Article Text

Abstract
Diffuse midline glioma is the leading cause of solid cancer-related deaths in children with very limited treatment options. A majority of the tumors carry a point mutation in the histone 3 variant (H3.3) creating a potential HLA-A*02:01 binding epitope (H3.3K27M26-35). Here, we isolated an H3.3K27M-specific T cell receptor (TCR) from transgenic mice expressing a diverse human TCR repertoire. Despite a high functional avidity of H3.3K27M-specific T cells, we were not able to achieve recognition of cells naturally expressing the H3.3K27M mutation, even when overexpressed as a transgene. Similar results were obtained with T cells expressing the published TCR 1H5 against the same epitope. CRISPR/Cas9 editing was used to exclude interference by endogenous TCRs in donor T cells. Overall, our data provide strong evidence that the H3.3K27M mutation is not a suitable target for cancer immunotherapy, most likely due to insufficient epitope processing and/or amount to be recognized by HLA-A*02:01 restricted CD8+ T cells.
- Immunotherapy
- Antigens, Neoplasm
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Diffuse midline glioma (DMG) is a devastating disease affecting primarily children and is the leading cause of solid cancer-related deaths in children, with a median survival of less than 1 year after diagnosis. Due to the proximity of the tumors to the brainstem, surgical removal is challenging and radiotherapy remains the only available treatment option.1
A point mutation in the histone H3 gene (H3F3A) causing a lysine to methionine amino acid exchange (K27M) leads to a reduced histone-3 trimethylation (H3K27me3) pattern and is found in the vast majority of DMG, including over 75% of diffuse intrinsic pontine glioma (DIPG) and 60% of non-brainstem midline tumors in the thalamus and spinal cord. The mutation is located at the N-terminus of the histone, a region which is highly conserved and crucial for the regulation and accessibility of chromatin.2 It has been reported that H3.3K27M peptides inhibit the enzymatic activity of the histone methyltransferase Polycomb repressive complex 23 and K27M mutated histones impede methylation at their cognate residues leading to aberrant epigenetic silencing. Tumors harboring the H3.3K27M mutation show a particularly aggressive progression.4
Cancer immunotherapy has developed into a promising approach for cancer therapies in the last decades. Adoptive T cell therapy (ATT) in particular, as in the case of using CAR T cells against the B cell antigen CD19, achieved remarkable regressions in leukemia and lymphoma patients.5 Translation of this concept to solid tumors has proven to be difficult, a major hurdle being the identification of suitable tumor-associated auto-antigens as T cell targets that mediate effective tumor-killing without causing dose-limiting pathology in normal somatic tissues.6 However, identification of truly cancer-specific antigens such as point-mutation-derived epitopes with good binding affinities to a frequent HLA molecule might prevent on-target toxicity and thus improve patient outcome.7 The H3.3K27M mutation has been reported to create an epitope (H3.3K27M26-35, RMSAPSTGGV) that binds HLA-A*02:01 and is recognized by a high-affinity T cell receptor (TCR) obtained from mutant peptide-stimulated HLA-A*02:01-positive donor PBMCs.8 In addition, this epitope is currently under investigation for peptide vaccination in an ongoing clinical trial for HLA-A*02:01 and H3.3K27M positive patients with DMG.9
Here, we discuss the identification of an H3.3K27M mutation-specific TCR after peptide immunization of transgenic mice expressing a diverse human TCR repertoire and human HLA-A2 transgene.10 We show that human PBMCs retrovirally transduced with the TCR possess high functional avidity specific to their target epitope, however, fail to recognize glioma cells naturally expressing or overexpressing the H3.3K27M mutation. We obtained similar results with the previously published TCR 1H5.8 Taken together, with this study we provide evidence that the H3.3K27M mutation is not a suitable target for HLA-A*02:01 restricted, CD8+ T cell-mediated immunotherapy.
