Article Text

Abstract
Background Immune checkpoint blockade (ICB) is a clinically proven concept to treat cancer. Still, a majority of patients with cancer including those with poorly immune infiltrated ‘cold’ tumors are resistant to currently available ICB therapies. Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is one of few clinically validated targets for ICB, but toxicities linked to efficacy in approved αCTLA-4 regimens have restricted their use and precluded full therapeutic dosing. At a mechanistic level, accumulating preclinical and clinical data indicate dual mechanisms for αCTLA-4; ICB and regulatory T cell (Treg) depletion are both thought to contribute efficacy and toxicity in available, systemic, αCTLA-4 regimens. Accordingly, strategies to deliver highly effective, yet safe αCTLA-4 therapies have been lacking. Here we assess and identify spatially restricted exposure to a novel strongly Treg-depleting, checkpoint-blocking, vectorized αCTLA-4, as a highly efficacious and potentially safe strategy to target CTLA-4.
Methods A novel human IgG1 CTLA-4 antibody (4-E03) was identified using function-first screening for monoclonal antibodies (mAbs) and targets associated with superior Treg-depleting activity. A tumor-selective oncolytic vaccinia vector was then engineered to encode this novel, strongly Treg-depleting, checkpoint-blocking, αCTLA-4 antibody or a matching surrogate antibody, and Granulocyte-macrophage colony-stimulating factor (GM-CSF) (VVGM-αCTLA-4).
Results The identified 4-E03 antibody showed significantly stronger Treg depletion, but equipotent checkpoint blockade, compared with clinically validated αCTLA-4 ipilimumab against CTLA-4-expressing Treg cells in a humanized mouse model in vivo. Intratumoral administration of VVGM-αCTLA-4 achieved tumor-restricted CTLA-4 receptor saturation and Treg depletion, which elicited antigen cross-presentation and stronger systemic expansion of tumor-specific CD8+ T cells and antitumor immunity compared with systemic αCTLA-4 antibody therapy. Efficacy correlated with FcγR-mediated intratumoral Treg depletion. Remarkably, in a clinically relevant mouse model resistant to systemic ICB, intratumoral VVGM-αCTLA-4 synergized with αPD-1 to reject cold tumors.
Conclusion Our findings demonstrate in vivo proof of concept for spatial restriction of Treg depletion-optimized immune checkpoint blocking, vectorized αCTLA-4 as a highly effective and safe strategy to target CTLA-4. A clinical trial evaluating intratumoral VVGM-αhCTLA-4 (BT-001) alone and in combination with αPD-1 in metastatic or advanced solid tumors has commenced.
- CTLA-4 antigen
- oncolytic virotherapy
- antibody specificity
- immunotherapy
Data availability statement
The datasets generated during and/or analyzed during this current study have been deposited or are available from the corresponding author on reasonable request. The assigned accession number for RNAseq data reported in this paper is GEO: GSE176052.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Treatment with immune checkpoint blocking antibodies has transformed survival of patients with advanced solid cancers including metastatic melanoma, non-small cell lung cancer and mismatch repair-deficient cancers.1–3 Still, a great unmet need remains since many patients fail to respond or acquire resistance to immune checkpoint blockade (ICB).4 Reasons for lack of efficacy are believed to include lack of, or inadequate, tumor-infiltrating lymphocytes (TILs), most notably CD8+ T cells.5 6 Paucity of chemotactic and inflammatory signals in the solid cancer tumor microenvironment (TME) is similarly thought to underlie resistance to chimeric antigen receptor (CAR) T cell therapy.7
Identification of therapeutics that induce recruitment of inflammatory immune cells into ‘immune desert’ or ‘immune-excluded’ tumors, translating into robust systemic adaptive antitumor immunity and CD8+ T cell infiltration with regression of primary and metastasized tumors, is therefore highly desired.
Intratumoral oncolytic virotherapy induces T cell infiltration and improves αPD-1 immunotherapy.8 Combination therapy with αCTLA-4 and αPD-1 antibodies enhances efficacy compared with single-agent ICB, likely through complementary mechanisms of systemic CD4+ and CD8+ T cell differentiation and tumor-localized modulation of T effector and regulatory T cells.9 10 However, tolerability issues with systemically administered αCTLA-4, including with the approved ipilimumab, have restricted clinical use.11
Efficacy and tolerability of systemic αCTLA-4 antibody therapy appear to be linked. Increasing ipilimumab dose enhanced both efficacy and side effects.12 Consistent with the central immune checkpoint function of CTLA-4, side effects may be severe and of systemic autoimmune nature.13 Interestingly, depletion of intratumoral Treg cells, which overexpress CTLA-4 relative to CD8+ and CD4+ effector T cells, was recently reported to contribute to ipilimumab therapeutic activity. Treg depletion-enhanced αCTLA-4 antibody variants showed improved therapeutic activity in tumor-bearing FcγR-humanized mice.10 These findings indicate that tumor-localized therapy with Treg-depleting αCTLA-4 antibodies may provide powerful therapeutic activity with reduced side effects compared with currently available αCTLA-4 therapies14 15 — in particular when combined with validated and safe immunomodulators, for example, blockers of the PD-1/PD-L1 axis or oncolytic viruses (OVs).
Here, we describe and preclinically characterize one such approach. A vaccinia virus (VV)-based oncolytic vector was designed to incorporate both GM-CSF and a novel full-length human recombinant αCTLA-4 antibody selected and characterized for its FcγR-dependent Treg-depleting efficacy (BT-001, VVGM-αhCTLA-4). Viruses encoding a matching Treg-depleting mouse surrogate antibody were additionally generated, enabling proof-of-concept studies in syngeneic immune competent mouse tumor models representing inflamed or immune-excluded TMEs sensitive or resistant to ICB.
Materials and methods
Cell lines
HEK293T, B16-F10, CT26, A20, EMT6, LL/2, LoVo, MIA PaCa-2, Hs-746 T, SK-OV-3, HCT 116, TF-1, and the NK-92 cell line were purchased from the American Type Culture Collection. Cells stably transfected with human CTLA-4 (293T-CTLA-4) were obtained from Crown Bio. The MC38 cell line was a gift from Mark Cragg.
Mice
Mice were maintained in local pathogen-free facilities. For all experiments, young adult mice were sex-matched and age-matched and were randomly assigned to experimental groups. C57BL/6 and BALB/c mice were obtained from Taconic, Janvier, or Charles River. Genetically altered strains used were C.129P2(B6)-Fcer1gtm1Rav (Fcer1g-KO on BALB/c background and BALB/cAnNTac wild type (WT) controls) purchased from Taconic; and B6.129S(C)-Batf3tm1Kmm/J16 ; Batf3-KO on C57BL/6J background and C57BL/6 J WT controls) purchased from Jackson Laboratories.
Human (clinical) samples and ethics
Patient samples were obtained through the Department of Obstetrics and Gynecology and the Department of Oncology at Skånes University Hospital, Lund, Sweden. Ascitic fluid was assessed as single cell suspension that had been isolated. Processing of human tissue is described in online supplemental material.
Supplemental material
Data availability
The assigned accession number for RNAseq data reported in this paper is GEO: GSE176052.
Statistical analysis
All statistical analyses were carried out using GraphPad Prism V.9.0 (GraphPad Software, La Jolla, California, USA). P values were calculated using Student’s t-tests or one-way analysis of variance. The survival periods to the humane end point were plotted using the Kaplan-Meier method with analysis for significance by the log-rank test. Significance was accepted when the p value was <0.05.
Supplementary information contains detailed method descriptions for antibody and viral vector generation, and in vitro, ex vivo, and in vivo characterization.
