Article Text

Abstract
Background Pancreatic ductal adenocarcinoma (PDAC) is one of the most malignant cancers worldwide. Despite the promising outcome of immune checkpoint inhibitors and agonist antibody therapies in different malignancies, PDAC exhibits high resistance due to its immunosuppressive tumor microenvironment (TME). Ameliorating the TME is thus a rational strategy for PDAC therapy. The intratumoral application of oncolytic herpes simplex virus-1 (oHSV) upregulates pro-inflammatory macrophages and lymphocytes in TME, and enhances the responsiveness of PDAC to immunotherapy. However, the antitumor activity of oHSV remains to be maximized. The aim of this study is to investigate the effect of the CD40L armed oHSV on the tumor immune microenvironment, and ultimately prolong the survival of the PDAC mouse model.
Methods The membrane-bound form of murine CD40L was engineered into oHSV by CRISPR/Cas9-based gene editing. oHSV-CD40L induced cytopathic effect and immunogenic cell death were determined by microscopy and flow cytometry. The expression and function of oHSV-CD40L was assessed by reporter cell assay. The oHSV-CD40L was administrated intratumorally to the immune competent syngeneic PDAC mouse model, and the leukocytes in TME and tumor-draining lymph node were analyzed by multicolor flow cytometry. Intratumoral cytokines were determined by ELISA.
Results Intratumoral application of oHSV-CD40L efficiently restrained the tumor growth and prolonged the survival of the PDAC mouse model. In TME, oHSV-CD40L-treated tumor accommodated more maturated dendritic cells (DCs), which in turn activated T helper 1 and cytotoxic CD8+ T cells in an interferon-γ-dependent and interleukin-12-dependent manner. In contrast, the regulatory T cells were significantly reduced in TME by oHSV-CD40L treatment. Repeated dosing and combinational therapy extended the lifespan of PDAC mice.
Conclusion CD40L-armed oncolytic therapy endues TME with increased DCs maturation and DC-dependent activation of cytotoxic T cells, and significantly prolongs the survival of the model mice. This study may lead to the understanding and development of oHSV-CD40L as a therapy for PDAC in synergy with immune checkpoint blockade.
- oncolytic viruses
- tumor microenvironment
- dendritic cells
- lymphocytes
- tumor-infiltrating
- cytotoxicity
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the dominant type of pancreatic neoplasm, with a current 5-year survival rate of only 10%.1 Despite the success of checkpoint inhibition immunotherapies, such as cytotoxic T-lymphocyte-associated antigen 4 or programmed cell death-1 (PD-1) blockade, poor responses are observed in PDAC due to its immunosuppressive tumor microenvironment (TME).2 Hence, it is highly demanded to develop an effective approach to convert the immunosuppressiveness of PDAC TME.
Oncolytic viruses provide a promising strategy that involves multiple mechanisms in one therapy.3 4 Herpes simplex virus-1 (HSV-1) is one of the viral vectors that has been developed into oncolytic viral therapeutics, especially for neurological diseases, such as glioblastoma.5 In recent years, HSV-1-derived viruses have been attempted to a wider range of solid tumors. For example, talimogene laherparepvec (T-VEC), a granulocyte-macrophage colony-stimulating factor (GM-CSF)-expressing HSV-1, showed clinical efficacy for solid tumors, such as metastatic melanoma and PDAC.4 6 7 Previous studies revealed that TME in PDAC is altered by the treatment of oncolytic herpes simplex virus-1 (oHSV) to become more immunoactive in respect of the transcription profile in an immune competent mouse model.8 Prospectively, the concomitant presence of immunomodulators renders higher efficacy of oHSV.
CD40-CD40L signaling pathway is recognized as an attractive target in the immunotherapy for PDAC.9 10 CD40, a member of the tumor necrosis factor receptor superfamily, is expressed in a variety of cell types, such as dendritic cells (DCs), B cells, T cells, monocytes as well as PDAC cells. Ligation of CD40 stimulates antigen-presenting and T-cell-mediated responses.11–13 The STING (stimulator of interferon (IFN) genes protein) agonist and CD40 agonist agents have been studied in patients with PDAC and showed encouraging result in early clinical trials.14 However, the moderate tumor regression was observed with CD40 agonists due to the lack of immune reactivity of DC in TME.15 We hypothesized that oHSV armed with CD40 ligand (CD40L) evoke innate response, and enhance DCs maturation and activation, which in turn activate T cells.
Herein, we constructed an HSV-1-derived CD40L-encoding oncolytic virus (oHSV-CD40L). With the immune competent syngeneic PDAC mouse model, we demonstrated that oHSV-CD40L effectively restrained tumor progression and prolonged the survival of tumor-bearing mice. In vivo analysis of the oHSV-CD40L-treated TME presented increased tumor infiltrating immune cells with elevated maturated DCs, activated T helper (Th) 1, and cytotoxic CD8+ T cells. This study addressed the immunological effects of oHSV-CD40L on TME, and may lead to the development of oncolytic therapeutics to reboot antitumor immunity and overcome tumor resistance to immune checkpoint inhibitors (ICIs).
Materials and methods
Cell lines
Vero cells from ATCC (VA, USA), HEK293FT-sgGFP cell line, and the murine pancreatic cancer Krasem4(LSL-G12D) Trp53em4(R172H) Pdx1em1(Avi-CreERT2) (KPC) cell line from Shanghai Model Organisms Center (Shanghai, China) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). HEK293FT-sgGFP cell line was stably transduced by lentiviruses with two lentiCRISPR-sgRNA plasmids targeting the GFP-coding sequence. The human HVEM overexpressing murine PDAC cell line, Pan02_HVEM, was originally established by our laboratory,8 and this cell line was maintained in RPMI-1640 containing 10% (v/v) FBS.
Viruses
oHSV was previously constructed on the backbone of wild-type HSV-1 (F strain), in which both copies of ICP34.5-coding sequences were replaced by the GFP gene, and the ICP47 gene was deleted.8 oHSV-CD40L was constructed from oHSV by CRISPR/Cas9-based gene editing. Donor DNA containing the coding sequence of murine CD40L extracellular domain (amino acids 120–260), 218 linker and PDGFR transmembrane domain and flanked with the homology arms of genomic regions near the ICP34.5-coding sequences, was subcloned in pCDH plasmid and transfected into HEK293FT-sgGFP cells. The transfected cells were then infected with oHSV at a multiplicity of infection (MOI) of 0.1 and cultured in the presence of 10 μM SCR7 (HY-12742, MCE). At 48 hours post infection (hpi), the produced viral particles were collected to infect Vero cells, and the GFP-negative CD40L-positive cells stained with CD40L antibody (157004, BioLegend) were sorted by flow cytometry with BD FACSAria Fusion. Viruses were amplified from single plaque, and repeated for five rounds. The plaque purified viral particles were verified by next-generation sequencing analysis.
Virus titration
Vero cells were plated in six-well plates and infected with serial diluted viral samples. At 36 hpi, plaques per parallel well were counted in triplicates and the mean value was calculated. Virus titers were presented as plaque forming unit (pfu)/mL.
Mice and PDAC model
All animals in this study were male C57BL/6 mice aged 6 weeks obtained from Vital River Laboratories (Beijing, China). Pan02_HVEM cells (5×105) in RPMI-1640 or KPC cells (5×105) in DMEM mixed with Matrigel (356237, Corning) were subcutaneously inoculated in left flank or both flanks of mice as indicated, and the day set as day 0. For rechallenged experiment, the oHSV-CD40L cured mice that survived for 80 days after the initial tumor graft and the age-matched mice were subcutaneously rechallenged with 5×105 Pan02_HVEM cells. Tumor volumes were calculated according to the formula: V (mm3)=length×width2/2. When tumor volume exceeded 2000 mm3 or the animal was in a state of distress or pain, the mouse was sacrificed.
In vivo treatments
When inoculated tumor volumes reached ~50 mm3, tumor-bearing mice were randomized into control or treatment groups, and unilateral intratumorally injected with viral suspension in phosphate buffered saline (PBS) (5×106 pfu/mouse) or PBS along every third day. For combined therapy, CD40 antibody (100 µg/dose, BE0016-2, BioXCell), PD-1 antibody (100 µg/dose, BE0146, BioXCell) or isotype antibody (BE0089, BioXCell) was injected intraperitoneally on the day of viruses or PBS injection. For CD8+ T cells depletion and cytokines neutralization experiments, CD8α antibody (250 µg/dose, BE0004-1, BioXCell), interleukin (IL)-12 antibody (500 µg/dose, BE0233, BioXCell), IFN-γ antibody (250 µg/dose, BE0055, BioXCell), or indicated isotype antibodies (BE0088, BE0089, or BE0090, BioXCell) was injected intraperitoneally 24 hours prior to each viral treatment and administrated in 7-day intervals after the last oHSVs treatment, respectively.