Results
The recurrent mutation in the histone 3 variant that results in a lysine to methionine amino acid exchange (K27M) creates an H3.3K27M26-35 decamer peptide epitope (RMSAPSTGGV) which is predicted to bind HLA-A*02:01 at substantial affinity (NetMHC v3.4: 285 nM) compared with the wild type counterpart (RKSAPSTGGV, NetMHC v3.4: 22 651 nM). In order to isolate functional TCRs specific for the H3.3K27M mutation, we immunized ABabDII mice, which express a diverse human TCR repertoire restricted to HLA-A*02:01.10 On H3.3K27M26-35 peptide re-stimulation of splenocytes isolated from immunized mice, a reactive CD8+ population secreting IFNγ was detected, CD8+IFNγ+ cells were bulk sorted (figure 1A) and each dominant α and β chain was identified in the reactive T cell population of mouse #27 27633. The human constant regions were replaced by the respective murine ones and the identified TCRαβ pair was synthesized as a bicistronic cassette separated by a P2A element. The TCR cassette was subsequently cloned into the MP71 vector, followed by retroviral transduction into primary human T cells of healthy donors. To better assess and compare the functionality of our mouse-derived human TCR (henceforth 27633), we also cloned and transduced a published human-derived TCR (henceforth 1H5),8 into T cells of the same donor, as well as a well-characterized TCR (TCR 14/35) against an irrelevant target.11 After a 10-day expansion of the PBMCs, we detected 36.4%, 30.6%, and 31.4% CD8+ TCR-transduced T cells, respectively (figure 1B).

H3.3K27M peptide is recognized by a high avidity human TCR isolated from TCR transgenic mice. (A) Representation of a reactive CD8+ cell population secreting IFNγ on H3.3K27M26-35 peptide (RMSAPSTGGV) restimulation of splenocytes isolated from immunized ABabDII mice, measured by intracellular staining 7 days after the last immunization. Stimulation without peptide (Ø) was used as a negative control. (B) Transgenic TCR expression levels in human PBMCs after retroviral transduction with H3.3K27M-specific TCRs 27633 and 1H5 (8), as well as CDK4-specific control TCR14/35 (11) and untransduced control, were measured by flow cytometry staining for the mouse TCRβ constant region. Percentage of positive transduced CD8+ T cells are shown in the top right quadrant in (A, B), cells were gated on CD3+. (C, D) Levels of IFNγ secretion of H3.3K27M-specific TCRs 27633 and 1H5-transduced PBMCs, cocultured with T2 cells loaded with decreasing concentrations of either H3.3K27M mutant (C) or H3.3 wild-type (WT) (D) peptide. A total of 104 CD8+ transduced-TCR+ cells were cocultured in a 1:1 E:T ratio with T2 cells. P/I represents maximum IFNγ secretion from PBMCs stimulated with PMA/Ionomycin. The experiment was performed three times with similar results and graphs represent means of quadruplicate cultures±SD. TCR, T cell receptor; PBMC, peripheral blood mononuclear cells; E:T ratio, effector to target ratio.
Functional avidity of both TCRs was analyzed by coculture with TAP deficient T2 cells, exogenously loaded with decreasing concentrations of the H3.3K27M26-35 or the wild type H3.326-35 peptide. Both TCRs showed strong IFNγ secretion down to a concentration of 10-10 M H3.3K27M26-35, suggesting a high functional avidity of both TCRs against the mutant peptide (figure 1C). On binding to the wild type peptide, the human-derived TCR 1H5 induced a robust IFNγ secretion down to 10−8 M peptide concentration, while the mouse-derived TCR 27633 was specific for the mutant peptide (figure 1D).