Results
Identification and characterization of Treg-depleting αCTLA-4 antibodies
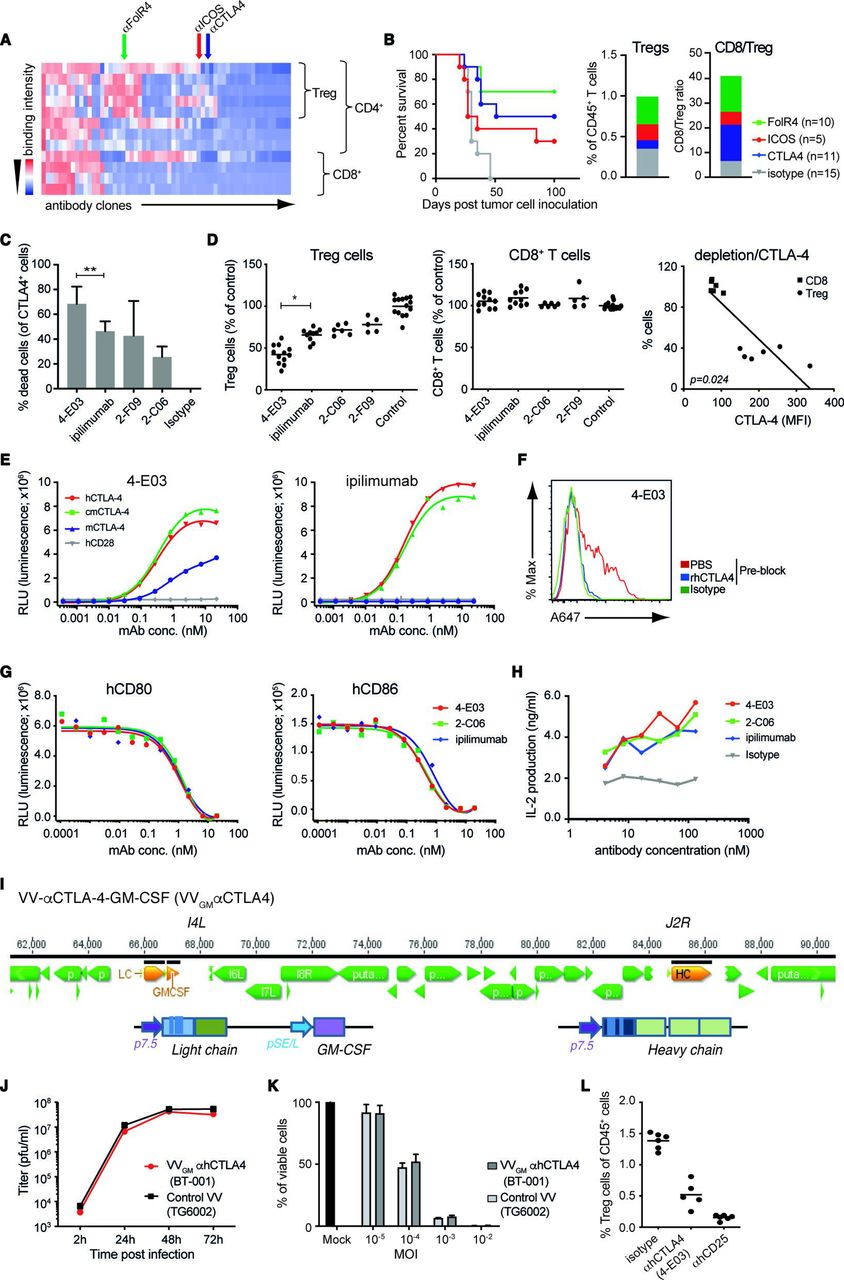
ICB and αCTLA-4 antibody therapy are clinically validated approaches, yet mechanisms underlying αCTLA-4 antibody efficacy are incompletely characterized. Accumulating data suggest that αCTLA-4 antibodies, besides acting to lower the threshold for T cells to recognize tumor antigen and reject tumors, may exert therapeutic activity through depletion of intratumoral Treg cells following antibody interactions with FcγR-expressing effector cells.17–19 We used the target agnostic F.I.R.S.T discovery platform to screen a large (>1010 members) human antibody library (n-CoDeR) for scFv antibodies that bound to target receptors upregulated on Treg, compared with non-Treg CD4 and CD8 effector TILs.20 Following conversion to full-length IgG format, antibodies were produced, purified, and evaluated for in vivo Treg depletion and antitumor efficacy. The targets of the most functional antibody clones were identified using complementary deconvolution approaches. An array of antibodies and their associated targets capable of depleting Treg cells and improving survival in the T cell-inflamed CT26 mouse tumor model were identified (figure 1A,B). CTLA-4 and αCTLA-4 antibodies were identified among these, and αCTLA-4 mIgG2a mAb depleted intratumoral Treg cells and conferred survival on treatment of animals bearing syngeneic CT26 tumors (figure 1B). Focused screening of human CTLA-4-specific IgG1 antibodies identified several clones with similar in vitro depleting activity of CTLA-4-expressing human T cells (figure 1C). One clone (4-E03) stood out based on its consistently stronger CTLA-4+ T cell-depleting efficacy compared with other clones when screened across multiple donors (figure 1C). To investigate the in vivo relevance of this clone’s apparent stronger depleting activity, we turned to a model where human peripheral blood mononuclear cells (PBMCs) are engrafted into NOD SCID interleukin (IL)-2R gamma−/− (NSG) mice. Owing to a graft-versus host type of interaction human Treg and CD8+ T cells are strongly activated, and show similar co-stimulatory and co-inhibitory molecule expression compared with that observed in human tumors21 (online supplemental figure 1A). Analyses of NOG-hPBMC CD4+CD25+CD127low Treg and CD8+ T cells indeed revealed similar CTLA-4 expression levels to that observed on T cells obtained from nine patients with ovarian cancer (online supplemental figure 1A). Furthermore, dosing of NOG-hPBMC mice with 10 mg/kg ipilimumab or additional αCTLA-4 antibody clones (2-C06 and 2-F09) demonstrated similar in vivo depleting activity of human CD4+CD25+CD127low Treg cells (figure 1D). However, 4-E03 evoked significantly greater depletion of human Treg cells. Importantly, and consistent with an observed lower expression of CTLA-4 on intratumoral and NOG-hPBMC CD8+ T cells compared with Treg cells (online supplemental figure 1A), and a strong correlation between CTLA-4 expression and antibody-mediated depletion, 4-E03 showed no depletion of human activated CD8+ T cells (figure 1D). Biochemical characterization, in particular HPLC-SEC analyses of 4-E03 antibody preparations, showed >95% monomeric IgG (data not shown), ruling out that 4-E03-enhanced Treg depletion resulted from aggregation of antibody. Consistent with our observation and others’ observations, αCTLA-4 mAb Treg depletion was shown to depend on antibody Fc–FcγR interactions. An FcγR binding-impaired variant of 4-E03 (IgG1N297Q) showed severely impaired Treg depletion compared with WT FcγR-proficient IgG1 (online supplemental figure 1B).
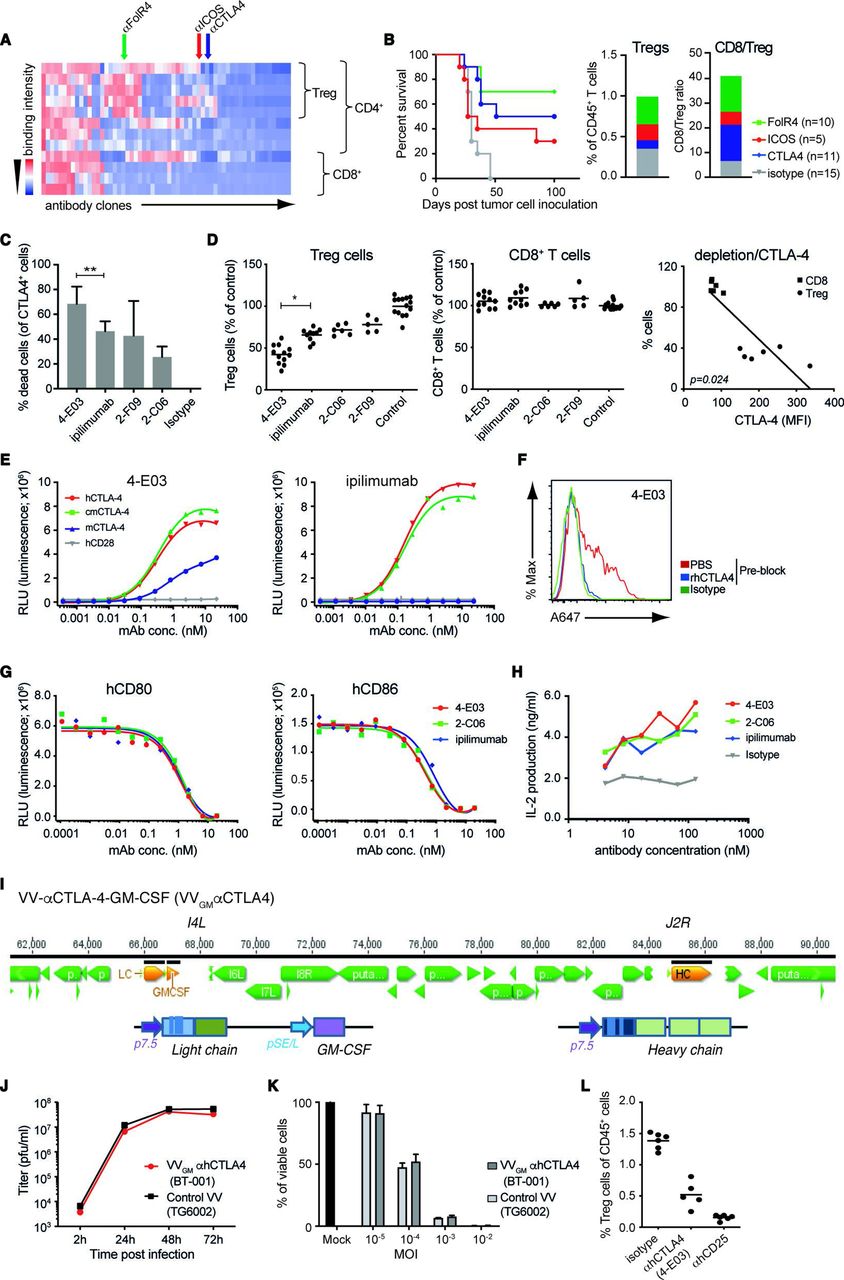
Generation and characterization of novel Treg-depleting αCTLA-4 mAbs and oncolytic VVs expressing Treg-depleting αCTLA-4 and GM-CSF. (A) Heatmap shows function-first isolated antibody clones (vertical lines) binding to T cells from CT26 tumor-bearing and naïve BALB/c mice. (B) Antibody-mediated survival (left panel) and TIL modulation (right panel) in CT26 tumor-bearing BALB/c mice. Animals with established tumors received four injections (10 mg/kg) of antibodies with indicated Treg-associated specificity or control mIgG2a antibody (n=5–15). (C) CTLA-4-specific mAbs induce ADCC of in vitro-activated CD4+ T cell. Lysed target T cells were identified by FACS. Figure shows mean±SD (n=4–8); **p<0.01 by Student’s t-test. (D) Anti-CTLA-4 (IgG1) mAbs mediate Treg depletion in vivo in PBMC-humanized mice. Clone 4-E03 shows enhanced depletion of human Treg cells (left panel) but not CD8+ T cells (right panel) compared with ipilimumab. Each dot represents one mouse. Graph shows mean data from two experiments. *p<0.05 by one-way analysis of variance. Right: Level of 4-E03-induced cell depletion plotted in relation to CTLA-4 expression as determined by flow cytometry. (E) 4-E03 hIgG1 and ipilimumab binding to human, mouse, and cynomolgus CTLA-4 and CD28 by ELISA. (F) 4-E03 IgG1 binding to in vitro-activated CTLA-4-expressing human T cells was preblocked with rhCTLA-4-Fc protein (blue line) (G) 4-E03 and 2-C06 block CD80 and CD86 binding to CTLA-4 by ELISA. (H) Functional ligand blockade in vitro. Graphs show interleukin-2 in supernatants following treatment of in vitro activated human PBMCs with αCTLA-4. A representative donor is shown (n=6). (I) Schematic illustration of the VV vectors used to encode heavy (at J2R locus) and light chains of the αCTLA-4 antibody and GM-CSF (at the I4L locus). (J) Replication kinetics in LoVo cells and (K) oncolytic activity on MIA PaCa-2 cells of VVGM-αhCTLA-4 (BT-001). TG6002 (recombinant J2R and I4L deleted VV) was added as control. (L) Functional assessment of αCTLA-4 mAb 4-E03 produced by BT-001-infected MIA PaCa-2 cells in vivo (Treg depletion) as in figure 1D. ADCC, antibody-dependent cell cytotoxicity; FACS, fluorescent-activated cell sorting; ICOS, inducible costimulatory molecule; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cells; PBS, phosphate-buffered saline; RLU, relative light unit; TIL, tumor-infiltrating lymphocyte; VV, vaccinia virus.
