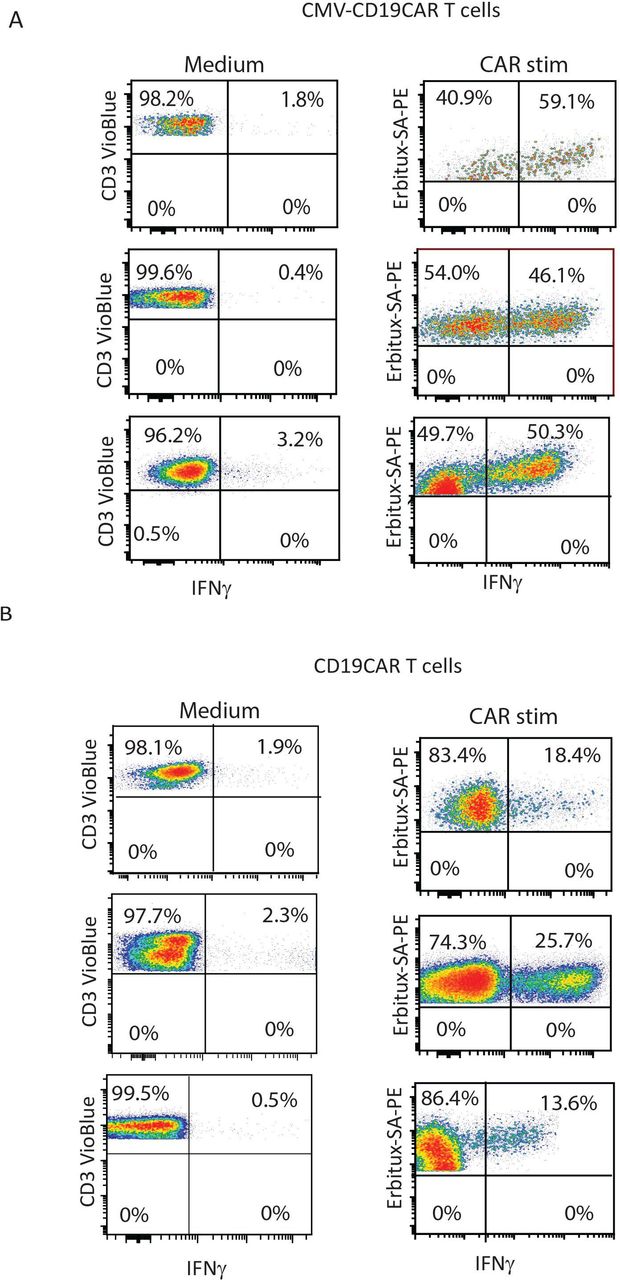
To directly compare the effector function of CMV-CD19CAR T cells and conventional monospecific CD19CAR T cells against CD19, we generated both versions of CAR T cells from the same donor. Interestingly, after the same period of ex vivo expansion (14 days), CMV-CD19CAR T cells secreted more IFNγ in response to stimulation with CD19+LCL target cells than conventional CD19CAR T cells. This suggests that CMV-CD19CAR T cells may be more potent than CD19CAR T cells (figure 6A,B), regardless of expansion via CMV stimulation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytomegalovirus (CMV)-specific CD19CAR T cells possess stronger effector function against CD19 +tumor compared with monospecific CD19CAR T cells. (A) CMV-CD19CAR T cells were also generated from the same donor and were expanded for 14 days. Both CD19CAR T cells and CMV-CD19CAR T cells were then stimulated with CD19+target cells (lymphoblastoid cell line (LCL)) overnight and intracellular interferon γ (IFNγ) was analyzed by flow cytometry. Data from three different donor-derived CD19CAR and CMV-CD19CAR T cells are presented. CMV-CD19 chimeric antigen receptor (CAR) T cells without stimulation were used as controls. (B) To generate conventional monospecific CD19CAR T cells, peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3/CD28 and transduced with the same CD19CAR lentivirus and expanded for 14 days. CD19CAR T cells were then stimulated with CD19+target cells (LCL) overnight and intracellular IFNγ was analyzed by flow cytometry. Data from three different donor-derived CD19CAR T cells are presented. CD19CAR T cells without stimulation were used as controls.