To explore whether cells endogenously harboring the mutation are recognized by our TCR—an essential requirement for a potential clinical target, we retrovirally transduced the H3.3K27M+ DIPG cell line SF8628 to express HLA-A*02:01 (SF8628-A2) (online supplemental figure 1A). Unexpectedly, T cells transduced with either H3.3K27M-specific TCR failed to secrete IFNγ during a 24-hour coculture with 1×104 tumor SF8628-A2 effector cells (E:T, 1:1) (figure 2A). To investigate whether this could be due to potential low (heterozygous) expression of the mutant allele in this cell line, we overexpressed either the H3.3K27M mutant or the wild type full-length cDNAs. Expression of the mutation was confirmed by western blot (figure 2B) and the level of overexpression of both mutant and wild-type cDNA was determined by qPCR (online supplemental figure 1B). However, even an 18-fold overexpression of the H3.3K27M cDNA in SF8628-A2 cells did not induce IFNγ secretion by the TCR-transduced T cells (figure 2A). In contrast, when a minigene encoding three copies of the H3.3K27M26-35 epitope (triple epitope), a robust IFNγ response was observed, confirming HLA-A*02:01-epitope binding and TCR specificity (figure 2A). To confirm that SF8628-A2 cells are in general capable of processing and presenting epitopes in an HLA-A*02:01 dependent manner, we transduced T cells from the same healthy donor with TCR 14/35, which specifically recognizes the R24L mutation in CDK4.11 As expected, overexpressed full-length CDK4R24L cDNA in the same SF8628-A2 target cells was recognized by the respective TCR, inducing strong IFNγ secretion (figure 2A). These results suggest that although both H3.3K27M-specific TCRs are of high functional avidity, they fail to lead to IFNγ production against a glioma cell line, which is functional for antigen processing and presentation. In addition, we also investigated the recognition of a second DIPG cell line, SF7761, harboring the H3.3K27M mutation, which we also transduced to express HLA-A*02:01 (SF7761-A2) (online supplemental figure 1A). Corroborating the above results, both TCRs were unable to recognize the naturally expressed mutant epitope, while the exogenously loaded mutant peptide elicited a robust immune response by both TCRs (online supplemental figure 1C). The heterozygous expression of the H3F3A alleles in the DIPG cell lines was confirmed by RT-PCR and Sanger Sequencing (online supplemental figure 1D). To further support this notion, we sought to use the well-established, processing and presentation competent HLA-A*02:01+ melanoma cell line Mel624. In order to assess recognition by our TCR, we transduced Mel624 cells to overexpress wild type or mutant H3.3 cDNAs, validated by western blot (online supplemental figure 2A) and qPCR analysis (online supplemental figure 2B). Here too, on coculture with H3.3K27M-specific TCR-transduced T cells, 27 633 and 1H5 TCR-transduced T cells failed to produce detectable amounts of IFNγ (online supplemental figure 2C). To exclude that recognition through the transduced TCRs leads to expression of cytokines