Article Text

Abstract
Background Dostarlimab is a humanized monoclonal antibody that binds with high affinity to PD-1, resulting in inhibition of binding to PD-L1 and PD-L2. We report interim data from patients with endometrial cancer (EC) participating in a phase I trial of single-agent dostarlimab.
Methods GARNET, an ongoing, single-arm, open-label, phase I trial of intravenous dostarlimab in advanced solid tumors, is being undertaken at 123 sites. Two cohorts of patients with EC were recruited: those with dMMR/MSI-H disease (cohort A1) and those with proficient/stable (MMRp/MSS) disease (cohort A2). Patients received dostarlimab 500 mg every 3 weeks for 4 cycles, then dostarlimab 1000 mg every 6 weeks until disease progression. The primary endpoints were objective response rate (ORR) and duration of response (DOR) per RECIST V.1.1, as assessed by blinded independent central review.
Results Screening began on April 10, 2017, and 129 and 161 patients with advanced EC were enrolled in cohorts A1 and A2, respectively. The median follow-up duration was 16.3 months (IQR 9.5–22.1) for cohort A1 and 11.5 months (IQR 11.0–25.1) for cohort A2. In cohort A1, ORR was 43.5% (95% CI 34.0% to 53.4%) with 11 complete responses and 36 partial responses. In cohort A2, ORR was 14.1% (95% CI 9.1% to 20.6%) with three complete responses and 19 partial responses. Median DOR was not reached in either cohort. In the combined cohorts, the majority of treatment-related adverse events (TRAEs) were grade 1–2 (75.5%), most commonly fatigue (17.6%), diarrhea (13.8%), and nausea (13.8%). Grade≥3 TRAEs occurred in 16.6% of patients, and 5.5% discontinued dostarlimab because of TRAEs. No deaths were attributable to dostarlimab.
Conclusion Dostarlimab demonstrated durable antitumor activity in both dMMR/MSI-H (ORR 43.5%) and MMRp/MSS EC (ORR 14.1%) with a manageable safety profile.
Trial registration number NCT02715284.
- immunotherapy
- programmed cell death 1 receptor
- clinical trials as topic
Data availability statement
Data are available on reasonable request. Anonymized individual participant data and study documents can be requested for further research (www.clinicalstudydatarequest.com).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Antibodies targeting programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1) have been studied in multiple hematologic and solid tumor types and in primary and recurrent settings.1 As a drug class, anti–PD-(L)1 pathway–targeted therapies have been shown to be well tolerated and exhibit consistent safety profiles.2
Endometrial cancer (EC) is the most common gynecologic malignancy in the developed world and has the highest rate of mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) status of any tumor type; up to 30% of all ECs are classified as dMMR/MSI-H.3 The major cause of MSI is a defect in the DNA MMR genes that repair mismatched bases. Loss of expression of one or more of the MMR proteins (MLH1, MSH2, MSH6, and PMS2) due to genetic mutation or epigenetic silencing is associated with an accumulation of DNA replication errors at microsatellite regions. Screening by immunohistochemistry (IHC) is used to test for MMR protein status, either dMMR (absence of one or more MMR proteins) or MMRp (presence of all MMR proteins). MSI is a consequence of dMMR and can be detected by either PCR or next-generation sequencing (NGS).
Molecular characterization of ECs has demonstrated four subgroups of EC with associated prognostic significance: POLε-mutated tumors with the most favorable prognosis, MMR-deficient tumors with intermediate prognosis, and MMR-proficient and p53-mutated tumors with the worst prognosis.4 Although dMMR tumors are more likely to be of a low-grade endometrioid histologic subtype, MMRp tumors, which constitute the majority of ECs (≈70%), comprise a variety of histologic subtypes associated with a poor prognosis and limited treatment options.5
Dostarlimab (JEMPERLI) is an IgG4-k humanized monoclonal antibody that binds with high affinity to PD-1, resulting in inhibition of binding to PD-L1 and PD-L2. In the USA, dostarlimab is approved as a monotherapy in adult patients with dMMR recurrent or advanced endometrial cancer that has progressed on or after a platinum-containing regimen.6 In the EU, dostarlimab is approved as a monotherapy in adult patients with recurrent or advanced dMMR/MSI-H EC that has progressed on or after treatment with a platinum-containing regimen.7
We report interim data on the antitumor activity and safety of dostarlimab in advanced or recurrent disease from two separate EC cohorts in GARNET: dMMR/MSI-H EC (cohort A1) and MMRp/MSS EC (cohort A2). Accrual in both cohorts has completed.
Methods
Study design
GARNET is a phase I, single-arm study of dostarlimab monotherapy in patients with advanced and recurrent solid tumors.
In parts 1 and 2A of the trial, the recommended therapeutic dose (RTD) was determined to be 500 mg intravenously Q3W for four cycles, then 1000 mg intravenously Q6W until disease progression.
Part 2B of the ongoing GARNET study (NCT02715284) is exploring antitumor activity and safety in prespecified tumor types using the RTD. Cohorts A1 and A2 are the two cohorts that enrolled patients with EC.
The key inclusion criteria for cohorts A1 and A2 were as follows: recurrent EC that progressed on or after platinum doublet therapy; ≤2 prior lines of treatment for recurrent or advanced disease; measurable disease at baseline confirmed by central radiology review; anti–PD-(L)1 naive. All histological subtypes except sarcoma and carcinosarcoma were eligible.