These findings indicated that 4-E03 binds to a functionally distinct epitope on CTLA-4. We therefore characterized the binding and ligand-blocking activity of 4-E03 relative to ipilimumab and other αCTLA-4 antibodies. All antibodies showed high specificity for the extracellular domain of human CTLA-4 and no observable binding to its closely related human homologue CD28 by ELISA (figure 1E). While 4-E03 and ipilimumab bound with similar potency and efficacy to hCTLA-4 (figure 1E and online supplemental figure 1C), only 4-E03 showed weak but clear cross-reactivity with mouse CTLA-4 (figure 1E). These findings are consistent with the two antibodies binding to distinct epitopes. Further comparative analyses of 4-E03 and ipilimumab assessing binding to endogenously expressed CTLA-4 on in vitro-activated human CD4+ T cells (online supplemental figure 1D and figure 1F), blockade of B7–CTLA-4 interaction (figure 1G), or inhibition of B7–CTLA-4-mediated Tcell suppression (figure 1H), revealed otherwise near identical efficacy and potency. In conclusion, our results indicated that 4-E03 binds to a functionally distinct CTLA-4 epitope, which is associated with stronger Treg depletion but equivalent blockade of B7–CTLA-4 induced T effector cell suppression to the epitope targeted by ipilimumab.
Since 4-E03 cross-reacted only weakly with mouse CTLA-4 (figure 1E), we next focused our screening to identify an appropriate surrogate for in vivo proof-of-concept studies in immunocompetent mouse tumor models. One clone (5-B07), which showed highly specific binding to mouse CTLA-4 transfected CHO cells (online supplemental figure 2A) and mouse CTLA-4 protein (online supplemental figure 2B), blockade of B7–CTLA-4 interactions (online supplemental figure 2C) and similarly strong depletion of intratumoral mouse Treg (online supplemental figure 2D) compared with 4-E03 in the human setting, was identified. Further, αmCTLA-4 (5-B07) conferred antitumor activity and improved survival of CT26 tumor-bearing BALB/c mice (online supplemental figure 2E). As observed with αhCTLA-4 (4-E03) on human cells, αmCTLA-4 (5-B07) depletion of mouse Treg was found to depend on Fc–FcγR interactions. Fc–FcγR binding-proficient but not Fc–FcγR binding-impaired variants of 5-B07 depleted intratumoral Tregs (online supplemental figure 2F).
The similarly pronounced Treg-depleting activity, alongside their similarly high specificity for CTLA-4 and blocking activity of CTLA-4–B7 family interactions, indicated the therapeutic potential of αhCTLA-4 (4-E03) and of αmCTLA-4 (5-B07) as a suitable mode of action (MoA)-matched surrogate.
Engineering of antibody-encoding OVs for tumor-selective CTLA-4 blockade and Treg depletion
The frequent side effects of αCTLA-4 antibody therapy are consistent with the well-established role of CTLA-4 acting as a central checkpoint to maintain T cell homeostasis and tolerance to self.13 22 Recent work by Quezada and colleagues, however, has indicated that intratumoral Treg depletion may significantly contribute to ipilimumab clinical activity,10 and intratumorally delivered Treg-depleting antibodies may afford substantial antitumor activity in mouse tumor models.15 23 This dual activity of αCTLA-4, acting in central and peripheral compartments, respectively, suggests that localizing αCTLA-4 therapy to tumors may be an attractive strategy to uncouple αCTLA-4 efficacy from toxicity.
We hypothesized that intratumorally delivered OVs engineered to express Treg-depleting αCTLA-4 would represent a particularly attractive means to achieve effective, yet safe, tumor-localized αCTLA-4 therapy. Besides enabling local antibody production and blockade of CTLA-4 receptors and Treg depletion in the TME on infection of tumor cells, OVs are thought to exert both direct and indirect antitumor activity and have been approved for cancer immunotherapy.24
We therefore engineered a VV vector, derived from an attenuated Copenhagen strain25 with clinically proven safety and strong immunomodulatory effects observed in global smallpox vaccination programs, and cytolytic and inflammatory cell infiltration-inducing properties in mouse experimental models of immune desert and immune-excluded cancer,26–29 with full-length αhCTLA-4 or αmCTLA-4 IgG antibody sequences. Variant vectors additionally encoding GM-CSF (VVGM-αCTLA-4) (figure 1I), a growth factor inducer and enhancer of myelopoiesis and innate immune cell chemotaxis, were also generated and evaluated for therapeutic efficacy. Following genetic reconstruction, recombinant αCTLA-4 encoding viruses were confirmed to preferentially infect, replicate in (figure 1J), and lyse (figure 1K) tumor cell lines. Tumor cell lines infected with engineered OVs were further shown to produce full-length IgG antibody and GM-CSF transgenes (online supplemental figure 3A,B) with equipotent binding to CTLA-4 receptors (online supplemental figure 2D) and support of GM-CSF-dependent cell proliferation (online supplemental figure 2C) compared with recombinantly produced proteins. 4-E03 produced by BT-001-infected MIA-PaCa-2 tumor cells was also shown to deplete human Treg cells in vivo (figure 1L).
Intratumoral VVGM-αCTLA-4 has antitumor activity associated with tumor-selective CTLA-4 receptor saturation and Treg depletion
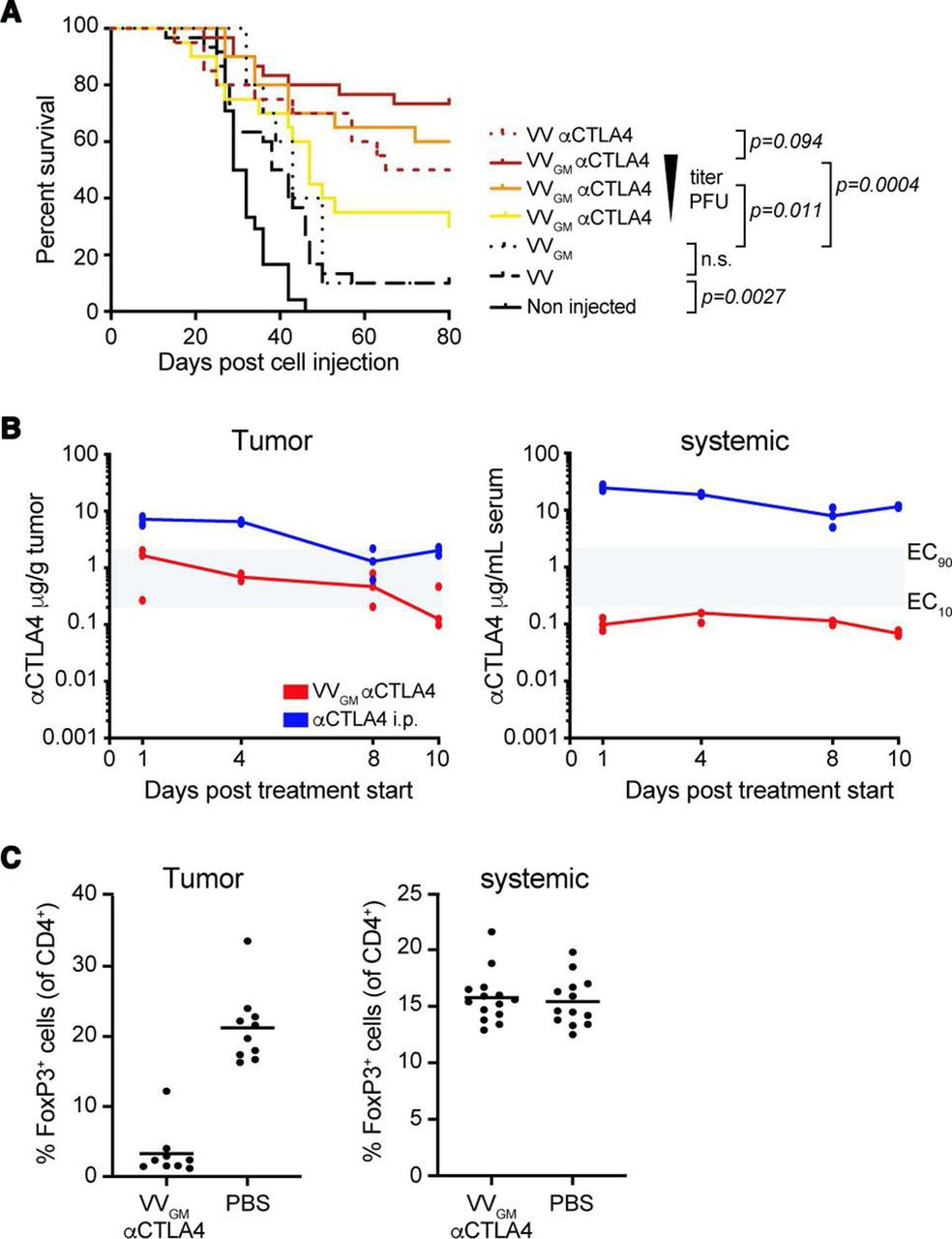
VVGM-αCTLA-4 antitumor activity was first assessed in the CT26 BALB/c model known to be highly infiltrated by T cells and sensitive to systemic αCTLA-4 antibody treatment.30 Three intratumoral injections with 7.5×104, 7.5×105, or 7.5×106 plaque-forming units (pfu) of VVGM-αCTLA-4 to CT26 tumor-bearing animals demonstrated a dose-dependent antitumor effect, which peaked at 106–107 pfu, with 6–7/10 animals cured (figure 2A). Treatment with control virus lacking αCTLA-4 antibody and/or GM-CSF transgenes demonstrated a strong dependence on αCTLA-4 antibody (1/10 mice surviving), and a marginal αCTLA-4 enhancing effect of GM-CSF, for therapeutic efficacy (7/10 vs 5/10 mice surviving). Based on the established dose-dependent, αCTLA-4-antibody-dependent, and GM-CSF enhancing effects, we therefore focused our further therapeutic and mechanistic evaluation and characterization on the double transgene-encoding OV (VVGM-αCTLA-4) using an intratumorally delivered dose of 1×107 pfu.