Discussion
CMV is a common virus for which 75% of adults in the USA test positive23 24 and is the first virus targeted by adoptive transfer strategies. Pioneering immunotherapy trials1–3 show that adoptive transfer of CMV-specific T cells is sufficient to reduce the incidence of CMV disease without toxicity, including graft-versus-host disease. We choose CMV-specific T cells for engineering with a CD19CAR to endow the potent and persistent virus-specific T cells with a second specificity for CD19 tumor antigen. This gives the CMV-CD19CAR T cells the potential to persist and expand in vivo via stimulation by CD19 + tumor cells and the endogenous TCR by viral antigens (from CMV virus or vaccine). Initially, the major obstacle to the clinical application of CMV-specific T cells was the lengthy process required for their selection and generation.1 25–28 Therefore, to enable cellular therapy on demand, a simple short-term ex vivo protocol was developed in which CMV-reactive CD4 + and CD8+ T cells were isolated based on CMVpp65-specific IFNγ secretion prior to CAR engineering using CliniMACS Prodigy CCS system. Here, we described a closed large-scale manufacturing process for reproducibly enriching CMV-specific T cells with uniform purity within a day (table 1), which allowed us to produce CMV-CD19CAR T cells in a cGMP manner within a therapeutically relevant time frame (figure 1).
The purified CMV-specific T cells consisted of different memory subsets (figure 1C) and had phenotypic characteristics suggesting that the enriched T cells are capable of responding to antigen stimulation and mediating persistent antiviral activity.29 30 Moreover, CMV-specific T cells could be engineered to express a CD19CAR (figure 2). In our planned clinical trial assessing the therapeutic potential of CMV-CD19CAR T cells, we plan to assess a flat dose of 10×106 CAR + T cells, since we will rely on in vivo expansion after CMV-CD19CAR T cell infusion via CMV vaccination. This dose level was also used in other virus-specific CAR T cell trials (eg, NCT01953900) and dose-escalated once safety was established. Our studies here demonstrated that we were able to generate sufficient CMV-CD19CAR T cells within a 14-day ex vivo expansion without further stimulation (figure 2). The shortened ex vivo expansion of CMV-CD19CAR T cells could prevent differentiation and promote effector functions after infusion. The CMV-CD19CAR T cell products generated from our qualification runs expressed the CD19 CAR (figure 2) and displayed CD19-mediated cytolytic function and cytokine production against CD19 +tumors (figures 4–5). Importantly, the CMV-CD19CAR T cells responded to CMVpp65 antigen with rapid proliferation and IFNγ secretion figures 4–6(figures 4-5). Although the final products were heterogeneous and contained CAR+, CMV+, CAR and CMV double positive, and CAR and CMV double negative subsets, the majority of the final products were able to secrete IFNγ on engagement with CMVpp65 antigen (figure 4), which represents the key population associated with CMV vaccine-boost expansion.