The trial was performed in accordance with the principles of the Declaration of Helsinki, Good Clinical Practices, and all local laws. Part 2B of the study was overseen by an independent data and safety monitoring committee. The study protocol and/or other relevant documents received approval by the institutional ethics committee, institutional review board, and/or relevant competent authorities at each site.
Biomarker screening
Patients were screened based on MMR/MSI testing results using IHC, PCR, or NGS performed in a certified local laboratory. In May 2019, the study was amended to specify that patients must have MMR IHC testing for eligibility. If local IHC testing was not available, central IHC testing was performed. Central confirmation of local IHC results was not required for eligibility. When results from more than one test (MMR or MSI) were available for a patient, the patient was classified by their MMR status. In cases where MMR testing was inconclusive (MMR unknown (MMRunk)), patients were classified by their MSI status. Patients with MSI-H and MMRunk EC were grouped with the patients with dMMR EC, and patients with MSS and MMRunk EC were grouped with the patients with MMRp EC. Patients with MMR IHC testing results were not required to have MSI testing performed.
Pathology
Histopathologic testing was performed by local laboratories. No central histopathological review was conducted.
Exploratory biomarkers
Biomarker testing was conducted to determine PD-L1 expression, tumor mutational burden (TMB), and POLε exonuclease domain mutations (POLεmut). PD-L1 expression was determined by combined positive score by Ventana assay. TMB status was determined by Foundation One test; TMB-high (TMB-H) was defined as ≥10 mutations/Mb. POLεmut status was determined by PCR amplification and Sanger sequencing; mutations between residues 268 and 471 were classified as POLεmut. All biomarker analyses are post hoc.
Enrolling sites
This is an international trial with 123 sites. Enrolling sites for cohort A1 (dMMR EC) and A2 (MMRp EC) are listed in the supplemental appendix.
Endpoints
The primary objective for each cohort (A1 and A2) was to evaluate the antitumor activity of dostarlimab per the objective response rate (ORR) and duration of response (DOR) by blinded independent central review (BICR) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
The prespecified secondary objectives for both cohorts A1 and A2 included immune-related ORR (irORR), immune-related disease control rate (irDCR), and irDOR based on investigators’ assessment using immune-related RECIST (irRECIST) and DCR based on BICR using RECIST v1.1.
Safety analyses
Safety analyses included incidence of treatment-emergent adverse events (TEAEs), immune-related AEs of interest (irAEIs), and serious adverse events (SAEs) occurring while patients were on treatment or up to 90 days after the end of treatment. Any changes in clinical laboratory parameters (hematology, chemistry, thyroid function, coagulation, urinalysis) and Common Terminology Criteria for Adverse Events (CTCAE) v4.03-graded laboratory toxicities, vital signs, Eastern Cooperative Oncology Group performance status, ECG parameters, physical examinations, and usage of concomitant medications were recorded.
No formal hypothesis-testing analysis of AE incidence rates was performed.
Potential sources of bias
In this interim analysis (data immature), an efficacy-evaluable subpopulation is being used from an ongoing single-arm study.
The enrollments for cohorts A1 and A2 were separate, and the number of patients enrolled in the cohorts does not reflect the natural distribution of dMMR/MMRp status in patients with EC.
Sample size
Cohort A1 was designed to enroll approximately 100 patients with dMMR/MSI-H EC, with the potential for up to 165 patients. Cohort A2 was designed to enroll approximately 125 patients with MMRp/MSS EC, with the potential for up to 250 patients.
For cohort A1 in part 2B, the null hypothesis that the true response rate is ≤20% (H0: p≤0.2) was tested against a one-sided alternative of ≥40% (Ha: p≥0.4). With 65 patients treated, the cohort has a 92% power to rule out a ≤20% ORR (null hypothesis) when the true ORR is 40% at the 2.5% type I error rate (one-sided). The sample size of cohort A1 was increased to 100 patients, which allowed the lower-limit boundary of the exact 95% CI to exclude a response rate of 25% or less assuming the observed ORR is 35%.
For cohort A2 in part 2B, a two-stage design was used. The null hypothesis that the true response rate is ≤5% (H0: p≤0.05) was tested against a one-sided alternative of ≥15% (Ha: p≥0.15). In the first stage, 25 patients were accrued. Because two or more responses were observed, approximately 40 additional patients were accrued for an approximate total of 65. This design yields a type I error rate of 10% (one-sided) and power of 87% when the true response rate is 15%.
Based on encouraging clinical activity seen from the first stage of the cohort A2, the sample size of cohort A2 was increased to 125 patients with the potential for up to 250 patients to allow for testing the estimate of ORR more precisely with the lower limit of the exact 95% CI, excluding a response rate of 15% or less.
Statistics
All statistical outputs were generated using SAS (V.9.4). Patient demographics, baseline characteristics, safety, and efficacy results were summarized descriptively. All patients who received at least one dose of dostarlimab by the data cut-off date were included in the safety analysis. All patients who received at least one dose of dostarlimab, had at least one BICR-confirmed measurable lesion at baseline, and had the opportunity to be followed for at least 6 months as of the data cut-off date were included in the efficacy-evaluable population, regardless of whether the patient had a postbaseline tumor assessment.