Flow cytometry analysis
For in vitro assays, cells were digested with Accutase (07920, STEMCELL) and blocked with αCD16/32 antibody (101320, BioLegend). Then cells were stained with indicated fluorescence-conjugated antibodies in PBS supplemented with 2% (v/v) FBS. For in vivo studies, stripped tumor tissues or tumor-draining lymph nodes (tdLNs) were minced, prepared in dissociation solution (1 mg/mL collagenase I, 0.5 mg/mL DNase I, and 0.05 mg/mL dispase Ⅱ dissolved in PBS), and resuspended in ACK lysis buffer (CS0001, Leagene). For multicolor flow cytometry analyses, samples were first stained with Viability dye (423106, BioLegend), blocked with αCD16/32 antibody, and then incubated with indicated fluorescence-conjugated antibodies including against mouse CD45 (103108, 103131, or 103126, BioLegend), CD3ε (100328, BioLegend), CD4 (100428 or 100430, BioLegend), CD8 (100752, BioLegend), CD25 (102036, BioLegend), CD69 (104514, BioLegend), Gr-1 (108428, BioLegend), CD11c (117334, BioLegend), CD40 (562847, BD; 157506, BioLegend), CD80 (104734, BioLegend), CD86 (105036, BioLegend), major histocompatibility complex (MHC)-I (116517, BioLegend), MHC-Ⅱ (107614, BioLegend), programmed death-ligand 1 (PD-L1) (124312, BioLegend), and human HVEM (318805, BioLegend). For intracellular and nuclear staining, samples were fixed and permeabilized by the True-Nuclear Transcription Factor Buffer Set (424401, BioLegend), and then proteins were stained with FOXP3 (320014, BioLegend), T-bet (644803, BioLegend), GATA3 (653813, BioLegend), IFN-γ (505846, BioLegend), IL-4 (504133, BioLegend), and granzyme B (GZMB) (372208, BioLegend). Single-stained samples were performed by BD FACS Calibur Ⅱ and multicolor samples were analyzed by flow cytometry with BD LSRFortessa X-20, respectively. Data were calculated by FlowJo X software.
Single cell RNA sequencing data analysis
Gene-cell count matrix files for 16 PDAC tissue samples were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155698.16 The count matrices were generated by Cell Ranger V.3.0.0 and imported into the R package Seurat V.3.2.3 for downstream analysis referred to the code from https://githubcom/PascaDiMagliano-Lab/MultimodalMappingPDA-scRNASeq. Data were normalized using the NormalizeData function with default parameters. Variable genes were identified using the FindVariableFeatures function. Data were scaled by regressing out the counts. Principal component analysis was performed by the RunPCA function using the previously defined variable genes. The R package Harmony V.1.0 was used to remove batch effects and integrate all the tissue samples data. Cell clusters were identified using the FindNeighbors and FindClusters functions, with a resolution of 1.2, and uniform manifold approximation and projection clustering algorithms were performed. One of the clusters was defined as the DC cluster based on the markers: HLA-DRA, LYZ, IRF7, and ITGAX.16 The expression levels of 22 genes by each cell within the DC cluster were visualized by the DoHeatmap function and the average gene expression by each tissue sample was calculated using the AverageExpression function. The heatmap was generated by the R package pheatmap V.1.0.12 (https://cran.r-project.org/web/packages/pheatmap/index.html), and clustering rows (genes) by the ward algorithm.
CD40L function assay
The reporter cell line Jurkat/NF-κB-GFP-CD40 was established as described previously.17 Briefly, Jurkat cell line was stably transfected with plasmids containing NF-κB-GFP reporter and full-length CD40-coding sequences. Pan02_HVEM cells were mock-infected or infected with HSV-1 or oHSV-CD40L at MOI=1. At 24 hpi, cells were co-cultured with Jurkat/NF-κB-GFP-CD40 cells at the ratio of 1:1 for 24 hours. The expression of GFP was determined by flow cytometry.
Cytopathic effect and immunogenic cell death analysis
Pan02_HVEM cells were mock-infected or infected with indicated viruses at MOI=1 for determination of immunogenic cell death (ICD) or MOI=5 for induction of cytopathic effect (CPE). At 24 hpi, cells were observed and photographed by microscopy with camera (Ts2, Nikon) setting for at least three fields. For ICD assay, cells were stained with anticalreticulin (CRT) antibody (bs-5913R-AF647, Bioss), and subjected to flow cytometry analysis. The culture media were collected to determine ATP concentration by the Enhanced ATP Assay Kit (S0027, Beyotime) following the manufacturer’s instruction.
Quantitative RT-PCR analysis
Total RNA was extracted from cells using NucleoZOL (740404, MN), and complementary DNA (cDNA) was synthesized using a GoScript Reverse Transcriptase Kit (A2790, Promega) according to the manufacturer’s instruction. cDNA samples were mixed with AugeGreen qPCR Master Mix (S2008, UE) and amplified with specific primers for IFN-β (5’-AATTTCTCCAGCACTGGGTG-3’ and 5’-AGTTGAGGACATTCCCACG-3’), and β-actin (5’-CATTGCTGACAGGATGCAGAAGG-3’ and 5’-TGCTGGAAGGTGGACAGTGAGG-3’). Real-time PCR was performed by the Real-time PCR Detection System (CFX96, Bio-Rad). The β-actin gene was used as the reference gene to normalize the expression level between samples, and samples were calculated using the cycle threshold (ΔΔCT) method.
DC maturation analysis
Bone marrow cells were collected from the tibia and femur of mouse, and erythrocytes were removed with the ACK lysis buffer. Progenitor cells were plated in RPMI-1640 supplemented with 10% (v/v) FBS, 20 ng/mL murine GM-CSF (315-03, Peprotech), and 20 ng/mL murine IL-4 (241-14, Peprotech), and fed with fresh medium every other day. On day 8, induced bone marrow-derived DCs (BM-DCs) were separated by CD11c Microbeads (130-125-835, Miltenyi Biotec). BM-DCs were co-cultured with mock or viruses-infected Pan02_HVEM cells at the ratio of 1:1 for 12 hours, then stained with indicated fluorescence-conjugated antibodies for flow cytometry analysis.
For DC functionality assay, cells were isolated from mouse spleen with mouse lymphocyte separation medium (7211011, DAKEWE), and mixed with the co-cultured BM-DCs and tumor cells at the equal ratio as mentioned above for 12 hours. T cell activation was monitored by flow cytometry using the markers as indicated.
Quantification of cytokines
For in vivo assays, tumor tissues were minced, homogenized, and subjected to beads heating in PBS with protease inhibitor cocktail (K1007, APExBIO) on day 3 or day 6 after completion of treatment. For in vitro tests, culture media were collected by centrifugation. Quantification of cytokines in tumors and media was performed by Mouse IL-12 ELISA Kit (M1270, R&D) and Mouse IFN-γ ELISA Kit (E-EL-M0048c, Elabscience) following the manufacturer’s instruction, respectively.
Cytotoxicity assay
Tumor infiltrating CD8+ T cells were separated by CD8a+ T Cell Isolation Kit (130-104-075, Miltenyi Biotec) following the manufacture’s instruction, and co-cultured with Pan02_HVEM cells prestained with 6 mM carboxyfluorescein diacetate succinimidyl ester (CFSE, 565082, BD) at the ratio of 10:1. After 24 hours, cells were stained with 7-aminoactinomycin D (7-AAD, 51-68981E, BD) and analyzed by flow cytometry.
Statistical analysis
Statistical analyses were performed by GraphPad Prism V.7.0.0 software. Differences between groups were analyzed by two-tailed Student’s t-test. Data for mice survival were presented using Kaplan-Meier survival curves and log-rank test was performed to determine statistical significance between treatment groups. Value of p<0.05 was considered to be statistically significant.