Intratumoral VVGM-αCTLA-4 has in vivo antitumor activity associated with tumor-restricted CTLA-4 receptor saturation and Treg depletion. (A) CT26 tumor-bearing mice were treated with VVGM-αCTLA-4 (7.5×106, 7.5×105, or 7.5×104 pfu), VV-αCTLA-4 (7.5×106 pfu), empty VV (7.5×106 pfu) (n=20–30 mice/group) or VVGM (7.5×106 pfu) (n=10 mice/group). Statistical analysis by log-rank test. (B) Pharmacokinetics of αCTLA-4 in tumors and serum of CT26 tumor-bearing mice after three intratumoral injections (days 0, 2, and 4) of VVGM-αCTLA-4 at 107 pfu or after single intraperitoneal injection of 3 mg/kg of αCTLA-4 mAb 5-B07 (n=3 mice/time point). Area in gray indicates EC10 to EC90 range of CTLA-4 receptor saturation (see online supplemental figure 2A). (C) Numbers of FoxP3+ cells were analyzed by FACS in tumors and spleen at day 10 post VVGM-αCTLA-4 injection. Graphs show pooled data from three independent experiments (n=13 mice/group).
We next assessed tumor and systemic concentrations of αCTLA-4 (figure 2B), GM-CSF (online supplemental figure 4A), and viral particles (online supplemental figure 4B) following intratumoral administration of our transgene-encoding vaccinia OV. Intratumoral injection of 107 VVGM-αCTLA-4 infectious particles into syngeneic mouse tumour-bearing immune competent mice generated intratumoral antibody exposure associated with sustained saturation of tumor, but not blood, CTLA-4-expressing cells (figure 2B and online supplemental figure 2A). Similarly, intratumoral administration of VVGM-αhCTLA-4 to human tumor xenograft-bearing immune-deficient mice generated orders of magnitude greater antibody concentrations in tumor compared with blood (online supplemental figure 4C–E). Consistent with intratumoral administration achieving receptor saturating concentrations in tumor but not systemic compartments (figure 2B), intratumoral VVGM-αCTLA-4 resulted in near-complete depletion of intratumoral Treg cells but did not affect Treg numbers in spleens of CT26 tumor-bearing BALB/c mice (figure 2C).
Collectively, our results demonstrated that intratumoral administration of αCTLA-4-encoding VV successfully achieved tumor-restricted CTLA-4 receptor saturation and Treg depletion in vivo, supporting its tumor-selective αCTLA-4 therapeutic nature and prompting testing of in vivo efficacy and tolerability in diverse cancer experimental models.
VVGM-αCTLA-4 has broad antitumor activity
We proceeded to assess the antitumor activity of intratumoral VVGM-αCTLA-4 in a range of immune competent mouse cancer models spanning hematological (A20) and solid cancers of different origin on different genetic mouse backgrounds (CT26 BALB/c colon; EMT6 BALB/c breast, MC38 C57BL/6 colon and B16 C57BL/6 melanoma; figure 3A), representing highly T cell-inflamed (CT26) to immune-excluded (B16) TMEs. The models included those sensitive or resistant to ICB with αCTLA-4 or αPD-1. Strikingly, intratumoral administration of VVGM-αCTLA-4 to C57BL/6 or BALB/c mice carrying established syngeneic tumors characterized by diverse immune inflamed types of TME cured the majority (A20=10/10, EMT6=8/10, MC38=8/10 and CT26=8/9 surviving mice) of animals (figure 3A). Equally impressively, in the B16 C57BL/6 model characterized by an immune desert type of TME and resistance to both αPD-1 and αCTLA-4, intratumoral VVGM-αCTLA-4 significantly delayed tumor growth and cured 3/10 animals. These results indicated a broad therapeutic potential of VVGM-αCTLA-4 in diverse cancer types, including in patients with diverse inflamed and immune-excluded types of TMEs.
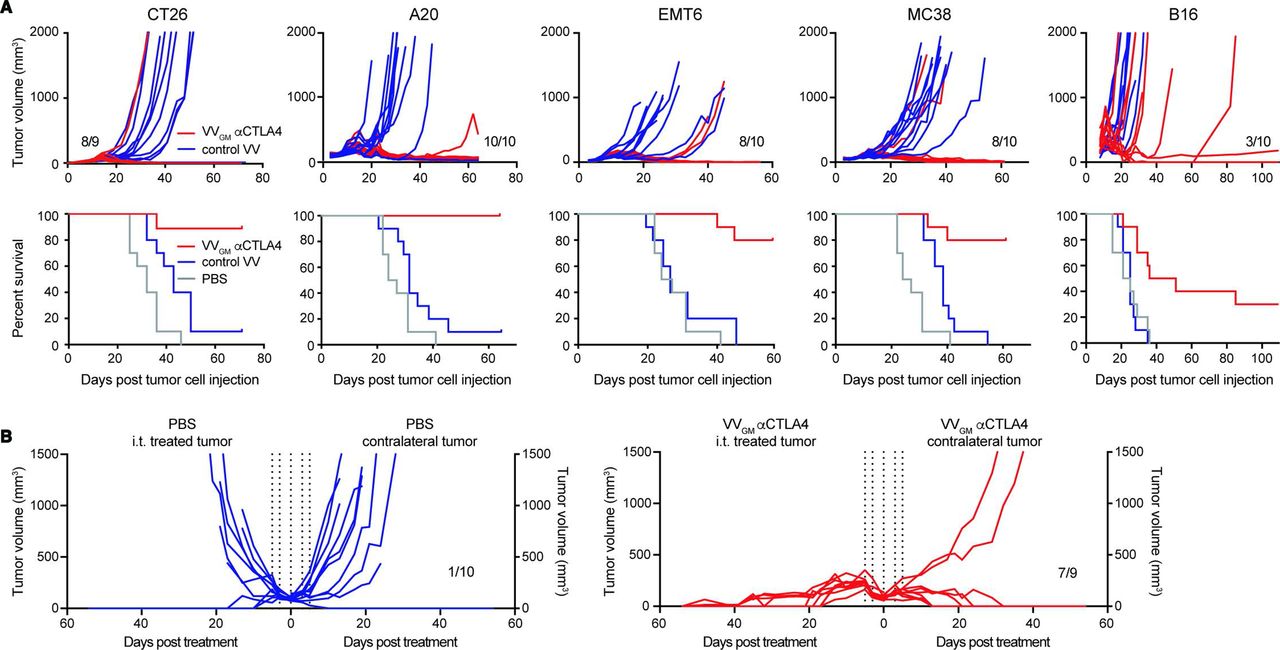
Intratumoral VVGM-αCTLA-4 has broad antitumor activity in syngeneic tumor models spanning inflamed and cold tumor microenvironments. (A) BALB/c mice bearing CT26, A20, or EMT6 tumors, or C57BL/6 mice bearing MC38 or B16 tumors received three intratumoral injections of VVGM-αCTLA-4, control virus lacking αmCTLA-4 mAb (VV empty for A20, EMT6, and MC38 or VVGM for CT26 and B16), or PBS. Treatment started when tumors had a volume of ~50 to 100 mm3. Graphs show tumor growth of individual mice and corresponding survival (n=10). (B) CT26 tumor cells were implanted into the right and left flanks of BALB/c mice. Intratumoral injections (vertical dotted lines, same as in A) in right flank tumors with VVGM-αCTLA-4 started when tumors reached a volume of ~100 mm3 (n=9–10). VV, vaccinia virus.
Intratumoral treatment with VVGM-αCTLA-4 induces long-lasting systemic antitumor immunity
Preclinical and clinical studies have demonstrated the therapeutic potential of tumor-localized cancer immunotherapy. Intratumoral oncolytic virotherapy, alone31 or combined with ICB,8 32 induces durable responses in patients with melanoma cancer. Mechanistically, intratumoral oncovirotherapy has been proposed to induce or enhance inflammatory cell infiltration into injected tumors, resulting in increased tumor antigen presentation, migration to draining lymph nodes, and, following priming, CD8+ T cell trafficking to distant (non-injected) tumor lesions to exert systemic antitumor ‘abscopal’ effects.33 In the clinic, such induction of systemic adaptive antitumor memory responses will be critical since patients with cancer may present with widespread disease characterized by metastasized, non-detectable, or uninjectable tumors.
We used a multipronged approach to assess whether intratumoral VVGM-αCTLA-4 induced abscopal effects and systemic antitumor immunity. First, using a ‘twin tumor model’ where tumor cells are subcutaneously grafted to the right and left flanks of each animal but only one tumor is injected with OV and the other is left untreated, abscopal effects can be evaluated and manifested as reduced tumor growth in uninjected tumors. Intratumoral injection of a maximally efficacious VVGM-αCTLA-4 dose in CT26 tumor-bearing mice resulted in complete (9/9) rejection of injected tumors and near-complete rejection (7/9) of uninjected tumors, indicating a strong abscopal effect (figure 3B). The true abscopal nature of intratumoral OV administration was confirmed twofold. First, non-injected tumors were analyzed and confirmed negative for viral particles (online supplemental figure 5A). Second, intratumoral administration conferred enhanced survival compared with intravenous administration both of therapeutically maximally efficacious (107 pfu) and suboptimal (105 pfu) doses of VVGM-αCTLA-4 (online supplemental figure 5C). In fact, intratumoral dosing of 105 pfu was at least as efficacious compared with the 100-fold greater intravenous injected dose in rejecting non-injected tumors (online supplemental figure 5B,C). Finally, and consistent with intratumoral OV administration inducing immunological memory characteristic of an adaptive antigen-specific immune response, cured animals were protected against rechallenge with the same (CT26), but not unrelated (Renca), tumors (online supplemental figure 5D).