other than IFNγ, we also tested for TNFα and IL-2 secretion, but we were unable to detect either of the two cytokines (online supplemental figure 2C). In contrast, overexpression of the CDK4R24L mutant cDNA in Mel624 cells led to recognition by the 14/35 control TCR and secretion of all three cytokines. These results show that the H3.3K27M TCR-transduced T cells fail to produce cytokines associated with T cell cytotoxicity on coculture with cells naturally expressing or overexpressing the H3.3K27M mutation.
Supplemental material

H3.3K27M TCR-transduced T cells fail to recognize cell lines naturally expressing or overexpressing the mutant H3.3 histone. (A) Levels of IFNγ secretion of 27633- and 1H5-TCR transduced PBMCs, cocultured with the human SF8628 DIPG cell line. SF8628 cells were retrovirally transduced to express HLA-A*02:01 (SF8628-A2) and additionally contained H3.3K27M full length cDNA (MP71_H3.3K27M-CDS-P2A-GFP), H3.3 wild type full length cDNA (MP71_H3.3-CDS-P2A-GFP), CDK4R24L full length cDNA (MP71-CDK4-R24L-P2A-GFP), H3.3K27M RMSAPSTGGV triple epitope (MP71_H3.3K27M-triple-GFP), or H3.3 wild-type RKSAPSTGGV triple epitope (MP71_H3.3-triple-GFP). A total of 104 CD8+ transduced cells were cocultured at a 1:1 E:T ratio. CDK4R24L specific TCR-transduced T cells (14/35) were used as positive control to assess processing and presenting capabilities of the cell line. P/I represents maximum IFNγ secretion from PBMCs stimulated with PMA/Ionomycin. The experiment was performed three times with similar results and graphs represent means of triplicate cultures±SD. (B) H3.3K27M protein was detected by Western blot. Top panel shows β-actin (45 kDa), lower panel shows H3.3K27M (17 kDa) in parental or transduced SF8628-A2. Size discrepancy between naturally expressed and overexpressed mutant protein in SF8628-A2 cells is due to 18 amino acids at the C terminal end, encoded by the P2A element in the latter. (C) U87MG glioblastoma cell line was retrovirally transduced with MP71_H3.3K27M-CDS-P2A-GFP, MP71_H3.3-CDS-P2A-GFP or MP71-CDK4-R24L-P2A-GFP and sorted for GFP expression to overexpress either H3.3K27M, wild type H3.3 or CDK4R24L full length cDNA, respectively. (D) Levels of IFNγ secretion of 27 633 and 1H5 TCR-transduced PBMCs, cocultured with FACS-sorted transduced U87MG are shown, the CDK4R24L-specific TCR (14/35) served as positive control for functional antigen processing and presenting machinery in the U87MG cell line as well as IFNγ detection by TCR-transduced T cells. (E) The mutated H3.3 peptide is not detectable by a LC-MS/MS-based immunopeptidomic screen in U87MG glioblastoma cell line overexpressing H3.3K27M cDNA. Samples 1–4 recombinantly express H3.3K27M, samples 5–8 express CDK4R24L. MS2 fragment intensities for the endogenous mutated CDK4 and H3.3 peptides eluted from HLA-A*02:01+/CDK4R24L+ and HLA-A*02:01+/H3.3K27M+ cells after W6/32 HLA pulldowns and their heavy synthetic counterparts are shown.V* and P* indicate a heavy amino acid with a mass shift of +6 on valine or proline, respectively; M: oxidized methionine; n.d.: not detected. An amount of 25 fmol for CDK4R2L or 5 fmol for H3-3K27M of the synthetic heavy peptides were injected along with each sample. Only charge state 2+ was used for the quantitation. LC-MS/MS, liquid chromatography tandem mass spectrometry.