Results
CD40 agonist combined with oHSV in suppression of PDAC
We first examined gene expression patterns of DCs from a publicly available single cell RNA sequencing (scRNA-seq) data of 16 PDAC tissue samples (online supplemental figure S1A).16 Considerable variability of DC function markers was observed across individual patients (figure 1A and online supplemental figure S1B). The expression levels of human leukocyte antigen (HLA) class I molecules (HLA-A, HLA-B, and HLA-C) and DC maturation markers (CD80, CD83, and CD86) were downregulated in some patients. It is noteworthy that IL12A was not detectable in the DC cluster from all patients (data not shown), which is considered to associate with poor T cell priming as revealed by the scRNA-seq data.16 Other common markers, such as IDO1 and LGALS1, which contribute to the generation of regulatory T cells (Tregs),18 19 were upregulated in 5 and 8 out of 16 PDAC samples, respectively. It was also shown that CD274 (known as PD-L1) was significantly elevated in half of the patients. Overall, the scRNA-seq analysis suggested impaired DC response in PDAC TME.
Supplemental material

Heatmap of gene expression and combinational therapy of PDAC model. (A) Heatmap of the average expression level of 22 selected genes in DC cluster from 16 PDAC tissue samples. Gene expression level was indicated by a color gradient, where red indicated high expression and blue indicated low expression. (B) Experimental regimen of combination therapy of oHSV and CD40 agonist antibody on tumor-bearing mice. Mice received subcutaneous inoculation of Pan02_HVEM cells in flank on day 0. On days 8, 11, and 14, mice were injected intratumorally with PBS or oHSV, and intraperitoneally with isotype antibody or CD40 antibody, respectively. (C) Tumor volumes were measured every third day (n=5). Arrows indicate the time points of treatment. Two-tailed Student’s t-test was performed on day 23. (D) The survival of tumor-bearing mice was plotted using Kaplan-Meier analysis and log-rank test (n=5). *p<0.05; **p<0.01. DC, dendritic cell; oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline; PDAC, pancreatic ductal adenocarcinoma.
Semi-maturation of DCs, with deficient expression of DC markers, has been reported in PDAC patients and mouse model.20 21 CD40-CD40L ligation is considered a key event to trigger the activation of DC.22 Among 16 patients with PDAC, 7 samples displayed increased expression of CD40 in their DCs (figure 1A). Accordingly, we set out the combination of oHSV and CD40 agonist antibody to treat the PDAC mouse model, which was inoculated with Pan02_HVEM cells as described previously (figure 1B).8 As shown in figure 1C and D, the combination therapy exhibited more tumor regression and longer survival than either monotherapy, suggesting that the activation of CD40 signaling coordinate with oHSV in the treatment of pancreatic cancer.
Oncolytic effects of oHSV-CD40L
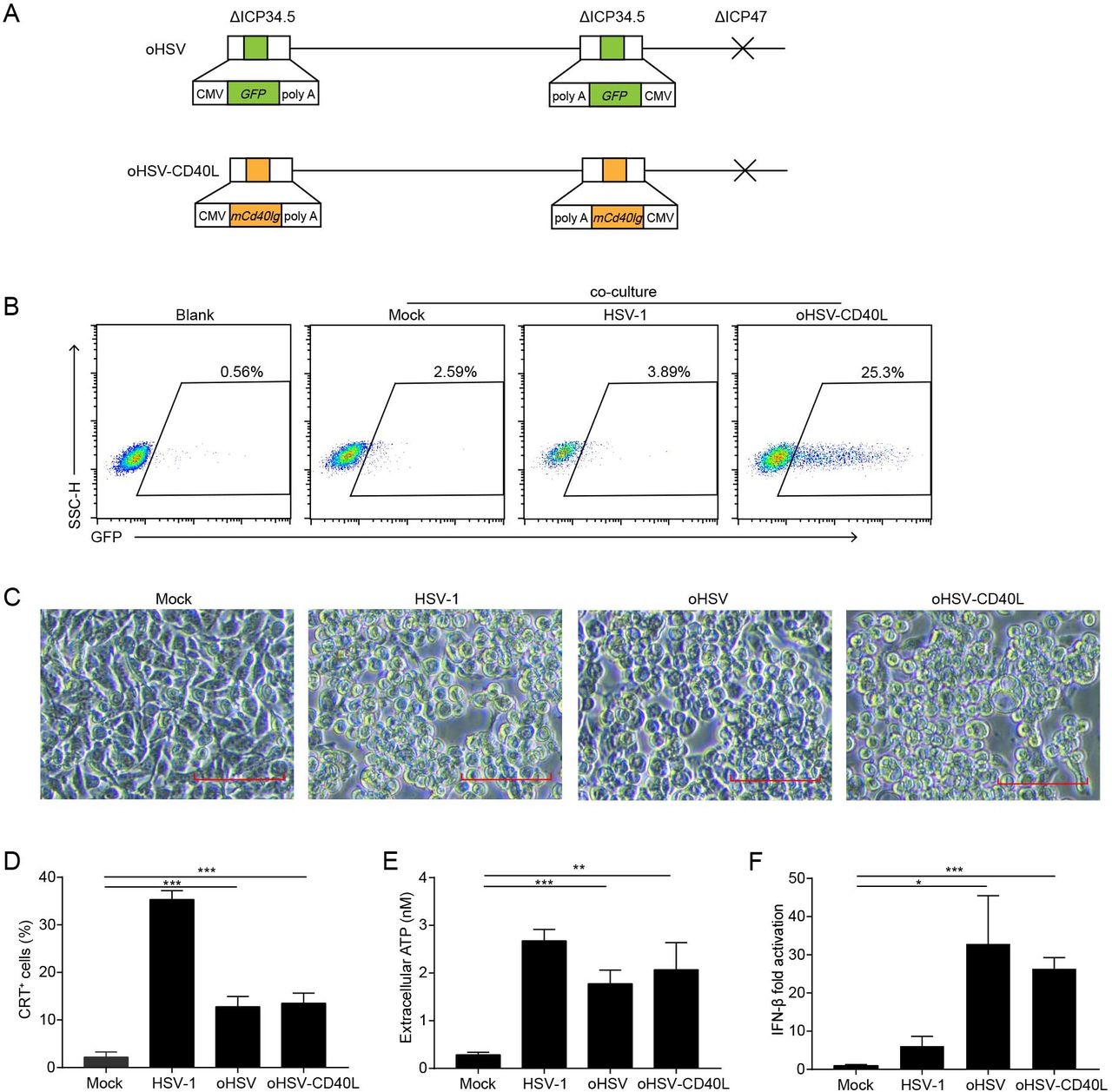
Despite the efficacy of CD40 agonist antibody, toxicity such as liver toxicity, cytokine release syndrome, or lymphopenia associated with CD40 agonist frequently occurs in clinical trials.23 24 While the soluble CD40L promotes the activity of myeloid-derived suppressor cells in patients with cancer,25 the transmembrane form is expected to enhance its stimulatory effects intratumorally.26 Herein, we modified oHSV to express the transmembrane form of CD40L, named oHSV-CD40L. As shown in figure 2A, the parental oHSV was constructed previously with replacement of ICP34.5 by the GFP gene and deletion of ICP47.8 For oHSV-CD40L, both loci of GFP in oHSV were substituted by sequences encoding the transmembrane murine CD40L. To examine the function of oHSV-CD40L carried CD40L, we established a reporter cell line Jurkat/NF-κB-GFP-CD40.17 CD40-CD40L signaling pathway was activated by the oHSV-CD40L-infected tumor cells with 25.3% reporter cells shown GFP-positive, in comparison with merely 2.59% by the mock-infected tumor cells (figure 2B).