Point estimates and exact two-sided 95% CIs were provided for ORR and irORR; DOR and irDOR were analyzed using the Kaplan-Meier method. Patients who did not achieve a confirmed response were excluded from the DOR and irDOR analysis. Median follow-up time was calculated using the reverse Kaplan-Meier method.
Role of the funding source
The trial was designed by GlaxoSmithKline (originally designed and funded by Tesaro, acquired by GlaxoSmithKline in 2018), in collaboration with the authors. The sponsor provided support for the statistical analyses of the data and funded a medical writer for the report. The authors performed the collection, analysis, and interpretation of the data. The authors had the final decision to submit the manuscript for publication. The manuscript was written by the authors with medical writing assistance funded by GlaxoSmithKline. GlaxoSmithKline had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. GlaxoSmithKline collaborated with the investigators in designing the trial, provided the study drug, coordinated the management of the study sites, funded the statistical analysis, and provided medical writing support. Authors employed by GlaxoSmithKline, in coordination with all authors, were involved in preparation, review, approval, and decision to submit the manuscript.
Results
Patients
The study was initiated on April 10, 2017, and enrollment in both cohorts is complete. Data analysis was performed using a data cut date of March 1, 2020.
In total, 129 patients with dMMR or MSI-H and MMRunk EC (cohort A1) and 161 patients with MMRp or MSS and MMRunk EC (cohort A2) were enrolled and dosed with dostarlimab (figure 1). Antitumor activity was assessed in the efficacy-evaluable population (figure 1), which includes patients enrolled on or before September 15, 2019 (24 weeks before the data cut-off date) and an additional three patients in the A1 cohort who had <24 weeks’ follow-up and had discontinued treatment prior to the data cut-off date (each with a best response of not evaluable). The median follow-up time was 16.3 months among the A1 efficacy-evaluable patients and 11.5 months among the A2 efficacy-evaluable patients, based on the reverse Kaplan-Meier method. Safety analyses were performed in all patients who received ≥1 dose of dostarlimab.
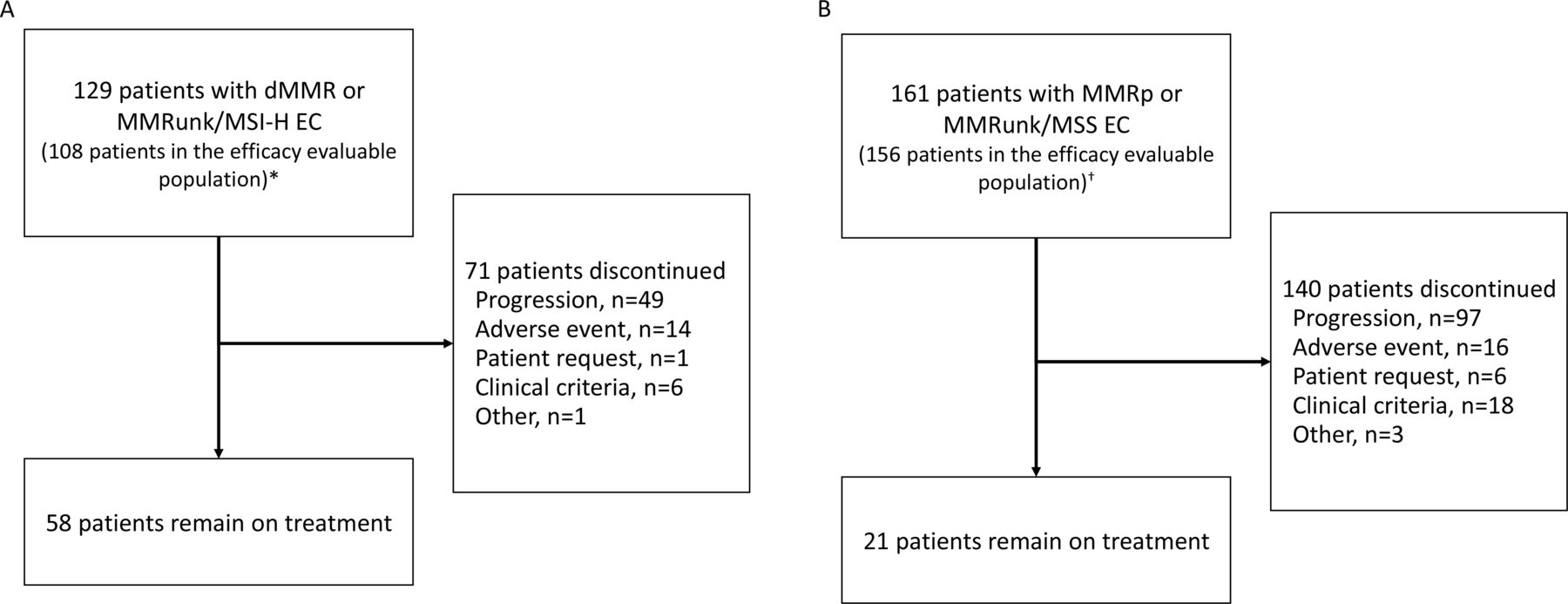
Enrollment and outcomes. *Twenty-one patients had no measurable disease by BICR at baseline (n=9) or had insufficient follow-up time (<6 months, n=12) and were excluded from this interim analysis efficacy-evaluable population; three patients with <6 months of follow-up time who had discontinued treatment (each with a best response of not evaluable) were included in the efficacy-evaluable population. †Sixteen patients had no measurable disease by BICR at baseline or had insufficient follow-up time (<6 months) and were excluded from this interim analysis efficacy-evaluable population. BICR, blinded independent central review; dMMR, mismatch repair deficient; EC, endometrial cancer; MMR, mismatch repair proficient; MMRun, mismatch repair unknown; MSI-H, microsatellite instability-high; MSS, microsatellite stable.