To account for possible differences between DMG/DIPG and glioblastoma (GBM) cell lines,12 we also overexpressed either H3.3K27M or H3.3, and as control, CDK4R24L full-length cDNAs in the GBM U87MG cell line used by Chheda et al (figure 2C). Again, only CDK4R24L expressing U87MG cells were recognized by the respective 14/35 TCR-transduced PBMCs, whereas neither mutant nor wild type H3.3-expressing U87MG cells induced IFNγ release in 27 633 or 1H5 TCR-transduced PBMCs, unless the cells were exogenously loaded with peptide (figure 2D). To further analyze if the predicted H3.3K27M epitope is naturally presented in HLA-A*02:01, we immunoprecipitated peptide/HLA-I complexes from the U87MG cells overexpressing either H3.3K27M or CDK4R24L and detected eluted peptides by liquid chromatography tandem mass spectrometry (LC-MS/MS). In contrast to spiked heavy synthetic H3.3K27M peptide, no endogenous mutant H3.3-derived peptides were detected in U87MG cells overexpressing the respective cDNA (figure 2E). As a control, mutant CDK4-derived peptides were detected in U87MG cells overexpressing CDK4R24L cDNA. The sequence of the eluted CDK4R24L peptide was validated by matching its LC-MS/MS retention time and spectra with that of its synthetic heavy peptide counterpart (figure 2E, online supplemental figure 3).
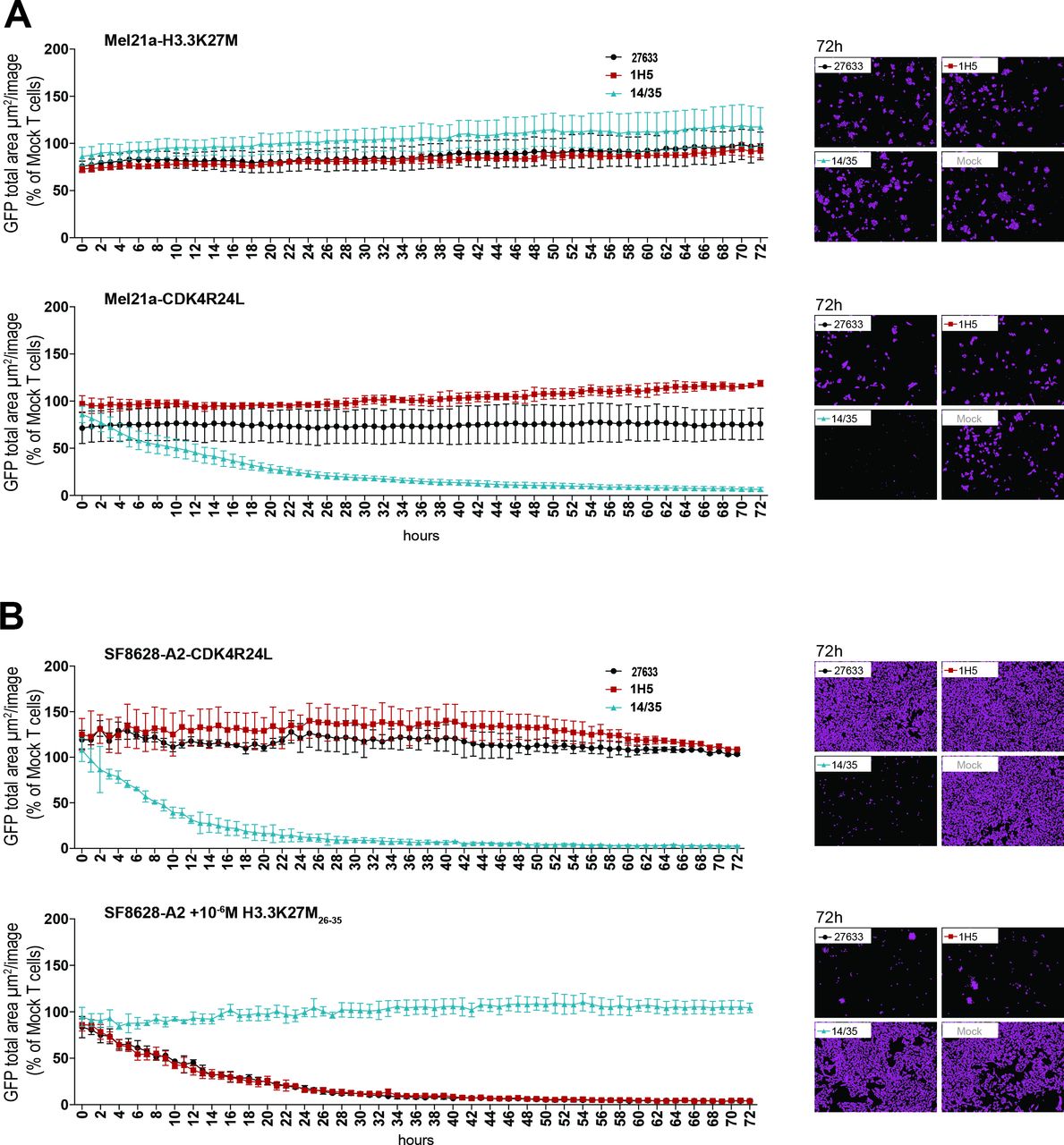
To investigate the cytotoxic potential of the H3.3K27M TCR-transduced T cells, we used a live-cell imaging-based cytotoxicity assay with target cells expressing GFP. On coculture of these fluorescent target cells with the respective T cells, the number of target cells surviving was determined relative to the number of control cells over a time period of 72 hours. Consistent with our previous results, no cytotoxic effect of H3.3K27M TCR-transduced T cells was observed, neither when the mutation was expressed naturally, nor when we overexpressed the H3.3K27M cDNA. T cells transduced with either 27 633 or 1H5 TCRs failed to lyse HLA-A*02:01 melanoma cell line Mel21a overexpressing full-length H3.3K27M cDNA (Mel21a-H3.3K27M; figure 3A, top panel). In contrast, the control T cells expressing the CDK4R24L-specific TCR14/35 killed 50% of their target cells, Mel21a-CDK4R24L, within 11 hours (figure 3A, bottom panel). In addition, natural expression of the H3.3K27M mutation in the SF8628-CDK4R24L glioma cell line did not lead to lysis by the two H3.3K27M-specific T cells, while coculture with the control T cells showed substantial and prompt killing of 50% tumor cells within 9 hours (figure 3B, top panel). As depicted in figure 3B (bottom panel), cytotoxicity mediated by the H3.3K27M TCR-transduced T cells was only observed against target cells loaded with the mutant peptide (50% killing within 10 hours), confirming the cytotoxic potential of the H3.3K27M TCR-transduced T cells used in the assay.