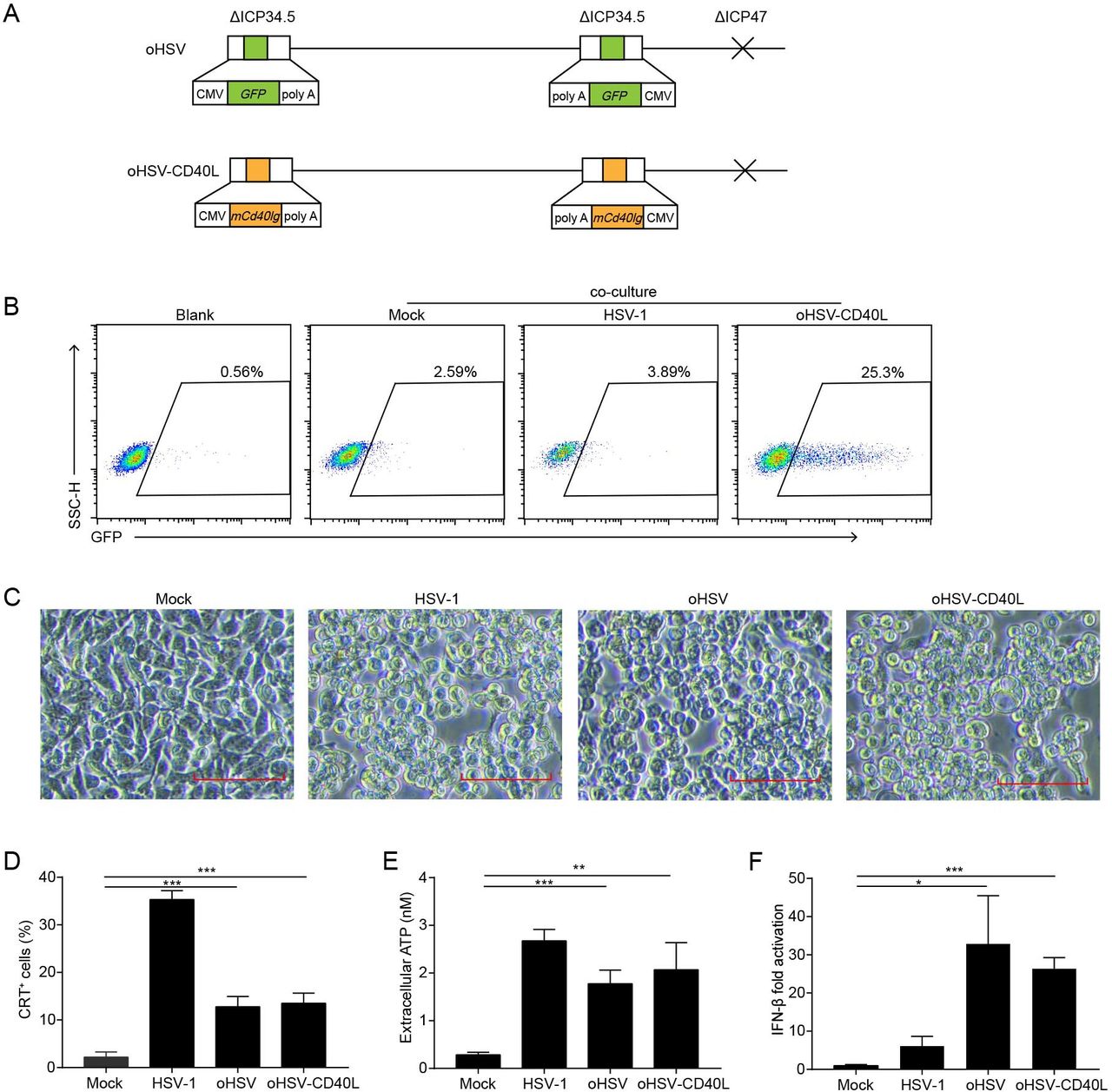
The construction and cytolytic effects of oHSV-CD40L. (A) Schematic representations of oHSV and oHSV-CD40L genomes. For oHSV (upper row), both ICP34.5 loci were replaced with GFP-coding sequences, and the ICP47 gene region was deleted. oHSV-CD40L (lower row) was derived from oHSV by replacing the GFP sites with murine CD40L-encoding sequence. (B) Pan02_HVEM cells were mock-infected or infected with HSV-1 or oHSV-CD40L, and co-cultured with Jurkat/NF-κB-GFP-CD40 reporter cells as described in the ‘Materials and methods’ section. These cells were then subjected to flow cytometry analysis. The histograms were shown with fluorescence intensity of GFP as the horizontal axis. The reporter cells that were not co-cultured with Pan02_HVEM cells were designated as blank. The percentage of GFP-positive cells was labeled inside. (C) Pan02_HVEM cells were mock-infected or infected with wild-type HSV-1, oHSV, or oHSV-CD40L (MOI=5), respectively, and the cytopathic effects were monitored under microscopy at 24 hpi. Scale bar denotes 100 μm. (D) Pan02_HVEM cells were mock-infected or infected with wild-type HSV-1, oHSV, or oHSV-CD40L (MOI=1) for 24 hours, respectively. Cells were then stained with calreticulin (CRT) antibody for flow cytometry and the percentages of CRT+ cells were plotted in columns. The media were harvested, and the levels of released ATP were determined and plotted in E. (F) Total mRNA was isolated from cells infected as indicated, and IFN-β mRNA was quantitated by real-time quantitative PCR and plotted as a ratio relative to that of β-actin. Data were representative of three independent experiments. All values were presented as mean±SD. Statistics were performed using two-tailed Student’s t-test (D–F). *p<0.05; **p<0.01; ***p<0.001. hpi, hours post infection; IFN, interferon; MOI, multiplicity of infection; oHSV, oncolytic herpes simplex virus-1.
The oncolysis effect of oHSV, oHSV-CD40L, and unmodified wild-type HSV-1 was assessed in vitro. As illustrated in figure 2C, oHSV-CD40L-infected cells showed similar CPE as that of HSV-1 and oHSV. The induction of ICD was also examined by the surface CRT and the release of ATP. Although much lower than wild-type HSV-1, the level of surface expressed CRT was 6.9-fold and 8.4-fold elevated in oHSV-infected and oHSV-CD40L-infected cells, respectively comparing with the mock group (figure 2D). The release of ATP also showed 5.3-fold to 6.4-fold higher in the oHSV-infected and oHSV-CD40L-infected cells than the mock group (figure 2E). Thus, the infection of oncolytic viruses induced the ICD of PDAC cells. On the other hand, both oHSV and oHSV-CD40L infection stimulated the production of IFN-β significantly higher than the wild-type HSV-1 (figure 2F).
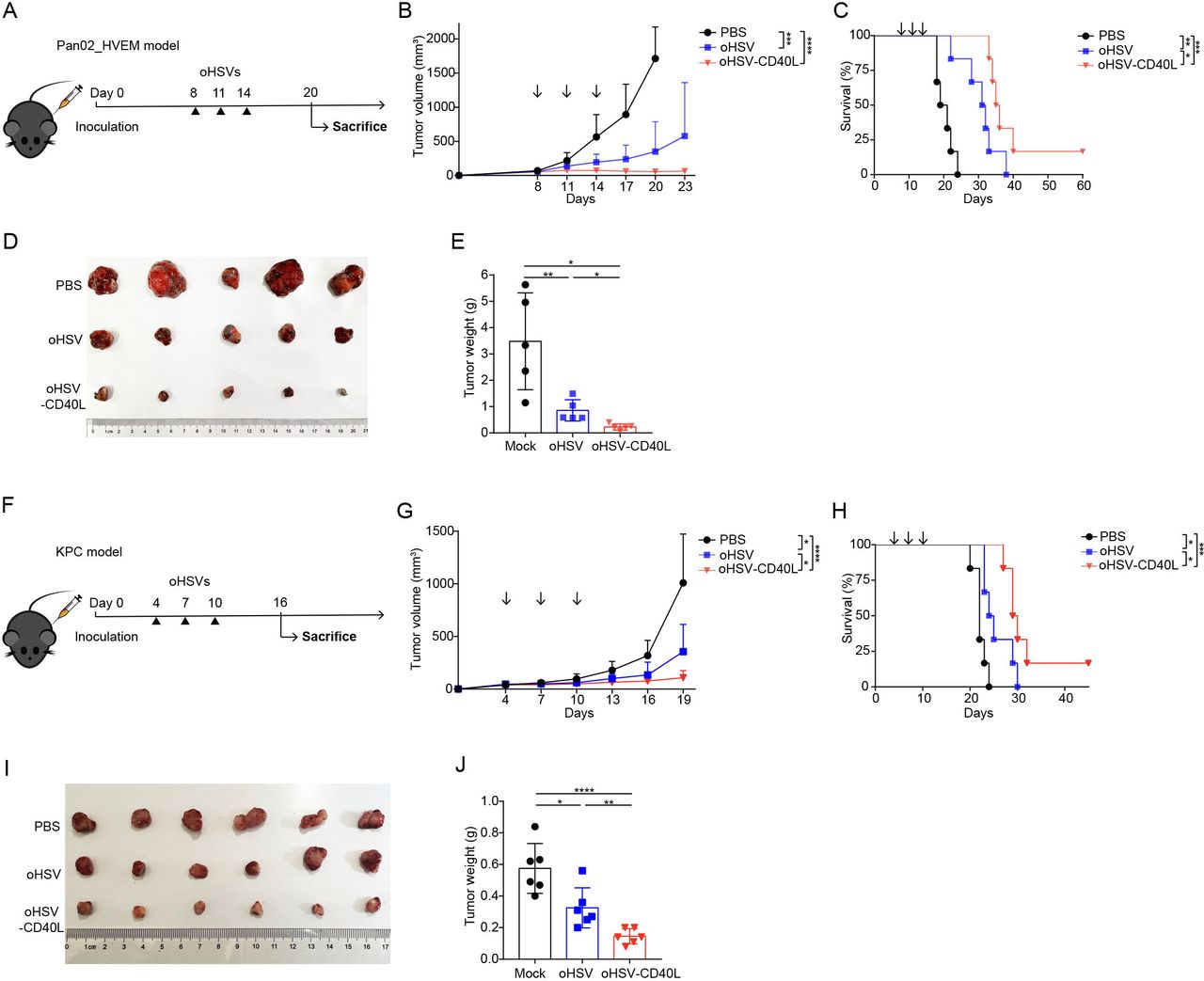
To evaluate the antitumor efficacy of oHSV-CD40L, we applied oHSVs (oHSV or oHSV-CD40L) to two different PDAC mouse models (figure 3A–E,F–J). oHSV-CD40L treatment significantly inhibited the progression of tumors and prolonged the median survival time of mice by 4 and 5 days compared with the oHSV groups, respectively (figure 3C and H). The individual measures were also presented in online supplemental figure S2. The results from both models suggested that oHSV-CD40L can be developed as a potential therapy for PDAC.