Broad TCR Vβ usage was observed in CMV-CD19CAR T cell products and CD19CAR T cell product from the same donor (online supplemental figure 1), suggesting that CMV-specific T cell selection, transduction and expansion did not result in a T cell pool with a skewed TCR repertoire.19 21 The CMV-CD19CAR T cells continued to express CD62L, CD27 and CD28 but not the exhaustion markers (figures 1C and 3A), characteristics that are associated with engraftment and persistence following adoptive transfer. Interestingly, compared with the same donor-derived monospecific CD19CAR T cells, CMV-CD19CAR T cells exhibited superior anti-CD19 effector function (figure 6). This observation may be due to the use of CMVpp65 antigen stimulation prior to transduction rather than CD3/CD28 supraphysiological activation that is required for the generation of conventional CAR T cells but drives T cell differentiation and exhaustion as well as activation-induced cell death of CMV-specific T cells.31 It is possible that the intrinsic difference between CMV-specific T cells and bulk T cells or the potential bias of using cells selected for their ability to secrete IFNγ to generate CMV-CD19CAR T cells may also have played a role in the difference in effector functions. To understand the potential characteristics of CMV-CD19CAR T cells following in vivo vaccination, we examined the expression of genes associated with persistence, memory and homing after stimulation through either the CMV-specific TCR or CD19CAR (figure 3BC). Interestingly, CMV-CD19CAR T cells had superior features of persistence, memory and homing following CMV stimulation compared with CD19 stimulation (figure 3C), suggesting that CMV-CD19CAR T cells may maintain memory function and possess superior migration capacity to tumors following in vivo CMV vaccine stimulation. However, in these experiments, CAR +IFNγ+cells after CD19 stimulation could include CAR+ and CMV-CD19CAR T cells, whereas CAR+IFNγ+cells after CMV stimulation are pure CMV-CD19CAR T cells, which is a limitation of this analysis. This observation could account for why virus-specific T cells persist better than tumor-specific CAR T cells that rely on synthetic receptor signaling rather than endogenous TCR signaling. These data may provide insight into avenues to improve CAR T cell therapy; CAR T cell expansion through the endogenous TCR may better maintain overall CAR T cell function, especially via enhanced expression of homing molecules. We therefore hypothesize that this manufacturing platform can be applied to CAR T cell therapy targeting multiple diseases and may also have potential to overcome problems associated with insufficient CAR T cell homing to solid tumors.32–35
The most frequent and severe complications after CD19CAR T cell therapy include CRS, immune effector cell-associated neurotoxicity syndrome, macrophage activation syndrome/hemophagocytic lymphohistiocytosis and tumor lysis syndrome,36–39 which occur early (day 0 to +30) post-CAR T cell infusion. These early toxicities may be followed by prolonged B cell aplasia (between days +30 and +100 postinfusion), which significantly contributes to infectious complications, including CMV infection.40 41 CMV-CD19 CAR T cells may therefore provide sufficient anti-CMV activity following CAR T cell therapy to prevent CMV infection without the need for antiviral drugs. In addition, the Triplex vaccine can specifically expand endogenous CMV-specific T cells, including pp65-specific, IE1-specific and IE2-specific populations, which would effectively prevent CMV reactivation postinfusion.
In summary, we have redirected CMV-specific T cells to recognize and lyse CD19+ tumor cells via a CD19CAR, while maintaining their ability to proliferate in response to CMV antigen stimulation. These results illustrate the potential for clinical application of CMV vaccination to augment the antitumor activity of adoptively transferred CMV-CD19CAR T cells in patients with B cell malignancies, as well as other CARs specific for different tumors. With our data from the investigational new drug (IND)-enabling studies, we plan to initiate our first clinical trial in Fall 2021 for patients with intermediate/high-grade B cell non-Hodgkin lymphoma (NHL) postautologous HCT followed by two CMV-MVA Triplex vaccinations 28 and 56 days after autologous CMV-CD19CAR T cell infusion for in vivo expansion of the T cells. The primary objective of this trial is to examine safety and persistence/expansion of CMV-CD19CAR T cells before and after CMV-MVA Triplex vaccine boost. This study serves as proof of principle for a method of enhancing effectiveness of CAR T cell therapy and can be applied to multiple diseases in both transplant and non-transplant settings, as well as in allogeneic settings using highly matched donors.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All human tissues and blood samples were obtained under protocols approved by the institutional review board (University Health Network Research Ethics Board). Surgical specimens were obtained from the UHN Biospecimen Program. Written informed consent was obtained from all donors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors XW, RN, and SJF contributed to study concept and design and data interpretation. MW, MG, TH, VV, S-HC, BG and RU performed experiments and generated figures. XW and RN wrote the manuscript. AC, DJD, and JZ, MCC, and SM reviewed and edited the manuscript. All authors contributed to critical revision of the manuscript for intellectual content. XW, as guarantor, accepts full responsibility for the work and the conduct of the study, had access to the data, and control the decision to publish.
Funding Lymphoma SPORE (P50CA107399), LLS (TRP 6611-20).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.