The median age of patients was 64.5 years in the dMMR/MSI-H cohort and 64.5 years in the MMRp/MSS cohort (table 1). The majority of patients with dMMR/MSI-H EC (65.7%; 71/108) had low-grade (grade 1 or 2) endometrioid carcinoma at primary diagnosis. Patients with MMRp/MSS EC demonstrated more variation in histologic subtype, and high-grade tumors were common, particularly serous histologic subtype (37.8%; 59/156).
Demographics and baseline characteristics
Antitumor activity
The ORR per RECIST v1.1 was 43.5% (95% CI 34.0% to 53.4%) in patients with dMMR/MSI-H, with 11 (10.2%) confirmed complete responses and 36 (33.3%) confirmed partial responses. The ORR was 14.1% (95% CI 0.1% to 20.6%) in patients with MMRp/MSS EC, with 3 (1.9%) confirmed complete responses and 19 (12.2%) confirmed partial responses (table 2). The median time to response was 11.9 weeks in the patients with dMMR EC and 12.1 weeks in the patients with MMRp EC. Among the responders, the median DOR was not reached in either cohort: 42 (89.4% of responders) patients with dMMR/MSI-H EC and 14 (63.6% of responders) patients with MMRp/MSS EC remain in response as of the data cut-off date. The DCR was 55.6% (95% CI 45.7% to 65.1%) in the patients with dMMR/MSI-H EC and 34.6% (95% CI 27.2% to 42.6%) in the patients with MMRp/MSS EC.
Antitumor activity
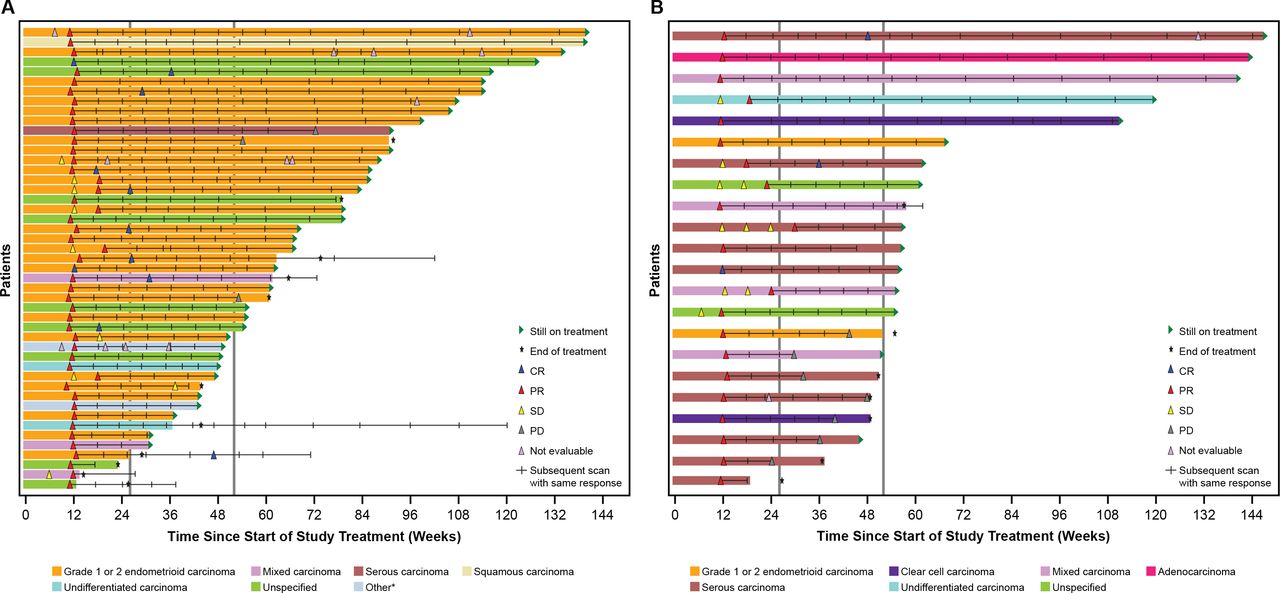
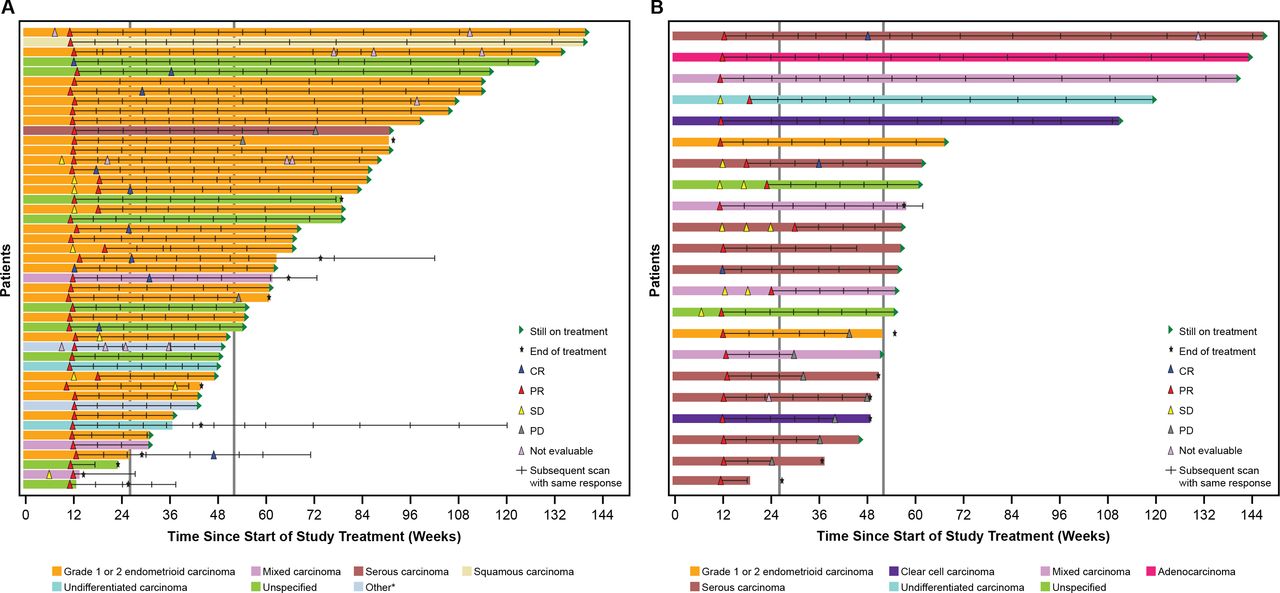
In both cohorts, responses were seen across histologic subtypes. The greatest percent change in target lesion size relative to baseline by histologic subtype is shown in figures 2 and 3. Similarly, DOR among responders showed consistency across histological subtypes (figures 2 and 3).
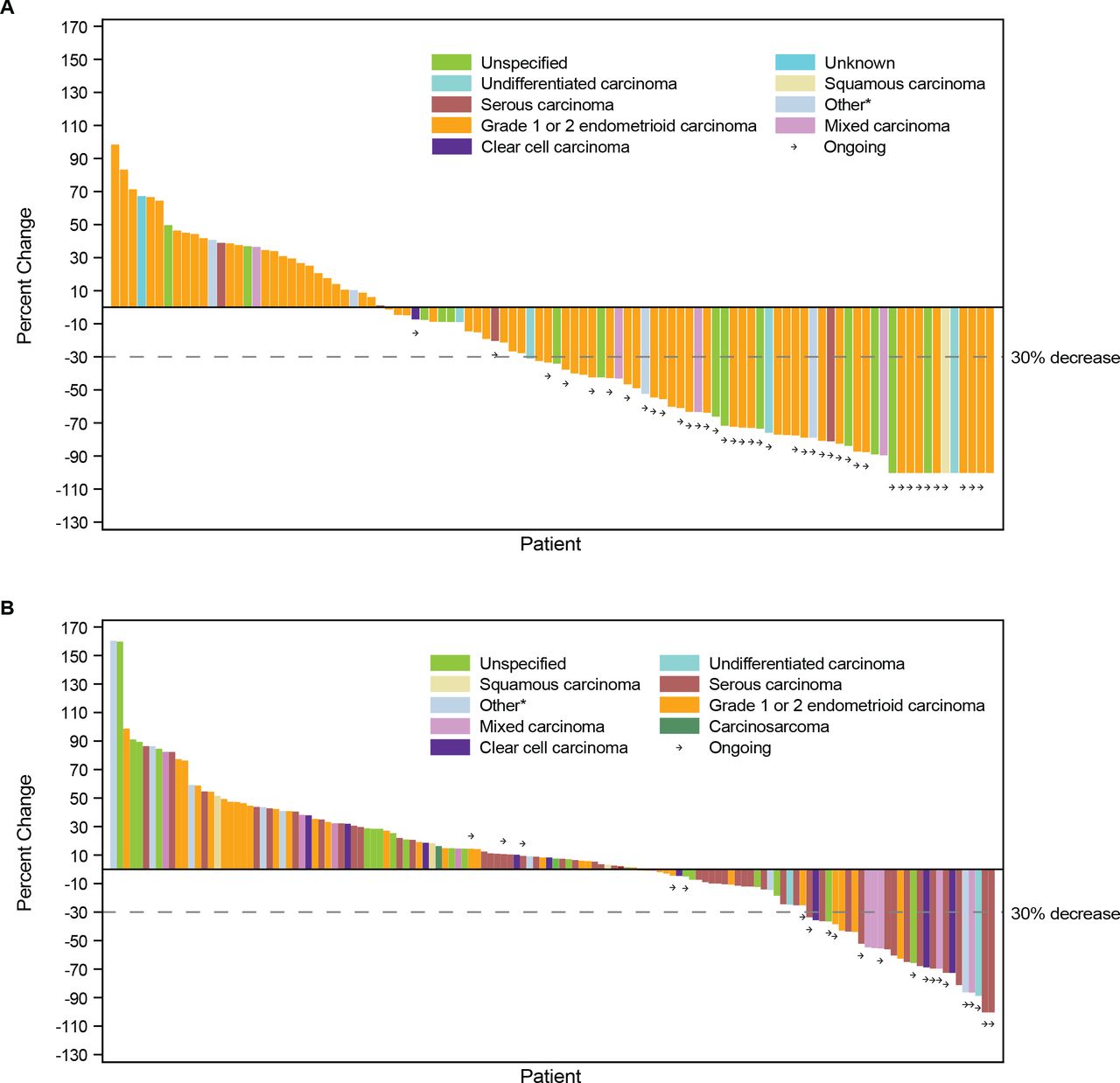
Best percent change from baseline in target lesion size. CR, complete response; dMMR, mismatch repair deficient; EC, endometrial cancer; MSI-H, microsatellite instability-high; PD, progressive disease; PR, partial response; SD, stable disease. *Includes adenocarcinoma and adenocarcinoma with ambiguous differentiation
{kind=link}
{kind=link}
{kind=link}