Antitumor effect of oHSV-CD40L. (A) Experimental layout of oHSVs therapy in Pan02_HVEM model. Mice were inoculated with Pan02_HVEM cells on day 0. The treatments were performed on days 8, 11, and 14 (arrows) with PBS, oHSV, or oHSV-CD40L, respectively. (B) Tumor volumes were measured every third day and plotted as the vertical axis (n=6). (C) The survival of Pan02_HVEM-bearing mice was plotted using Kaplan-Meier analysis and log-rank test (n=6). (D) On day 20, mice were executed and Pan02_HVEM tumors were isolated from individual mice and pictured with the treatments labeled on the left. The weight of each tumor sample was presented in E (n=5). (F) Experimental layout of oHSVs therapy in subcutaneous KPC model. The treatments were performed on day 4, 7, and 10 with PBS, oHSV, or oHSV-CD40L, respectively. (G) Tumor volumes were monitored and plotted as the vertical axis (n=6). (H) The survival of KPC-bearing mice was subjected to Kaplan-Meier analysis and log-rank test (n=6). (I) On day 16, KPC tumors were isolated from individual mice and pictured. The weight of each tumor sample was presented in J (n=6). Data were presented as mean+SD (B and G) or ±SD (E and J). Two-tailed Student’s t-test was performed in B (on day 20), E, G (on day 19), and J. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline.
oHSV-CD40L increased tumor infiltration of immune cells and stimulated the maturation of DCs
We collected the tumor infiltrating immune cells on day 20 and analyzed the cells by multicolor flow cytometry with the gating strategy shown in online supplemental figure S3. As shown in figure 4A, tumor infiltrating leukocytes (CD45+ cells) were presented the highest in oHSV-CD40L-treated tumors (30.92%) among the three regimens. In such cell group, the percentages of CD11c+ DCs were moderately elevated in the oHSV-CD40L group (figure 4B), whereas T cells (marked by CD3+) in oHSV-CD40L samples were 1.7-fold to 2.6-fold of that seen in the oHSV and mock groups (figure 4C). This proportion suggested that the intratumoral application of oHSV-CD40L increased T cells recruitment to TME, although the amount of DCs remained comparable across the sample groups. Whether the oncolytic virus treatments resulted in more active DCs?

The maturation of DCs in tumors and lymph nodes. (A–C) Cell suspensions from treated Pan02_HVEM tumors were analyzed by flow cytometry after stained with indicated fluorescence-conjugated antibodies. The tumor infiltrating immune cells were presented as the percentages of CD45+ cells in total viable cells (A). DCs (B) and T cells (C) were expressed as the percentages of CD11c+ and CD3+ in CD45+ cells, respectively (n=5). (D–G) Pan02_HVEM cells were mock-infected (black bars), or infected with oHSV (blue bars) or oHSV-CD40L (red bars) at MOI=1. At 12 hpi, bone marrow-derived DCs (BM-DCs) were added and co-cultured for 12 hours. The expression of CD40 (D), CD80 (E), CD86 (F), and MHC-II (G) was analyzed by flow cytometry and presented as the percentages of CD11c+ cells, respectively. (H) The concentration of IL-12 in above co-culture media was quantitated by ELISA and plotted in columns. (I–L) The percentages of CD40+ (I), CD80+ (J), CD86+ (K), and MHC-II+ (L) in CD11c+ cells from treated tumors (on day 20) were plotted in scatter graphs (n=5). (M–P) Cell suspensions from tumor-draining lymph node (tdLN) of treated mice were analyzed by flow cytometry after stained with indicated fluorescence-conjugated antibodies on day 20. The DCs among tdLNs were defined as CD11c+ in CD45+ cells (M), and the percentages of CD40+ (N), CD80+ (O), and CD86+ (P) in CD11c+ cells were plotted in scatter graphs (n=5). (Q) Mice were sacrificed on day 3 or day 6 after completion of treatment. The concentration of IL-12 in the tumors was determined by ELISA (n=3). (R) Mice-bearing Pan02_HVEM tumors received PBS or oHSV-CD40L treatment, and IL-12 was blocked using anti-IL-12 antibody before each virus treatment. The survival of treated mice was plotted using Kaplan-Meier analysis and log-rank test (n=5). Other values were presented as mean±SD and statistics were performed using two-tailed Student’s t-test. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. DC, dendritic cell; IL, interleukin; MHC, major histocompatibility complex; oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline.
CD40 engagement stimulates the maturation and antigen presentation of DC that triggers the adaptive immunity against cancers.27 To this measure, we first examined the effect of oHSV-CD40L on DC maturation in vitro. The immature BM-DCs were incubated with oHSV-CD40L-infected Pan02_HVEM cells and the increased expression of several markers of mature DCs was detected. As shown in figure 4D–G, while a slight increase of CD86 and MHC-II was seen in the oHSV group (blue bars), oHSV-CD40L infection rendered significantly higher expression of CD40, CD80, CD86, and MHC-II (red bars). Besides the secretion of IL-12 was much higher in the DCs co-cultured with oHSV-CD40L-infected tumor cells than that of control or oHSV-infected cells (figure 4H).
Next, we performed in vivo assays to evaluate the maturation of DCs isolated from tumor tissues as well as lymph nodes. First, the proportion of CD40+ DCs was remarkably higher in DCs from the oHSV-CD40L-treated tumors (figure 4I). In contrast, the co-stimulators CD80 and CD86 were slightly higher in the samples of oHSV-CD40L treatment than that of oHSV (figure 4J and K). MHC-II expression was significantly enhanced by both oHSVs (figure 4L). We also observed a minimally increased DC populations in tdLNs (figure 4M), likely due to the migration of stimulated intratumoral DCs into the lymph nodes. DC maturation markers, CD40, CD80, and CD86 were consistently expressed at a relatively higher level in samples from oHSV-CD40L-treated mice (69.06%–79.06%, figure 4N–P), whereas the mean values, though scattered, of control or oHSV-treated mice appeared lower than oHSV-CD40L-treated mice.
Furthermore, the intratumoral Th1 cytokine, IL-12, was detectable on day 3 and dramatically increased on day 6 after completion of oHSV-CD40L therapy, while there was no difference of IL-12 in the oHSV group relative to the PBS group (figure 4Q). Depletion of IL-12 by neutralizing antibody dampened the efficacy of oHSV-CD40L (figure 4R), indicating that the antitumor effect is mediated by IL-12.