VVGM-αCTLA-4 elicits robust systemic CD8+ T-cell antitumor immunity
We proceeded to investigate the nature of the systemic antitumor immune response by assessing VVGM-αCTLA-4 therapeutic activity in immune intact compared with CD4+ T cell-depleted or CD8+ T cell-depleted CT26 tumor-bearing mice (figure 4A and online supplemental figure 7A). Strikingly, CD8+ T cell depletion completely eliminated VVGM-αCTLA-4 antitumor activity. CD4+ T cell depletion reduced, but did not ablate, VVGM-αCTLA-4 effects. These data demonstrated that VVGM-αCTLA-4 antitumor activity critically depended on CD8+ T cells. We therefore next assessed if intratumoral VVGM-αCTLA-4 induced or expanded tumor-specific and virus-specific CD8+ T cells in tumors and in the periphery. CT26 tumor-bearing BALB/c mice were treated intratumorally with VVGM-αCTLA-4 or, to mimick clinically available αCTLA-4 regimens, systemically (intraperitoneal) with αmCTLA-4 mAb 5-B07 (3 mg/kg). CT26 tumor-specific and vaccinia-specific CD8+ T cells in tumor and central compartments (spleen) were quantified by two approaches; direct quantification of tumor-specific CD8+ T cells in harvested spleens using CT26 tumor antigen (AH-1)-specific and VV-specific multimers, and by assessment of IFN-γ+TNF-α+ CD8+ T cells following ex vivo stimulation of splenocytes (figure 4B) or of TILs with CT26-derived tumor peptide AH-1 and vaccinia-derived peptide S9L8, respectively. Treatment with PBS or VV encoding only GM-CSF were included as controls. As expected, and consistent with intratumoral VVGM-αCTLA-4 inducing robust systemic CD8+ T cell-dependent antitumor immunity, intratumoral VVGM-αCTLA-4 induced tumor-specific CD8+ T cells both in injected tumors and in peripheral (non-injected tumor and spleen) compartments (figure 4B–D and online supplemental figure 7B) as assessed by ex vivo stimulation of splenocytes or dextramer staining. Impressively, intratumoral VVGM-αCTLA-4 expanded tumor-specific CD8+ T cells more effectively compared with systemic αCTLA-4, despite the latter achieving complete saturation of intratumoral CTLA-4 (figure 2B). Control treatment with PBS or virus lacking αCTLA-4 did not induce tumor-specific CD8+ T cells by either read-out. Interestingly, intratumoral treatment with VVGM-αCTLA-4 also induced vaccinia-specific CD8+ T cells albeit in low numbers.
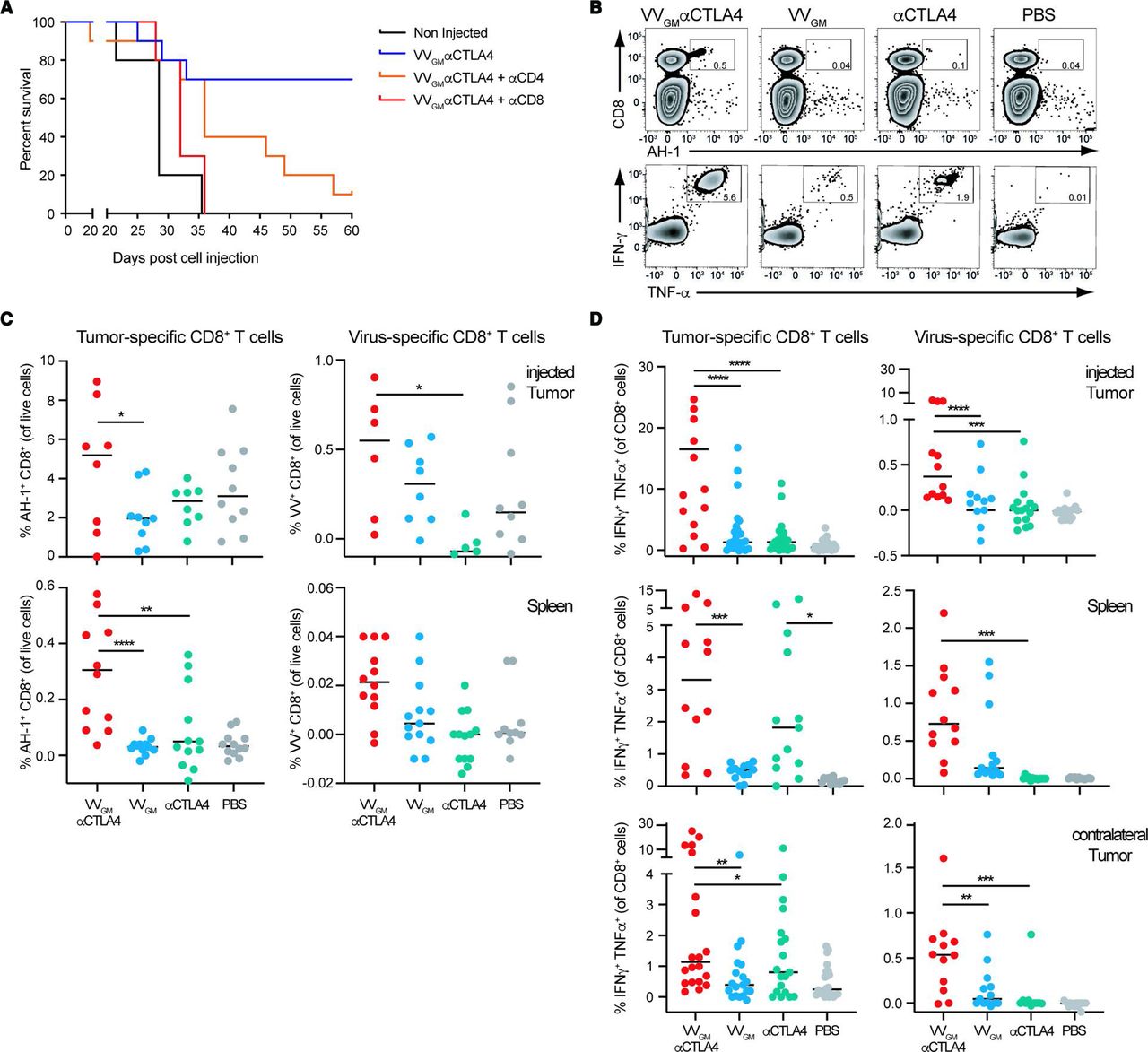
Intratumoral VVGM-αCTLA-4 elicits robust systemic CD8+ T cell-dependent antitumor immunity. (A) BALB/c mice were treated with CD8 or CD4 depleting antibody pre- and post-subcutaneous challenge with CT26 tumor cells. When tumors reached a volume of ~20 to 50 mm3, treatment as in figure 3A commenced. One representative experiment (out of two) is shown with 10 mice per group. (B–D) CT26 tumor-bearing mice were treated intratumorally with VVs or intraperitoneally with αCTLA-4 mAb (clone 5-B07 at 3 mg/kg). Tumor cell suspensions and splenocytes were restimulated ex vivo with VV-specific or CT26 (AH-1)-specific peptide and the percentage of IFN-γ+ and TNFα+ CD8+ T cells, or MHC class I-labeled multimer positive CD8+ T cells was quantified by FACS. (B) Flow-cytometry dot plots of AH-1 peptide-positive (upper panel) or cytokine-positive (lower panel) splenocytes. Quantification of (C) antigen-specific and (D) IFN-γ+/TNFα+ CD8+ T cells in indicated organs. Each dot represents one mouse (n=3–6 experiments). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way analysis of variance. IFN-γ, interferon gamma; VV, vaccinia virus.
Collectively, these data demonstrated that intratumoral VVGM-αCTLA-4 induced robust systemic CD8+ T cell-dependent antitumor immunity.