Duration of response among responders. Graphs show time since treatment initiation, and scans prior to the first response (SD or NE scans) are shown. The first response is shown, and hash marks indicate follow-up scans with the same response. In cases where a patient converted from a PR to a CR, the first PR is shown, with hash marks indicating further scans with a PR, then the first CR scan is shown, and subsequent hash mark(s) indicate follow-up scans with a result of CR. CR, complete response; EC, endometrial cancer; MMRp, mismatch repair proficient; MSS, microsatellite stable; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease. *Includes adenocarcinoma and adenocarcinoma with ambiguous differentiation
The key secondary endpoints—irORR, irDCR, and irDOR by investigator assessment—showed antitumor activity consistent with the assessments by BICR per RECIST v1.1 (online supplemental table 1).
Supplemental material
At the time of data cut-off, progression-free survival (PFS) data for the efficacy populations (n=108 and n=156) were immature, with 47.2% and 17.9% of the patients’ data being censored.
For the subset of 72 patients with dMMR/MSI-H EC who had a minimum of ≥13.5 months of follow-up (median follow-up was 19.2 months), median PFS was 12.2 months. The median overall survival had not been reached in this same population.
Exploratory subgroup analyses by number of prior lines of therapy, tumor mutational burden, PD-L1 status, and MMR protein loss (dMMR cohort only) status can be found in online supplemental figure 1. Across subgroups in each cohort, response rates were generally similar to the overall response for that cohort. The ORR of 45.5% (95% CI 76.6% to 16.7%) in TMB-H MMRp/MSS EC is interesting, but it represents only a small number of patients within the cohort (5 responders among 11 patients with TMB-H status). POLε exonuclease mutations were detected in 3 patients with dMMR/MSI-H EC (2 responders) and 2 patients with MMRp/MSS EC (0 responders).
Safety
The safety profile of dostarlimab was consistent with prior reports, with most TRAEs being grade 1 or 2 (table 3). Treatment-related adverse events (TRAEs) were consistent between the dMMR/MSI-H and MMRp/MSS cohorts. Accordingly, these patients are presented as a combined group (N=290). For the combined patient population, the most common any-grade TRAEs were fatigue (17.6%), diarrhea (13.8%), and nausea (13.8%).