{kind=link}
{kind=link}
{kind=link}
Lack of cytotoxicity against cells naturally expressing or overexpressing mutant H3.3 histone. (A) 15×103 transduced CD8+ cells were cocultured at a 5:1 E:T ratio with HLA-A*02:01+ Mel21a cells, retrovirally transduced to express either H3.3K27M full length cDNA (MP71_H3.3K27M-CDS-P2A-GFP) (top panel) or CDK4R24 full length cDNA (MP71-CDK4-R24L-P2A-GFP) (bottom panel). Cytotoxicity was observed over 72 hours using the live cell imaging system IncuCyte Zoom (Essen bioscience). Representative images are shown in the right panels. Values are calculated by normalizing the average GFP total area (µm²/image) in the target cells cocultured with the respective TCR-transduced T cells to the average of that cocultured with mock transduced T cells. The experiment was performed three times with similar results and graphs represent means of triplicate cultures±SD. (B) 15×103 CD8+ transduced cells were cocultured at a 5:1 E:T ratio with SF8628-A2 cells, naturally expressing the H3.3K27M mutation and additionally either retrovirally transduced to express CDK4R24L full length cDNA (top panel) or loaded with 10-6 M mutant H3.3K27M peptide (bottom panel). Cytotoxicity was observed over 72 hours using the live cell imaging system IncuCyte Zoom. Representative images are shown in the right panels. Values are calculated as in (A). The experiment was performed three times with similar results and graphs represent means of triplicate cultures±SD. GFP, green fluorescent protein.
These experiments unequivocally show that the putative mutant H3.3K27M epitope is not recognized by two independently isolated TCRs, regardless of whether the mutant gene is naturally expressed or overexpressed. In addition, we analyzed eight ERAP1 polymorphisms in the genomic DNA of the cell lines Mel21a, Mel624, SF8628, and U87MG used in this T cell recognition analysis and identified different ERAP1 allotypes among the cell lines (online supplemental table 1). Thus, we exclude the possibility that lack of recognition is caused by differential epitope trimming or destruction due to specific ERAP1 allotypes present in all cell lines.13
To exclude that T cell recognition of H3.3K27M was hampered by the presence of the endogenous human TCR chains resulting in mixed dimers, we knocked out the endogenous TCR in the transduced human T cells by CRISPR/Cas9. To confirm the functionality of TRAC/TRBC-specific gRNAs, mock-transduced T cells were targeted with either RNP complex, which led to a knockout efficiency of 73.3% and 29.5%, respectively (online supplemental figure 4A). CRISPR targeting with both gRNAs simultaneously in mock-transduced PBMCs led to a marked decrease in the number of hTCRα/β+ cells, from 90.3% to 17.4% (online supplemental figure 4B). When 27 633 and 1H5 TCR-transduced PBMCs were targeted, the hTCRα/β+mTCR+ double-positive populations, representing the population with potential mixed dimers, decreased to 5.8% from 11.6% and to 5.7% from 12.7%, respectively (online supplemental figure 4B). CRISPR/Cas9-edited TCR-modified T cells were cocultured with SF8628-A2 cells, either naturally expressing the H3.3K27M mutation, or overexpressing fulllength H3.3K27M cDNA, as well as with H3.3K27M26-35 peptide-loaded T2 cells. As depicted in online supplemental figure 4C, no alteration in the capacity of T cells lacking the endogenous human TCR to recognize the expressed mutation was observed: The endogenous as well as the overexpressed mutation were still not recognized by the TCR-modified T cells, whereas H3.3K27M26-35 peptide-pulsed T2 cells-induced IFNγ secretion by TCR-modified T cells to previously observed levels. Again, 1H5 TCR-modified T cells also recognized wild type H3.326-35 peptide-pulsed T2 cells, as already shown in figure 1D. When analyzing binding of H3.3K27M-specific MHC I dextramer no significant difference in staining intensity was detected for CD8+ T cells expressing H3.3K27M-specific TCRs. Thus CD8+Dextramer+ cells modestly decreased on CRISPR/Cas9 knockout from 23.8% to 19.9% for 27633 TCR-transduced T cells and increased from 27.3% to 30% for 1H5 TCR-transduced T cells. In addition, no change in the mean fluorescence intensity was observed between both panels. Surprisingly, 22.6% (-Cas9 RNP) and 21.8% (+Cas9 RNP) of CD4+ T cells expressing the human-derived TCR 1H5 were also stained with the dextramer, suggesting that MHC-peptide binding might also be occurring in a CD8 independent manner14 (online supplemental figure 4D). With these CRISPR/Cas9 modifications, we excluded that potential mispairing between endogenous and transduced human TCR chains might be affecting the recognition of the glioma target in any way, further strengthening the notion that the H3.3K27M26-35 is not a suitable target for TCR gene therapy in HLA-A*02:01+ patients with DMG.