Intratumorally induced CD8+ T-cell antitumor immunity is FcγR-dependent and cDC1-dependent
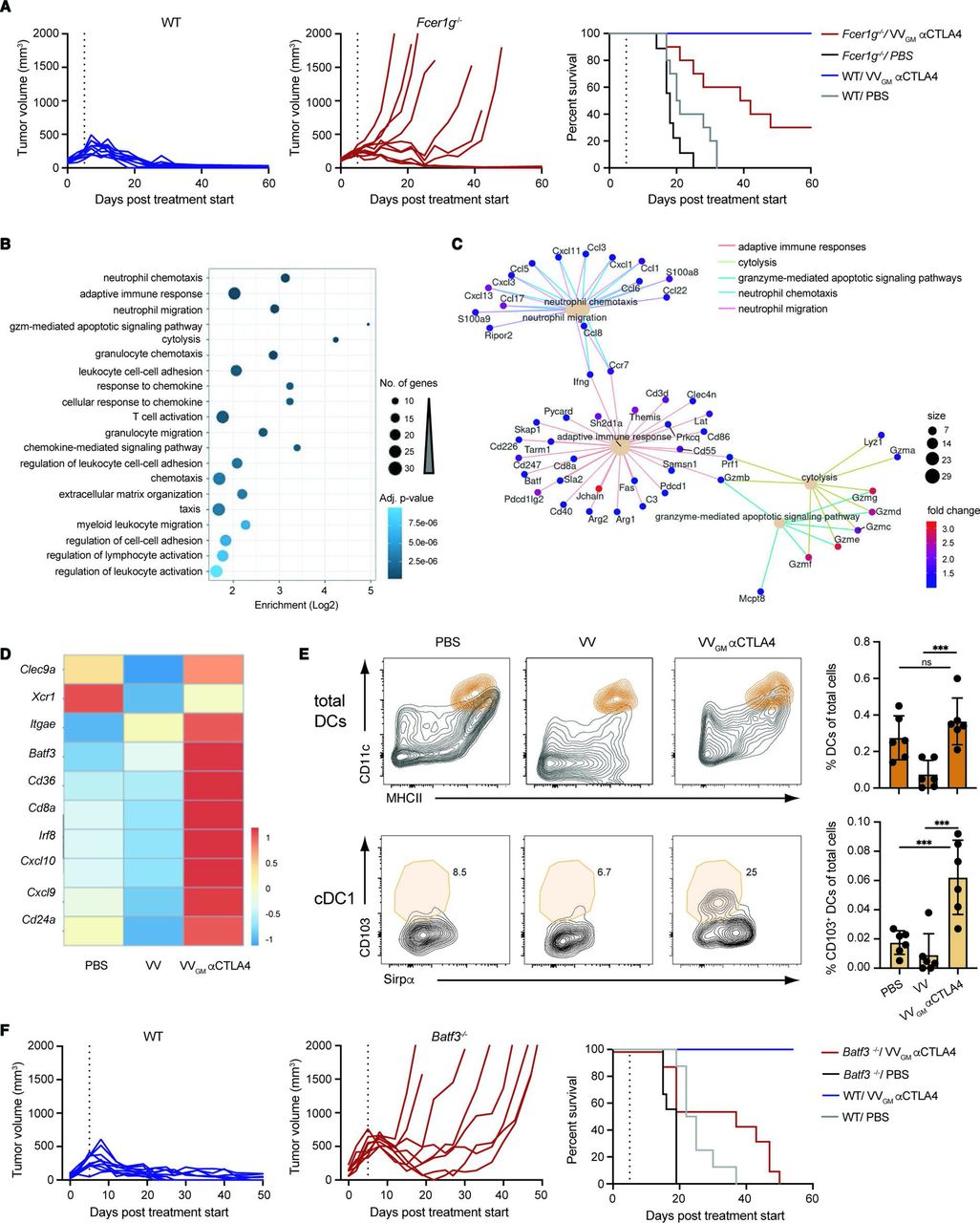
The broad antitumor activity, strong expansion of tumor-specific CD8+ T cells in tumor and periphery, and tumor-restricted depletion of Treg cells supported a highly effective and safe treatment with intratumoral VVGM-αCTLA-4. To further assess and confirm a role for antibody-mediated Treg depletion underlying antitumor immunity, we compared antitumor effects of intratumoral VVGM-αCTLA-4 in CT26 tumor-bearing WT and common gamma chain-deficient (Fcer1g−/−) BALB/c mice. Fcer1g−/− mice lack functional activating Fc gamma receptors and αCTLA-4 antibody in vivo Treg depletion, and associated antitumor activity was previously shown to be activating FcγR-dependent.10 18 Consistent with αCTLA-4-induced Treg depletion critically underlying intratumoral VVGM-αCTLA-4 antitumor immunity, WT (10/10), but not FcγR-deficient animals (3/10), were completely protected and cured from their cancer (figure 5A). The limited but significant antitumor activity observed in FcγR-deficient animals was consistent with our own observation and others’ observations that the viral vector (figure 3A) and CTLA-4:B7 blockade per se — in the absence of Treg depletion (online supplemental figures 2 and 6) — delayed tumor growth but contributed only limited survival advantage.

Intratumorally induced CD8+ T cell antitumor immunity is FcγR-dependent and cDC1-dependent. (A) CT26 tumor-bearing WT and Fcer1g−/− BALB/c mice received intratumoral injections of VVGM-αCTLA-4 or PBS as in figure 3A. Graphs show tumor volume (left and center panels) and mouse survival (right panel). Vertical lines indicate the end of the treatment. (n=10 mice/group) (B) GO terms enriched in the set of 352 differentially expressed genes, either upregulated or downregulated, in CT26 tumors treated with VVGM-αCTLA-4 versus VV empty. The 20 enriched terms with the lowest adjusted p value are shown. (C) Network view of the differentially expressed genes associated with the five most enriched GO terms from (B). Only genes upregulated were found associated with these five enriched GO terms. (D) Heatmap representation of cDC1-associated transcripts differentially expressed after treatment with VVGM-αCTLA-4 or VV empty. (E) Representative FACS plots and summarized quantitation of total DCs and cDC1s in tumors following treatment. DCs were gated as described in supplemental information and further defined as CD103+/Sirpα− cDC1s. ***p<0.001 by one-way analysis of variance. (F) MC38 tumor-bearing WT and Batf3−/− C57BL/6 mice received intratumoral injections of VVGM-αCTLA-4 or PBS as in figures 3A and 5A. Graphs show tumor volume (left and center panels) and mouse survival (right panel). Vertical lines indicate the end of the treatment. (n=8–10 mice/group).DC, dendritic cell; ns, not significant; VV, vaccinia virus; WT, wild type.
Besides affording immune effector-mediated ADCC and antibody-dependent cellular phagocytosis (ADCP) of antibody-coated target cells, FcγRs have been shown to promote tumor antigen cross-presentation,34 broadening and enhancing the CD8+ T cell antitumor response to encompass normally excluded major histocompatibility complex class II (MHCII)-restricted extracellular tumor antigens. Our findings that intratumoral VVGM-αCTLA-4 antitumor immunity was FcγR-dependent and induced more robust expansion of tumor-specific CD8+ T cells compared with systemic αCTLA-4 indicated that it might promote tumor antigen cross-presentation. This notion was reinforced by differential gene expression analyses of tumors harvested from VVGM-αCTLA-4-injected compared with viral backbone (VV)-injected and untreated CT26 tumor-bearing mice (figure 5B–D). Besides upregulating Batf3 and Irf8 per se, differential gene expression analyses demonstrated upregulation of type I interferon responses and markers (eg, CD8a and Itgae), which are associated with cDC1 dendritic cell antigen cross-presentation (figure 5D). FACS analysis data confirmed a significant increase in CD103+Sirpα− cDC1s in tumors of VVGM-αCTLA-4-treated mice compared with control groups (figure 5E). Additional signatures supported the aforementioned characterized CD8+ T cell-dependent (and potentially NK-cell-mediated granzyme-dependent) tumor cytolysis underlying triggering and actuation of VVGM-αCTLA-4 antitumor immunity (figure 5B,C).
To assess a role for antigen cross-presentation in VVGM-αCTLA-4-induced antitumor immunity, we used mice lacking the transcription factor Batf3 (Batf3−/− mice). Batf3−/− mice lack CD8α+ cDC1 dendritic cells and as a consequence show defective antigen cross-presentation and severely impaired CD8+ T cell responses to viruses during infection and to tumor antigens in mouse experimental models of cancer.16 Further, cDC1s and antigen cross-presentation are known to mediate therapeutic activity of immune checkpoint blockers including αCTLA-4.35 We therefore compared antitumor activity of intratumoral VVGM-αCTLA-4 in Batf3+/+ and Batf3−/− C57BL/6 mice transplanted with MC38 tumors. Strikingly, Batf3-deficiency abrogated intratumoral VVGM-αCTLA-4 antitumor immunity as demonstrated by 0/8 Batf3−/− compared with 9/9 WT mice surviving (figure 5F). Collectively, these results demonstrated that VVGM-αCTLA-4 has both FcγR-dependent and cDC1-dependent antitumor activity, identifying intratumorally induced Treg-depletion and tumor antigen cross-presentation as major mechanisms, and intratumoral CTLA-4–B7-blockade and oncolysis as supporting mechanisms, underlying intratumoral VVGM-αCTLA-4 induced CD8+ T cell antitumor immunity.