Taken together, the above data strongly suggested that oHSV-CD40L treatment promote the maturation of DCs in tumors and tdLNs.
oHSV-CD40L promoted Th1 differentiation and enhanced cytolytic T cell activity
The maturation of DCs provides sufficient signal molecules for the priming and activation of T cells.12 22 Similar to agonist CD40 mAb, mice treated with oHSV-CD40L elevated the expression of MHC-II and co-stimulatory molecules of DCs (figure 4I–L). In addition, oHSV manipulates the polarization of tumor-associated macrophages towards pro-inflammatory phenotype with antigen presentation ability.8 Therefore, we hypothesized that the infection of tumor cells by oHSV-CD40L potentiate T cell activation and Th1 shift differentiation. In vitro test was performed by adding splenocytes into the co-culture of BM-DCs and the PDAC cells pre-infected by oHSV or oHSV-CD40L, respectively. The activation of CD4+ and CD8+ T cells was indicated by the expression of CD69 (figure 5A and B) and the production of IFN-γ (figure 5C). As shown, the infection of tumor cells by oHSV-CD40L stimulated more T cells than those by mock or oHSV. Consistently, in vivo assay showed that the intratumoral IFN-γ in oHSV-CD40L-treated mice was 2.7-fold of the oHSV group, and 4.7-fold of the mock group on day 6 after the last treatment (figure 5D).
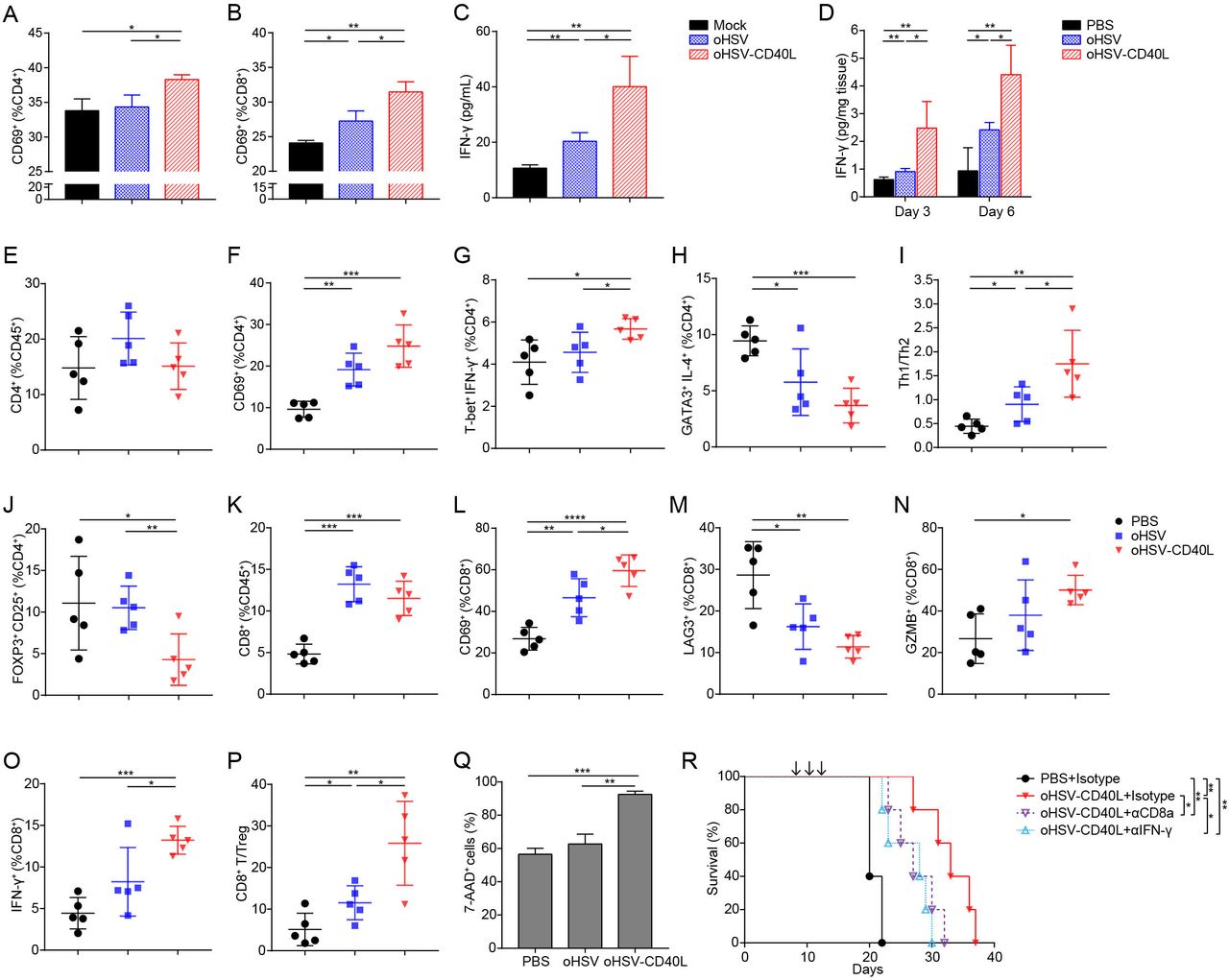
Effects of oHSV-CD40L treatment on T cell response. (A, B) Pan02_HVEM cells infected with different viruses were co-cultured with BM-DCs followed by adding lymphocytes isolated from spleen of mouse to the co-cultures. The activated CD4+ (A) and CD8+ (B) T cells were determined by the expression of CD69 after 12 hours of incubation and presented as percentages in columns. The secretion of IFN-γ was determined by ELISA and plotted in C. (D) Pan02_HVEM-bearing mice were sacrificed on day 3 or day 6 after completion of treatment. The concentration of IFN-γ in the tumors was determined by ELISA (n=4). (E–P) Cell suspensions from treated Pan02_HVEM tumors (on day 20) were collected and indicated cell proportion was labeled with fluorescence-conjugated antibodies (E–H and J–O). The ratios of Th1 to Th2 cells were presented in I, and the ratios of CD8+ T cell to Treg were shown in P. The percentages of the indicated group of cells or the ratios of indicated cells subsets were plotted in the scatter graphs (n=5). (Q) CD8+ T cells isolated from tumors of mice that had been treated by PBS, oHSV, or oHSV-CD40L three times. Pan02_HVEM cells were prestained with CFSE and then co-cultured with CD8+ T cells. After 24 hours, cells were stained with 7-AAD and the percentages of Pan02_HVEM cell death were assessed by flow cytometry. (R) Mice bearing Pan02_HVEM tumors received PBS or oHSV-CD40L treatment. To detect the role of CTLs and IFN-γ in antitumor efficacy of oHSV-CD40L, CD8+ T cells were depleted by anti-CD8 antibody, or IFN-γ was blocked using anti-IFN-γ antibody before each treatment. The survival of treated mice was plotted using Kaplan-Meier analysis and log-rank test (n=5). Other values were presented as mean±SD and statistics were performed using two-tailed Student’s t-test. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. 7-AAD, 7-aminoactinomycin D; BM, bone marrow; DC, dendritic cell; IFN, interferon; MHC, major histocompatibility complex; oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline; Th, T helper.
Since IFN-γ production associates with the activation of Th1 cells, we thus measured the proportion of intratumoral CD4+ T cells and their activation status under each indicated therapy. The total intratumoral CD4+ T cells were comparable (figure 5E), and the activated CD4+ T cells (CD4+ CD69+) in both oHSVs-treated groups were 9.52% to 15.19% more than the control (figure 5F). In regard to effector CD4+ T cells, the oHSV-CD40L treatment increased the populations of Th1 (T-bet+ IFN-γ+) (figure 5G), and reduced the proportion of Th2 (GATA3+ IL-4+) by 3.68%–5.76% (figure 5H), thus indicating a Th1 shift by the higher Th1/Th2 ratio compared with the oHSV and control group (figure 5I). Additionally, Tregs (FOXP3+ CD25+) were significantly reduced in oHSV-CD40L group (figure 5J).
The tumor infiltrating CD8+ T subset was elevated by the treatment of oHSV or oHSV-CD40L, respectively (figure 5K), implicating that the virotherapy recruit T cells to the tumor tissues or stimulate T cell proliferation. While comparing the active populations of the effector CD8+ T cells marked by CD8+ CD69+, the active cytotoxic T lymphocytes (CTLs) were elevated by the treatment of oHSV-CD40L, which were 1.3-fold that of oHSV-treated samples (figure 5L). On the other hand, the exhausted CD8+ T cells (CD8+ LAG3+) were much lower in mice receiving oHSV treatment, and further suppressed in oHSV-CD40L-treated tumors (figure 5M). In correlation with the functional CTLs, the expression of GZMB and IFN-γ was higher in oHSV-CD40L treatment than oHSV and placebo groups (figure 5N and O). Besides, the ratios of effector CD8+ T cells to Tregs were remarkably raised by the oHSV-CD40L treatment than that of oHSV (figure 5P). Together, both in vivo and in vitro assays demonstrated that oHSV-CD40L therapy induced a Th1 shift of CD4+ T cells, potentiated cytotoxic T cells, and reduced the proportion of Tregs.