Discussion
Although we confirmed that the TCRs generated against the H3.3K27M mutation show high affinity to their cognate peptide in vitro, they do not recognize endogenously expressed mutation, which is a prerequisite for TCRs to possess a clinical potential. Overall, these findings are in contrast to those reported by Chheda et al, where 1H5 TCR-modified T cells were shown to mediate reactivity in a LDH cytotoxicity assay on coculture with DIPG cells naturally expressing the H3.3K27M mutation. Since we tested the 1H5 TCR side by side with a high-affinity H3.3K27M peptide-specific TCR, obtained from huTCR transgenic mice, in coculture experiments measuring IFNγ, TNFα, and IL2 secretion as well as cytotoxicity, and found no signs of reactivity even to overexpressed H3.3K27M, the applicability of this adoptive T cell targeting approach in HLA-A*02:01+ DMG patients is questionable. Detection of H3.3K27M-specific vaccine responses in a study with DMG patients by mass cytometry supports the notion that the H3.3K27M epitope may be presented by HLA-A*02:01 to induce a T cell response, if the vaccine consists of the decamer mutant peptide.9 However, it is unlikely that the CD8+ T cells in this clinical trial were effective because we show here that the epitope is not presented in sufficient amounts to be recognized by T cells. In line with our findings, the H3.3K27M epitope vaccination treatment strategy (PNOC007) did not improve the overall outcome in H3.3K27M+ patients with DMG.9 We can rule out that factors such as different ERAP1 activity account for lack of recognition by using cell lines with different ERAP1 polymorphisms.13 We excluded presence/absence of methionine oxidation in the H3.3K27M/HLA-A*02:01 epitope15 to account for the different results with recognition of cell lines by showing recognition of triple H3.3K27M25-35 epitope speaking against a TCR specificity for the oxidized methionine only. Moreover, the discrepancy between our findings and previously published ones did not resolve, even when the same cell line (U87MG), authenticated by STR profiling, was used for antigen presentation. Here, a mass spectrometry-based immunopeptidomics screen further indicated that the predicted H3.3K27M epitope may lack sufficient T cell immunogenicity as it could not be detected in HLA-A*02:01+ U87MG cells overexpressing mutant H3.3 cDNA. Closing, we have to conclude that H3.3K27M is unlikely a suitable target for TCR gene therapy in HLA-A*02:01+ patients with DMG. We think it is important to share these negative data with the community, because clinical trials based on H3.3K27M could raise wrong hope at least in HLA-A*02:01-positive patients and would lead to the wrong conclusion that an in principle ideal target cannot be made use of for immunotherapy. To open an immunotherapeutic perspective for DMG patients, further analysis of targeting H3.3K27M by either CD8+ T cells restricted to HLAs other than HLA-A*02:01 or CD4+ T cells is therefore required. In general, immunogenicity of neoantigens predicted to bind the one or the other HLA need to be validated first by T cell assays or HLA ligandomics before these can be considered valuable targets for (peptide) vaccination or ATT.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Sabrina Horn, Kathrin Borgwald and Mathias Pippow for excellent technical support. We thank Xiaojing Chen for providing a modified HLA pull-down protocol. The authors declare no competing financial interests.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors GW designed the study, LI, GP and OP conducted experiments. LI, GP, PM, TB and GW analyzed data and wrote the manuscript. TB and GW are joint last authors.
Funding Funding was provided by grants from the German Research Foundation (SFB-TR36), the Berlin Institute of Health (CRG-1), Deutsche Krebshilfe (111 546 and 70113456), DKTK joint funding (NEO-ATT), European Union (ERC Advanced Grant 882963) and the Helmholtz-Gemeinschaft, Zukunftsthema ‘Immunology and Inflammation’ (ZT-0027).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Letter
- Letter