Safety
The most common grade ≥3 TRAEs were anemia (2.8%), alanine aminotransferase (ALT) increased (1.4%), diarrhea (1.4%), fatigue (1.4%), amylase increased (1.4%), and lipase increased (1.4%) for the combined patient population. Immune-related TRAEs are shown in online supplemental table 2.
There were 16 (5.5%) discontinuations due to TRAEs; the most common TRAEs leading to discontinuation were ALT increase (1.0%), aspartate transaminase (AST) increase (0.7%), and transaminase increase (0.7%). The protocol mandates discontinuation of dostarlimab when a grade ≥3 AST or ALT increase is observed. Further data on discontinuations due to TRAEs are shown in online supplemental table 3. Grade ≥3 TRAEs that occurred in ≥2 (0.5%) patients by grade (combined A1+A2 cohorts, N=290) are shown in online supplemental table 4; the majority of grade ≥3 TRAEs were grade 3.
No deaths were attributable to dostarlimab.
The safety profile in EC cohorts was consistent with the safety profile seen across other tumor types in GARNET. Pooled TEAE data for the 515 patients enrolled and dosed in GARNET part 2B cohorts are shown in online supplemental table 5.
Discussion
Dostarlimab demonstrated durable antitumor activity in patients with dMMR/MSI-H EC and patients with MMRp/MSS EC. The ORR in dMMR/MSI-H EC was higher than in MMRp/MSS EC. This finding was consistent with the known characteristics of dMMR/MSI tumors, whose mutations are associated with increased tumor neoantigen load, tumor-infiltrating lymphocytes, and increased PD-1 and PD-L1 expression, which can stimulate responses to immune checkpoint inhibition.8 Among responders (43.5% in patients with dMMR/MSI-H EC and 14.1% in patients with MMRp/MSS EC), durable responses were seen in both dMMR/MSI-H and MMRp/MSS EC; 89.4% of responders with dMMR/MSI-H EC and 63.6% of responders with MMRp/MSS EC remain in response as of the data cut-off date. The safety profile was manageable, consistent with prior experience, and similar to that of other anti–PD-1 antibodies.