Next, we determined the cytotoxic effects of CD8+ T cells isolated from the placebo (PBS), oHSV, and oHSV-CD40L-treated mice, respectively. When co-cultured with Pan02_HVEM cells, the isolated CD8+ T cells in the oHSV-CD40L group exhibited the most effective killing of tumor cells among these three groups (figure 5Q), suggesting that the oHSV-CD40L treatment induce strong cytotoxicity towards tumor cells. It is further confirmed by in vivo study in which the depletion of CD8+ T cells or neutralization of IFN-γ dramatically impeded the efficacy of oHSV-CD40L treatment (figure 5R), demonstrating an indispensable role of CD8+ T cells and IFN-γ in the mechanism of oHSV-CD40L therapy.
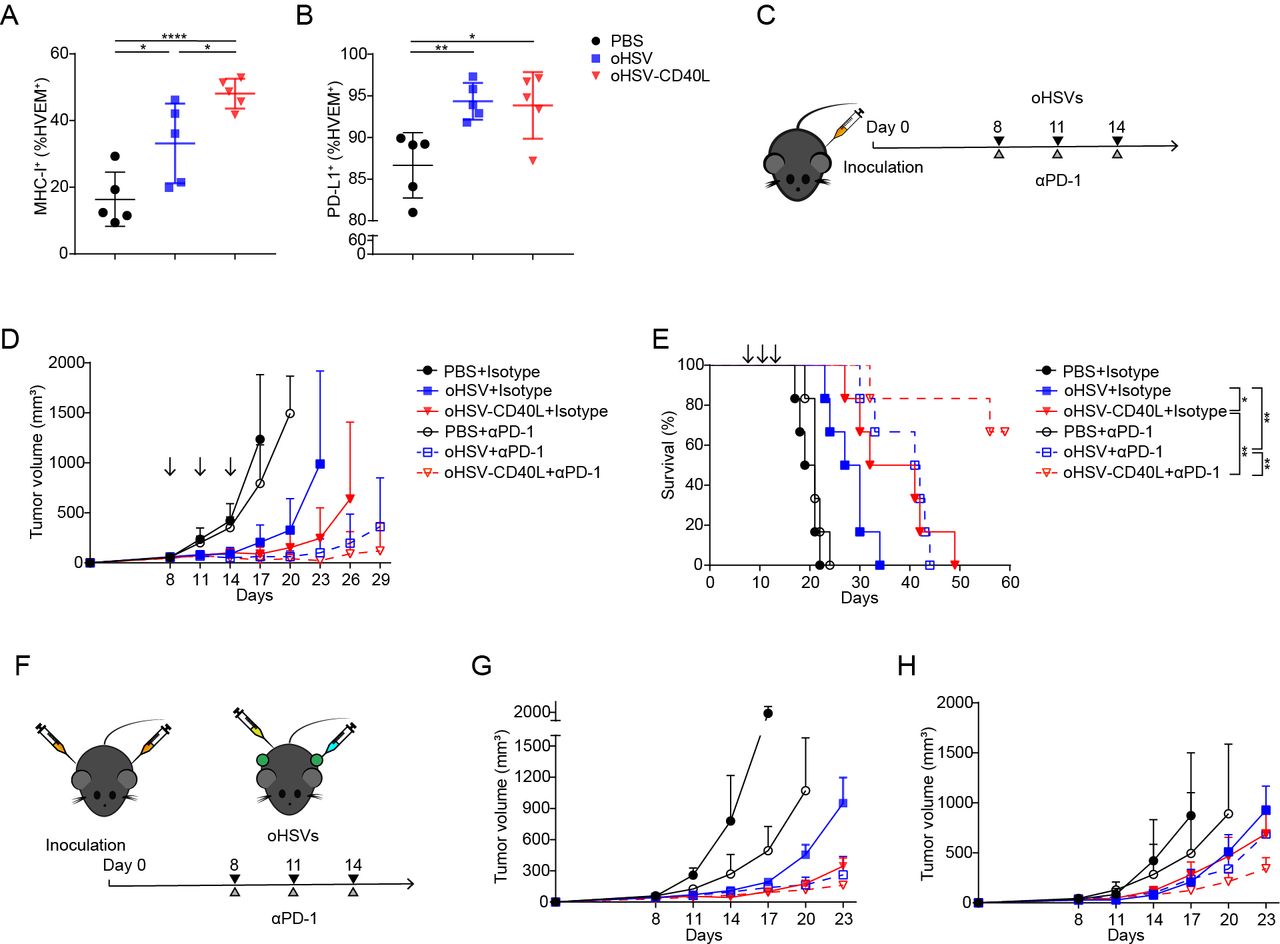
Together, the above data strongly suggested that oHSV-CD40L promote Th1 differentiation and cytolytic T cell function to eliminate tumor cells. Meanwhile, oHSV-CD40L treatment also upregulated the MHC-I expression of tumor cells (figure 6A), which facilitated antigen presentation and the activation of tumor-specific CTLs.

Combination therapy by oHSV-CD40L and PD-1 blockade. (A, B) Cell suspensions from treated Pan02_HVEM tumors were stained with indicated fluorescence-conjugated antibodies on day 20. The expression of MHC-I (A) and PD-L1 (B) on tumor cells was analyzed by flow cytometry and plotted in scatter graphs (n=5). Statistical analysis was performed by two-tailed Student’s t-test. (C) Scheme of combination therapy of oHSVs and PD-1 antibody. (D) After indicated treatments, tumor volumes were measured every third day and plotted as the vertical axis (n=6). (E) The survival of mice was subjected to Kaplan-Meier analysis and log-rank test (n=6). (F) Experimental layout of combination therapy of oHSVs and PD-1 antibody for the bilateral tumor mouse model. The growth of tumor in the treated flank (G) and the contralateral flank (H) was monitored and plotted with volumes in the vertical axis (n=4). Data were presented as mean±SD (A and B) or mean+SD (D, G, and H). *p<0.05; **p<0.01; ****p<0.0001. MHC, major histocompatibility complex; oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline; PD-1, programmed cell death-1; PD-L1, programmed death-ligand 1.
The combination of oHSV-CD40L and anti-PD-1 exhibited beneficial effect
PD-L1 expression was increased in Pan02_HVEM cells when treated by oHSVs (figure 6B), and that encouraged the strategy to combine oHSV-CD40L and PD-1 antagonist antibody. As described in figure 6C, the combination of oHSV-CD40L plus PD-1 antibody significantly inhibited tumor growth and extended the survival period of mice as compared with monotherapy by oHSV-CD40L or PD-1 antibody (figure 6D and E), suggesting a promising regimen to combine oHSV-CD40L and ICIs for PDAC treatment.
Furthermore, the bilateral model was tested for the effect of oHSV-CD40L. As shown in figure 6F, mice were inoculated at both sides, and tumor sizes were shrinking on oHSVs-treated flank (figure 6G) as well as contralateral flank (figure 6H). Particularly, the tumor growth was seen efficiently inhibited in contralateral by the oHSV-CD40L treatment with or without PD-1 antibody, suggesting that the oHSV-CD40L stimulate the systematic antitumor effects, which was supported by the more matured DCs in tdLNs (figure 4N–P). Besides, the above results may implicate a synergistic effect of oHSV-CD40L and PD-1 blockade.
Repeated dosing of oHSV-CD40L improved efficacy and long-term protection from tumor relapse
Though effective inhibition of tumor growth by oHSV-CD40L, the complete eradication of tumor cells is yet to be achieved. In our experiments so far, only 16.67% of treated mice showed tumor-free survival after three doses of oHSV-CD40L (figure 3C). Thereby, we implemented a repeated dosing strategy of oHSVs as depicted in figure 7A. As shown in figure 7B and C, repeated administration of virotherapy improved antitumor response and extended the overall lifetime of mice. Strikingly, four out of six mice were long survived after successive oHSV-CD40L treatments. These findings may promote the further investigation of the repeated dosing regimen of oHSVs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Repeated dosing therapy by oHSV-CD40L. (A) Repeated dosing schedule of oHSVs therapy. Mice were treated with oHSVs for 3 or 9 times in a row of every third day. Tumor volumes were monitored and plotted as the vertical axis in B, and the survival of tumor-bearing mice was monitored by Kaplan-Meier analysis in C. PBS, oHSV, or oHSV-CD40L×3 (n=5); oHSV or oHSV-CD40L×9 (n=6). (D) Scheme of rechallenged experiment. The oHSV-CD40L cured mice on the 80th day after the initial tumor graft or age-matched mice were inoculated with Pan02_HVEM cells subcutaneously (renamed as day 0). The survival of rechallenged mice (red) and age-matched mice (blue) was monitored by Kaplan-Meier analysis (n=5). Data were presented as mean+SD in B. Log-rank test on mice survival was performed in C and E. *p<0.05; **p<0.01; ***p<0.001. oHSV, oncolytic herpes simplex virus-1; PBS, phosphate buffered saline.