Intratumoral VVGM-αCTLA-4 expands peripheral effector CD8+ T cells and reduces Treg and exhausted CD8+ T cells
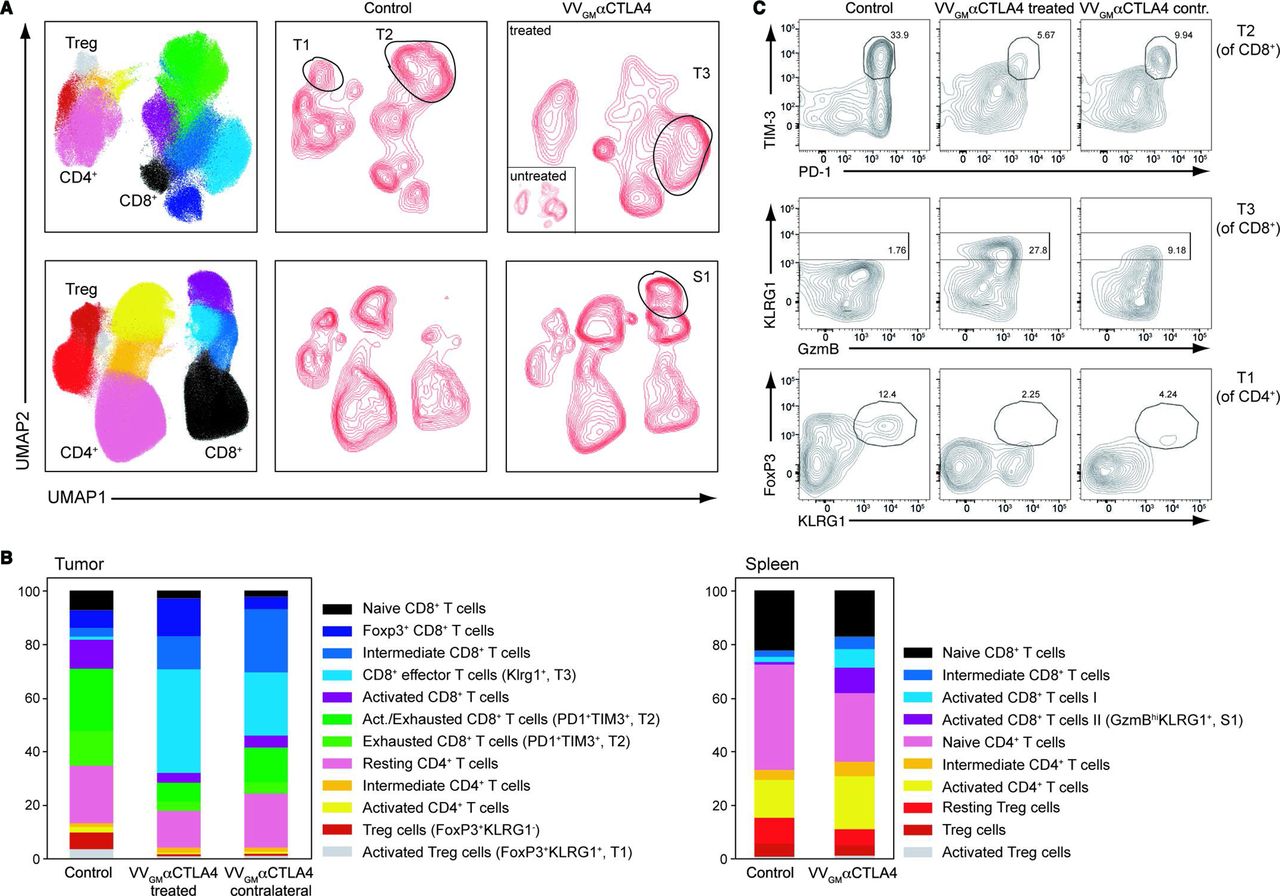
We proceeded to qualitatively characterize how intratumoral VVGM-αCTLA-4 modulates TIL responses in injected and flanking tumors, and in the periphery. Using multicolor flow-cytometry and a high-dimensional antibody panel designed to identify functionally distinct antitumor and protumor TIL subsets, 12 T cell clusters across treatment groups were identified (figure 6 and online supplemental figure 7C). Strikingly, intratumoral VVGM-αCTLA-4 eliminated exhausted (PD-1+TIM-3+) CD8+ T cells and robustly expanded non-exhausted KLRG1+ effector CD8+ T cells in injected tumors compared with mock-treated animals (figure 6). At the same time, and consistent with our previously mentioned findings, intratumoral VVGM-αCTLA-4 effectively depleted CTLA-4+ intratumoral Tregs, including KLRG1+ Tregs, which are known to express high levels of CTLA-4 and to be particularly suppressive (figure 6).36

Intratumoral VVGM-αCTLA-4 expands peripheral effector CD8+ T cells and reduces Treg and exhausted CD8+ T cells. CT26 ‘twin’ tumor-bearing BALB/c mice were treated intratumorally (right flank tumors only) with VVGM-αCTLA-4 or PBS. Spleens and injected and contralateral tumors were collected on day 10 post-treatment and stained with a high-dimensional panel designed to identify T-cell populations. (A) Intratumoral VVGM-αCTLA-4 reduced activated CD4+ Treg cells (FoxP3+KLRG1+, ‘T1’), reduced exhausted CD8+ T cells (PD1+TIM3+, and ‘T2’), and expanded activated effector CD8+ T cells (KLRG1+ and ‘T3’) in injected and uninjected tumors (upper panel) and expanded activated CD8+ T cells in spleen (S1, lower panel) (B) shows quantification of data illustrated in A. One representative experiment (out of three) with five mice/group is shown. (C) Flow cytometry plots show characteristic markers of selected intratumoral T-cell clusters. VV, vaccinia virus.
Assessment of flanking distal tumors, which had not been injected with antibody-encoding virus, revealed similar but less profound modulation of TIL by intratumoral VVGM-αCTLA-4. Further, and in keeping with our observations that intratumoral administration of the αCTLA-4-encoding OV expanded tumor-specific CD8+ T cells in the periphery (figure 4B–D), intratumoral VVGM-αCTLA-4 induced activated granzyme B+ (KLRG1+) CD8+ T cell subsets in spleen (figure 6A,C). Finally, and consistent with the antibody-encoding virus achieving tumor-restricted Treg depletion, Treg populations that were depleted in tumor beds were largely unaltered in spleen by intratumoral VVGM-αCTLA-4 (figure 6A).
Intratumoral VVGM-αCTLA-4 combines with αPD-1 to reject ‘cold’ distal tumors
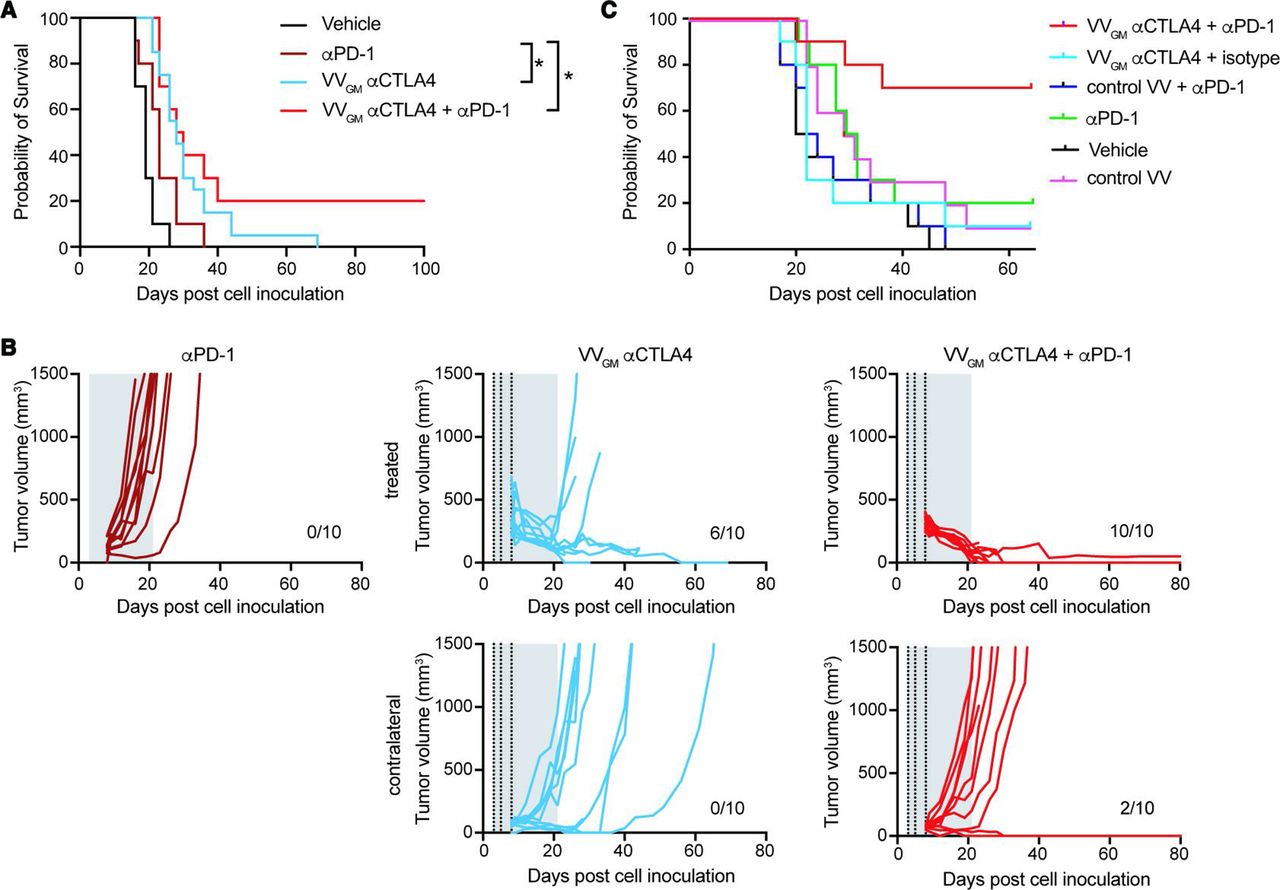
Our observations demonstrated that VVGM-αCTLA-4 acted locally in injected tumors, principally by mechanisms involving αCTLA-4 mAb-dependent tumor antigen cross-presentation and Treg-depletion, to ‘ignite’ systemic adaptive antitumor immunity and robust peripheral tumor-specific CD8+ T cell expansion. These findings indicated that VVGM-αCTLA-4 might synergize with therapeutic agents that help mobilize CD8+ T cells to the tumor. αPD-1 is thought to act principally by reversal of T cell exhaustion37 38 and possibly by mobilizing stem-like memory CD8+ T cells to tumors.39 40 Despite αPD-1’s documented ability to improve survival in multiple solid cancers of different origin, it does not improve outcome in patients with poorly immune infiltrated ‘cold tumors’,41 which perhaps represent the greatest unmet medical need in cancer therapy today. Based on their apparently different and potentially complementary mechanism of action, we therefore next examined synergizing effects of αPD-1 and VVGM-αCTLA-4 with a focus on ICB-resistant, poorly immune infiltrated and poorly immunogenic cold cancers using B16 C57BL/6 as a model system. Our previous data had demonstrated that B16 tumors responded poorly to ICB therapy, including to clinically relevant systemic administration of αPD-1 (10 mg/kg), αCTLA-4 (10 mg/kg) or the combination thereof (online supplemental figure 8A). So as to mimick the clinical situation where palpable large tumors will be injected with the antibody-encoding virus, but small or undetectable metastasized lesions cannot be injected, we established a twin-tumor B16/C57BL6 model where the animals carry one ‘large’ and one ‘small’ tumor, and where only the large tumor was injected intratumorally with VVGM-αCTLA-4. Resistance to αPD-1 was confirmed by lack of tumor growth inhibition or survival benefit following systemic treatment with a maximally efficacious dose of 10 mg/kg (figure 7A). As previously observed, single-agent treatment with VVGM-αCTLA-4 significantly reduced tumor growth of the primary injected tumor (figures 3 and 7B). However, intratumoral VVGM-αCTLA-4 only induced a slight delay of uninjected tumor’s growth (figure 7B), which did not translate into animal survival (figure 7A). In contrast, combined treatment with intratumoral VVGM-αCTLA-4 and systemic αPD-1 significantly inhibited injected and uninjected tumor growth, resulting in ~20% of animals bearing cured in this ICB treatment-resistant model of cold cancer (figure 7A). Further consistent with VVGM-αCTLA-4 being able to convert cold, ICB-resistant tumors toward an inflamed, ICB responsive, phenotype, combined treatment with VVGM-αCTLA-4 (but not αPD-1 alone) induced a strong influx of T cells into B16 tumors, which became similarly densely T cell rich compared with inflamed CT26 tumors (online supplemental figure 8B). Finally, and consistent with a broad potential to treat cold tumors, VVGM-αCTLA-4 significantly improved survival also of Lewis Lung tumor-bearing mice (online supplemental figure 8D). Like B16, this model was poorly T cell infiltrated and did not respond to clinically relevant, systemic, dual checkpoint αCTLA-4/αPD1 blockade (online supplemental figure 8C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intratumoral VVGM-αCTLA-4 synergizes with αPD-1 to reject ‘cold’ immune checkpoint blockade-resistant tumors. (A,B) C57BL/6 mice carrying two B16 tumors, one large (5×105 cells, treated tumor) and one small tumor (1×105 cells, contralateral side) received three intratumoral injections with VVGM-αCTLA-4 (vertical dotted lines) and/or intraperitoneal αPD-1 (29F.1A12, 10 mg/kg; two times per week for 3 weeks, gray area). (A) Survival (n=10–20), *p<0.05 by log-rank test. (B) Tumor growth curves of intratumorally injected and contralateral tumors. (C) A20 tumor-bearing BALB/c mice were treated thrice with VVGM-αCTLA-4 intratumorally (at a suboptimal dose of 1×105 pfu), αPD-1 intraperitoneally (RMP1-14, full dose of 10 mg/kg) or the combination of both when tumors had reached a volume of ~135 mm3. Graph shows animal survival (n=10). VV, vaccinia virus.