To our knowledge, these cohorts represent the largest populations of patients with dMMR/MSI-H and MMRp/MSS EC treated with an anti–PD-1 antibody monotherapy reported for other anti–PD-1 antibodies in solid tumors, including studies in EC of pembrolizumab,9 10 avelumab,11 and durvalumab,12 13 all with smaller sample sizes.
The results for patients with dMMR/MSI-H EC (cohort A1) from an earlier interim data cut have been previously published and were restricted to patients with confirmed dMMR status.14 The updated data presented here provide an additional 8 months of follow-up on the initial patients and increase the number of patients with available data. Conclusions on antitumor activity and safety are consistent with that report.
The ORR was lower in patients with MMRp/MSS EC than in patients with dMMR/MSI-H EC, but the responses were durable, and the DCR of 34.6% seen in MMRp/MSS EC suggests an encouraging clinical benefit from dostarlimab.
During the last decades, treatment options in the advanced/recurrent EC setting have included single-agent chemotherapy (paclitaxel or doxorubicin); re-treatment with platinum-based chemotherapy, single-agent bevacizumab; and hormonal therapies, each with limited benefit.15 For patients with advanced/recurrent dMMR/MSI-H EC, additional treatments are available. In the USA, pembrolizumab and dostarlimab monotherapy are both approved. In the EU, dostarlimab is the only anti–PD-1 agent approved for patients with dMMR/MSI-H EC who have failed platinum therapy. Lenvatinib and pembrolizumab combination therapy is currently approved for patients with EC who are not dMMR/MSI-H in the USA, Canada, and Australia. While antitumor activity of this combination is compelling, the safety profile is challenging, with a 66.9% incidence of grade ≥3 TRAEs and a 17.7% discontinuation rate of one or both drugs.16
Treatment for patients with advanced/recurrent EC MMRp/MSS remains an area of high unmet need.
Limitations
The limitations include independent enrollment of the two EC cohorts, which prevented time coordination in patients being assigned to the A1 or A2 cohorts. Robust translational analyses including PD-L1 expression, tumor mutational burden, and other biomarkers are limited to exploratory post hoc analyses. The study did not assess whether MMR testing by IHC, or MSI testing by PCR or NGS was superior for identifying responders; however, the results do demonstrate that MMR testing by IHC is predictive of response to dostarlimab.
In addition, the data are immature as this is an interim analysis. GARNET is a single-arm trial of dostarlimab and was not designed to assess superiority or equivalence with other therapies. Although responses were seen across EC histologies, the study is not powered to assess response rate by histologic subtype.
An ongoing randomized phase III trial of dostarlimab in combination with chemotherapy versus standard-of-care chemotherapy in advanced or recurrent EC is enrolling (RUBY; NCT03981796); patients with either dMMR/MSI-H or MMRp/MSS EC are eligible.
Conclusion
The presented cohorts are the largest prospective evaluation of a PD-(L)1 monotherapy in EC to date. Dostarlimab demonstrated durable antitumor activity in both dMMR/MSI-H and MMRp/MSS advanced/recurrent EC. Consistent with previous reports, confirmed dMMR status by IHC or MSI status by PCR or NGS was associated with a higher response rate.17 In EC, IHC analysis for MLH1, PMS2, MSH2, and MSH6 protein expression on paraffin-embedded tumor samples is established as the preferred testing method because of its low cost and wide availability.18
Dostarlimab demonstrated a notable DCR in patients with MMRp/MSS EC, a cohort that has more patients with high-grade ECs, a characteristic associated with a worse prognosis. Further classification of the MMRp responders is ongoing and may provide useful insights on the patients who responded to dostarlimab. No new safety signals were detected, and safety profile was consistent among patients with dMMR/MSI-H and MMRp/MSS EC. Only 5.5% of patients discontinued dostarlimab because of a TRAE, and no treatment-related deaths were reported.
Supplemental material
Data availability statement
Data are available on reasonable request. Anonymized individual participant data and study documents can be requested for further research (www.clinicalstudydatarequest.com).
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and the ethics committee at each investigational site approved the protocol. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
This study (NCT02715284) was sponsored by GlaxoSmithKline (Waltham, MA). Writing and editorial support, funded by GlaxoSmithKline (Waltham, MA, USA) and coordinated by Heather Ostendorff-Bach, PhD, of GlaxoSmithKline, were provided by Nicole Renner, PhD, and Jennifer Robertson, PhD, of Ashfield MedComms, an Ashfield Health company (Middletown, CT, USA). Trademarks are owned by or licensed to the GSK group of companies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors EI and WG, with input from the clinical investigators (authors and investigators acknowledged in the supplemental appendix), designed the trial. AO, LG, AT, JB, CM, JP, RS, DMO'M, VS, VB, LD, SG, PG, RK, CL, BP, and investigators acknowledged in the supplemental appendix collected the data. WG, TD, and XH completed the statistical analysis. JV and SZ provided logistical management of the trial. All authors interpreted the data, contributed to the writing, and provided final approval to submit for publication. AO and JV are responsible for the overall content as guarantors.