Furthermore, to test the long-term effect of oHSV-CD40L on the mouse model, we rechallenged those survived mice with Pan02_HVEM cells on the 80th day after the first tumor inoculation (figure 7D). Concurrently, age-matched naïve mice were implanted with an equivalent number of tumor cells as comparison. Excitingly, three out of five rechallenged mice showed complete rejection of tumor, and the other two mice displayed impeded tumor growth (figure 7E). This result led to our hypothesis that oHSV-CD40L therapy may potentially establish tumor-specific immune memory, which is considered to be critical to prevent tumor recurrence.
Discussion
Oncolytic viruses have been extensively developed as an alternative therapy for advanced malignancies.28 In recent years, several versions of oncolytic viruses have been designed with additional immune stimulation.29 For example, T-VEC, an engineered HSV-1, is composed of GM-CSF to promote the DC differentiation and has been proved effective and well-tolerated immunotherapy for patients with unresectable advanced melanoma.30 As of PDAC, an immunologically ‘cold’ tumor, valid strategies are to focus its immunosuppressive TME. Our previous study demonstrated that the basic oncolytic HSV, with the deletions of ICP34.5 and ICP47, increases the tumor infiltrating immune cells and reshapes the TME to be more inflammatory.8 Oncolytic virotherapies for PDAC are to be further improved by arming viruses with immunostimulatory molecules, including various cytokines or costimulatory modulators.
The promising results of T-VEC in several solid tumors are encouraging for the development of HSV-1-based oncolytic viruses. However, GM-CSF carried by T-VEC may potentially promote the proliferation of PDAC cells by inducing the immunosuppressive Gr-1+ CD11b+ cells, and impair the response of CD8+ T cells.31 Here, we reported a modified oHSV featured to deliver the immunostimulatory factor CD40L to trigger the responses mediated by the CD40-CD40L signaling axis. In recent years, CD40 pathway has been recognized as a promising target for cancer immunotherapy, and the agonistic CD40 antibody has been developed in early phase trials, particularly for PDAC.32 However, the systematic administration of CD40 agonist alone is not sufficient to provoke T cell-dependent immunity and it seems to inhibit tumor growth in T cell-independent manner by the degradation of stromal cells.23 Besides, the systematic administration of CD40 agonist antibody has been depreciated with the adverse effects,23 24 thus it is relevant to develop intratumoral administration approaches. Although adenovirus-carrying CD40L manifests to evoke tumor-specific immunity in some cancers including PDAC,33 34 the HSV-1 genome has the greater capacity to accommodate multiple genes,35 and HSV-1-derived oncolytic virus is the only approved oncolytic viral therapy by the US Food and Drug Administration. Herein, with CRISPR/Cas9 technology, we have designed oHSV-CD40L to treat PDAC, which is recognized as the immunological ‘cold’ tumor. Previous report described the modified oncolytic HSV-1 with different immune stimulators, including mCD40L, and the tumor growth was successfully hindered in the mCD40L-modified oHSV-treated glioma and lymphoma model.36 37 In support, our study here showed that oHSV-CD40L inhibited PDAC in vivo, and further demonstrated the increased CD8+ T cells and Th1 cytokines in tumor killing (figure 5R). Promisingly, the oHSV-CD40L cured mice presented enhanced overall survival when rechallenged by Pan02_HVEM cells (figure 7E).
The engagement of CD40 on DCs triggers the maturation of these cells with increased antigen-presenting, upregulation of co-stimulatory receptors and secretion of cytokines,22 and CD40L delivered by oHSV-CD40L enabled to directly stimulate these responses before the CD40L expressing on activated Th cells (figure 4D–H). Indeed, oHSV-CD40L treatment elevated the expression of markers for DC maturation (figure 4K and L). As noted, CD80 was rather lowered in oHSV-treated samples (figure 4E and J), perhaps by reduced transcription of CD80 as an immediate-early protein of HSV-1, ICP22, binds to CD80 promoter.38 Nevertheless, the knockout studies indicated that CD86 is more preferred for T cell activation, whereas CD80 is dispensable in maintaining T cell response.39 In this count, further design of oHSV may eliminate the binding region of ICP22 to CD80 promoter.
CD40-mediated signaling triggers DCs activation, and consequently induces the secretion of IL-12, which is known as an important cytokine in bridging the innate and adaptive immunity.12 40 Consistently, a remarkable increase of IL-12 and IFN-γ secretion was detected on oHSV-CD40L treatment (figures 4Q and 5D), in response to the activation of CTLs in PDAC. Although IL-12 has been recognized as an effective antitumor agent in preclinical studies,40 the clinical experiment was unsatisfactory as patients develop severe adverse reactions.41 In our study here, the IL-12 was elevated locally by the administration of oHSV-CD40L (figure 4H and Q), and plays the antitumor role in context of active DCs and T cells. As expected, neutralization of IL-12 renders reduced survival of PDAC mice (figure 4R). It may attribute to the systematic responses as seen in the bilateral tumor model (figure 6F–H).
The immunosuppressive feature of PDAC hindered the development of therapy. The previous report observed that the treatment of oHSV changes the immune character of TME by upregulating pro-inflammatory macrophages, as well as downregulating the immunosuppressive anti-inflammatory macrophages.8 Our study here highlighted changes in DC and T cell heterogeneity induced by oHSV-CD40L. As DC has been demonstrated to play a crucial role in initiating durable T cell response,42 oHSV-CD40L treatment leads to the maturation of DCs in TME, in consequence, contributes to (1) increase in Th1/Th2 (figure 5I) and (2) potent and less exhausted CTLs (figure 5L–Q). These results are supported by a line of evidence from adenovirus-carrying CD40L studies, which demonstrated an enhancement of antigen-presenting cells.43 Besides, our previous scRNA-seq analyses of oHSV therapy revealed highly expressed transcripts in correlation with proliferating CD8+ T cells, reflecting potent cytotoxicity in TME of PDAC upon oHSV treatment.8
In summary, we engineered oncolytic HSV-1 armed with murine CD40L and applied it to the syngeneic PDAC mouse model. As the replicating viral therapeutic, oHSV-CD40L exacerbated ICD of tumor cells. The expression of the armed CD40L increased the infiltration of immune cells and repolarized the TME toward a less immunosuppressive phenotype, leading to effective antitumor immune responses. We demonstrated that oHSV-CD40L therapy enhanced DC maturation in TME, promoted Th1 differentiation, and stimulated CTL activation. Furthermore, repeated dosing of viral therapy prolonged the tumor-free survival of the PDAC mice. These findings provided confidence in the clinical testing of oHSV-CD40L in combination with ICI against the PD-1/PD-L1 pathway for the treatment of PDAC.
Supplemental material
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Institute Research Ethics Committee of Nankai University, Tianjin, China (2021-SYDWLL-000125).
Acknowledgments
The authors would like to thank the center facility of Nankai University and animal facility for their timely support during the pandemic.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors YC, HZ, and CZ conceived and planned the experiments. HZ and RW designed animal experiments. CZ and RW planned and managed HSV-1 manipulation. RW constructed oHSVs. JC and ZZ assisted with the viral preparation and characterization. RW, JC, HW, SL, FL, and YL performed the cellular and animal experiments. YW, JY, and RW helped on flow cytometry. WW performed bioinformatic analyses. YC, HZ, CZ, and RW validated and interpreted the results. YC, HZ, and CZ supervised and acquired funding. YC primarily edited and revised the manuscript, and is responsible for the overall content as guarantor. All authors provided critical feedback and comments on the manuscript.
Funding This work was supported by the National Natural Science Foundation of China (grant numbers 81661148051 and 81672010 to YC, and 32171468 to CZ), the Shenzhen Fundamental Research Program (grant number JCYJ20210324121814039 to YC), the Fundamental Research Funds for the Central Universities Nankai University (grant number 63213070 to HZ), and the Key Laboratory of Immune Microenvironment and Disease Open Funding (grant number 20180102 to HZ).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.