The indicated synergizing effects of combined intratumoral VVGM-αCTLA-4 and systemic αPD-1 were confirmed in BALB/c mice transplanted with syngeneic A20 tumors. This model was shown to be semiresponsive to αPD-1 with ~20% of animals being cured by full therapeutic intraperitoneal dosing (10 mg/kg) (figure 7C). While optimal therapeutic dosing with intratumoral VVGM-αCTLA-4 was fully protective (10/10 animals cured, figure 3), suboptimal treatment with 1/100 of the optimal dose showed insignificant tumor growth inhibition and no survival advantage. When therapeutic intraperitoneal αPD-1 and subtherapeutic intratumoral VVGM-αCTLA-4 were combined, the majority of animals (7/10) were cured (figure 7C).
Discussion
We here provide in vivo proof of concept that intratumoral administration of oncovirally encoded Treg-depleting αCTLA-4 has stronger and broader antitumor activity compared with approved systemic αCTLA-4 regimens yet, through its tumor-restricted nature of exposure, is indicated to be safe and well tolerated. Intratumoral VVGM-αCTLA-4 induced stronger expansion of tumor-specific CD8+ T cells compared with systemic recombinant αCTLA-4 and had antitumor activity in poorly immune infiltrated cold syngeneic mouse tumor models resistant to clinically relevant dosing with systemic αCTLA-4 and αPD-1. Remarkably, our observations suggest that the potent systemic antitumor immunity induced by oncoviral αCTLA-4 derived strictly from ‘immune-igniting’ effects in injected tumors; intratumoral VVGM-αCTLA-4 was not associated with virus spread or antibody exposure to distal uninjected tumors, but rather achieved tumor-restricted CTLA-4 receptor saturation and Treg depletion. These observations have important implications both for expected clinical efficacy and tolerability of intratumoral VVGM-αCTLA-4. From an efficacy perspective, they demonstrate that local administration of oncovirally encoded αCTLA-4 may provide greater therapeutic benefit compared with available (ipilimumab) and Treg-depletion-optimized10 or ‘masked’,42 systemic αCTLA-4 antibody regimens, as well as compared with previously described OV approaches encoding non-Treg depletion-optimized αCTLA-4.43 At a mechanistic level, VVGM-αCTLA-4-induced FcγR-dependent Treg depletion and cDC1 antigen cross-presentation are likely to underly both the observed robust CD8+ T cell expansion and synergism with αPD-1 to reject cold tumors. Besides mediating induction of endogenous antitumor immune responses16 and efficacy of systemic checkpoint blockade therapy,35 44 45 cDC1s promote the proliferative response of intratumoral CD8+ TILs, expand the pool of TCF1+ stem-like precursors, and induce generation of TIM-3+ terminal effectors during αPD-1 therapy.46 Similarly, Treg depletion achieved with mAb to costimulatory or coinhibitory receptors, for example, IL-2R and CTLA-4, may promote CD8+ effector function and synergize with αPD-1.47 48 With regard to development of ‘dual activity’ immune modulatory antibodies that reduce Treg and expand antitumor CD8+ T cells, accumulating data demonstrate the importance of both target biology, fine-tuning of effector CD8+ T cell-enhancing and Treg-depleting properties, as well as delivery regimen. For example, it was recently demonstrated that FcγR-competent non-ligand blocking antibodies to IL-2R, which deplete Treg but do not starve CD8+ effector T cells of critical (IL-2-mediated) growth survival signaling, have superior therapeutic potential in cancer therapy compared with ligand-blocking αIL-2R antibodies.47 Analogously, but differently, we recently reported that antibodies to 4-1BB can be made to deplete Tregs or promote effector T cell expansion by antibody isotype switching (altering FcγR-engagement), but that harnessing both mechanisms required sequential administration or hinge-engineering.21 As described herein, spatial restriction of Treg-depleting, checkpoint blocking, αCTLA-4 to injected tumors appears to be a particularly promising approach for harnessing maximal therapeutic activity of immune modulatory αCTLA-4 antibodies, when used alone or in combination with synergizing checkpoint blockade therapy for example, αPD-1/L1.
Several observations support optimizing Treg depletion in αCTLA-4 for tumor-localized therapy. First, independent studies have established that therapeutic efficacy of αCTLA-4 depends on and correlates with Treg depletion.10 18 Herein presented data on therapeutic activity of Treg-depleting recombinant and oncovirally encoded αCTLA-4 antibodies, which showed strong curative effect in FcγR-proficient (Treg-depleting) antibody Fc formats and hosts compared with their FcγR-deficient (non-depleting) counterparts, support this notion. Second, while clinical outcome of patients with melanoma treated with ipilimumab was recently reported to correlate with FcγR-engagement and Treg-depletion, data from our T cell humanized mouse model suggests ipilimumab has limited depleting activity compared with the herein vectorized αCTLA-4 antibody 4-E03 against human Treg cells expressing intratumorally relevant levels of CTLA-4. Further, whereas clinically tolerated doses of ipilimumab (1–3 mg/kg, depending on indication and regimen) are associated only with subsaturating CTLA-4 receptor occupancy and submaximal effect,12 49 our data demonstrate that oncolytic vectorization and intratumoral administration can generate therapeutically optimal exposure in an apparently safe manner, even with Treg depletion-enhanced αCTLA-4. Finally, and supporting our vectorization of a Treg depletion-enhanced and checkpoint blocking ‘dual activity’ αCTLA-4 antibody, antibody-mediated CTLA-4 blockade was recently shown to synergize with FcγR-dependent depletion in improving tumor-specific CD8+ T cell responses. Antibody blockade of CTLA-4 functionally destabilized intratumoral Treg and promoted B7–CD28 costimulation and antitumor CD8+ T effector function through processes involving altered glycolysis and competition for B7 ligands.50
The fact that full therapeutic dosing achieved tumor-restricted αCTLA-4 exposure indicates that severe toxicities associated with sustained systemic Treg depletion, for example, those observed in the FoxP3-DTR mouse model,51 are unlikely to manifest. Similarly, since αCTLA-4 checkpoint blockade effects will be restricted to TILs with tumor-antigen specificity, untoward self-reactivity associated with systemic αCTLA-4 should be minimal. In contrast, but consistent with the well-documented central immune checkpoint nature of CTLA-4,52 53 αCTLA-4 side effects associated with systemic administration, that is, body-wide antibody exposure, may be of severe autoimmune nature and can have fatal consequences.13 22
Taken together, therefore, while efficacy and tolerability of available αCTLA-4 regimens are considered to be linked and dose-dependent precluding use of full therapeutic dosing, our findings strongly suggest that spatial restriction of vectorized Treg-depleting αCTLA-4 is able to overcome these current limitations, uncoupling efficacy from tolerability. A clinical study investigating intratumoral VVGM-αhCTLA-4 (BT-001) alone and in combination with αPD-1 in metastatic or advanced solid tumors is open and enrolling patients (NCT04725331).
Data availability statement
The datasets generated during and/or analyzed during this current study have been deposited or are available from the corresponding author on reasonable request. The assigned accession number for RNAseq data reported in this paper is GEO: GSE176052.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human subjects. All procedures were approved by the local ethical committee for experimental animals (Malmö/Lunds Djurförsöksetiska Nämnd, at BioInvent under permit numbers 17196/2018 or 2934/2020, or at Transgene APAFIS Nr21622 project 2019072414343465) and performed in accordance with local ethical guidelines. Ethical approval was obtained by the ethics committee of Skåne University Hospital. Informed consent was provided in accordance with the Declaration of Helsinki.
Acknowledgments
We thank Drs Mark Cragg and Stephen Beers (University of Southampton) for their valuable advice and critical review of the manuscript. We thank Huguette Schultz, Chantal Hoffmann, Eline Winter, Fadi Azar, Julie Hortelano, Dominique Villeval, Murielle Gantzer, Virginie Nourtier, Juliette Kempf, Mathilda Kovacek, and Ann-Helen Fischer for their technical contributions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MS, J-BM, EQ, and BF conceived the project and wrote the manuscript. MS, J-BM, LF, MR, CR, PH, NS, LM, JF, CS, PK, JD, MB and KLC designed, performed, and analyzed the experiments. FJ contributed to the data analysis. IT contributed scientifically. BF accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding All support for this article was entirely provided by BioInvent International AB and Transgene SA.
Competing interests MS, MR, PH, LM, CS, FJ, MB, IT, and BF are employees, MS, MR, LM, FJ, MB, IT, and BF are shareholders of BioInvent International. KLC received funding from BioInvent. J-BM, LF, CR, NS, PK, JD, JF, and EQ are employees and shareholders of Transgene.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.