Competing interests AO reports consulting fees from AstraZeneca, Bristol Meyers Squibb, Deciphera Pharmaceutical, Genmab, GlaxoSmithKline, ImmunoGen, Mersana Therapeutics, SUTRA, and Roche; institutional grants from Abbie Deutschland, Ability Pharmaceuticals, Advaxis Inc, Aeterna Zentaris, Amgen SA, Aprea Therapeutics AB, Clovis Oncology Inc, Eisai Ltd, F. Hoffmann-La Roche Ltd, GlaxoSmithKline, ImmunoGen Inc, and Merck Sharp & Dohme de Espana SA, Millennium Pharmaceuticals Inc, PharmaMar, and Regeneron Pharmaceuticals, and travel support from AstraZeneca, Clovis Oncology, PharmaMar, and Roche. LG reports institutional grants from AstraZeneca, Pfizer, and Merck Sharp & Dohme, Karyopharm, Tesaro, IMV, Alkermes, Clovis, ImmunoGen Inc, Roche, Mersana, Esperas, Novocure GmbH, OncoQuest Pharmaceuticals; consulting fees from Merck; honoraria from AstraZeneca, GlaxoSmithKline, Eisai, Eisai-Merck, and Alkermes. AVT reports institutional grants from AstraZeneca and personal fees from AstraZeneca and Eisai. JB reports honoraria from Olympus; consulting or advisory role at Caris, GlaxoSmithKline, Clovis, AstraZeneca, and Genentech; and speakers’ bureau at Clovis. CM reports institutional grants from GlaxoSmithKline. JP has nothing to disclose. RS reports institutional grants from AstraZeneca and Eisai; personal fees from AstraZeneca, GlaxoSmithKline, Novartis, Pfizer, and Roche; and nonfinancial support from Amgen, AstraZeneca, Pfizer, and Roche. DMO'M reports personal fees from Agenus, Array Biopharma, Eisai, GlaxoSmithKline, and ImmunoGen; consultant/advisory board for Abbvie, Ambry, Amgen, Clovis, EMD Serono, Ergomed, Janssen/J&J, Myriad Genetics, Novacure, Regeneron, Tarveda, and VentiRx; steering committee for Genentech/Roche and Merck; institutional funding from Ajinomoto Inc, Bristol Myers Squibb, Cerulean Pharma, GOG Foundation, INC Research Inc, Inventiv Health Clinical, Iovance Biotherapeutics Inc, Ludwig Cancer Research, New Mexico Cancer Care Alliance, PRA International, Serono Inc, Stemcentrx Inc, Tracon Pharmaceuticals, and Yale University. VS has nothing to disclose. VB reports consulting or advisory roles at Guidepoint Global and OncoArt; speakers’ bureau fees from Solti; travel support from START; honoraria from Loxo and IDEAYA Biosciences; and institutional grants from Abbvie, Adaptimmune, Alkermes, Amgen, Array BioPharma, AstraZeneca, Bayer, BioNTech AG, Boehringer Ingelheim, Boston Biomedical, Bristol Myers Squibb, CytomX Therapeutics, Genmab, GlaxoSmithKline, Incyte, Janssen Oncology, Kura Oncology, Lilly, Loxo, Menarini, Merck, Merus, Novartis, Pfizer, Pumo Biotechnology, Roche/Genentech, Sanofi, Seattle Genetics, Synthon, and Zenith Epigenetics. LD reports personal fees from Advance Medical, ASCO, AstraZeneca, British Journal of OB/GYN, ClearView Health Care, Cue Biopharma, Elsevier, Genentech/Roche, Innovio, JB Learning, Merck, MorphoTek, National Cancer Institute, Parexel, State of California, and UpToDate; and institutional grants from Abbvie, Advaxis, Aduro BioTech, Cerulean/NextGen, Eisai, Genentech/Roche, GlaxoSmithKline, Inovio, LEAP Therapeutics, Ludwig, Lycera, Morab, Morphotek, Merck, Novartis, Pfizer, and Syndax. SG reports consulting or advisory role from Seattle Genetics; speakers’ bureau fees from GlaxoSmithKline; and institutional research funding from Abbvie, Advaxis, Bristol Myers Squibb, Clovis, Genentech, GlaxoSmithKline, Merck, Roche, Seattle Genetics, and Takeda. PG has nothing to disclose. RK reports personal fees from GlaxoSmithKline. CL reports contracted research for GlaxoSmithKline and scientific advisory board fees from GlaxoSmithKline, Eisai, Clovis Oncology, Seattle Genetics, and AbbVie. WG is a former employee of GlaxoSmithKline. EI is a former employee of GlaxoSmithKline. SZ, XH, TD, and JV are employees of GlaxoSmithKline. BP reports institutional grants support from Tesaro/GSK, AstraZeneca, Merck, Genentech/Roche, Celsion, Mersana, Karyopharm, and Clovis Oncology; and advisory board fees from Tesaro/GSK, AstraZeneca, Merck, Eisai, Toray, Mersana, Elevar Therapeutics, Arquer Diagnostics, Sutro Biopharma, and Clovis Oncology.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.