Article Text

Abstract
Background Despite numerous therapeutic options, safe and curative therapy is unavailable for most patients with chronic lymphocytic leukemia (CLL). A drawback of current therapies such as the anti-CD20 monoclonal antibody (mAb) rituximab is the elimination of all healthy B cells, resulting in impaired humoral immunity. We previously reported the identification of a patient-derived, CLL-binding mAb, JML-1, and identified sialic acid-binding immunoglobulin-like lectin-6 (Siglec-6) as the target of JML-1. Although little is known about Siglec-6, it appears to be an attractive target for cancer immunotherapy due to its absence on most healthy cells and tissues.
Methods We used a target-specific approach to mine for additional patient-derived anti-Siglec-6 mAbs. To assess the therapeutic utility of targeting Siglec-6 in the context of CLL, T cell-recruiting bispecific antibodies (T-biAbs) that bind to Siglec-6 and CD3 were engineered into single-chain variable fragment–Fc and dual-affinity retargeting (DART)–Fc constructs. T-biAbs were evaluated for their activity in vitro, ex vivo, and in vivo.
Results We discovered the anti-Siglec-6 mAbs RC-1 and RC-2, which bind with higher affinity than JML-1 yet maintain similar specificity. Both JML-1 and RC-1 T-biAbs were effective at activating T cells and killing Siglec-6+ target cells. The RC-1 clone in the DART–Fc format was the most potent T-biAb tested and was the only anti-Siglec-6 T-biAb that eliminated Siglec-6+ primary CLL cells via autologous T cells at pathological T-to-CLL cell ratios. Tested at healthy T-to-B cell ratios, it also eliminated a Siglec-6+ fraction of primary B cells from healthy donors. The subpicomolar potency of the DART–Fc format was attributed to the reduction in the length and flexibility of the cytolytic synapse. Furthermore, the RC-1 T-biAb was effective at clearing MEC1 CLL cells in vivo and demonstrated a circulatory half-life of over 7 days.
Conclusion Siglec-6-targeting T-biAbs are highly potent and specific for eliminating Siglec-6+ leukemic and healthy B cells while sparing Siglec-6− healthy B cells, suggesting a unique treatment strategy for CLL with diminished suppression of humoral immunity. Our data corroborate reports that T-biAb efficacy is dependent on synapse geometry and reveal that synapse architecture can be tuned via antibody engineering. Our fully human anti-Siglec-6 antibodies and T-biAbs have potential for cancer immunotherapy.
Trial registration number NCT00923507.
- Antibodies, Neoplasm
- Antigens, Neoplasm
- B-Lymphocytes
- Epitope Mapping
- Hematologic Neoplasms
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Sialic acid-binding immunoglobulin-like lectin-6 (Siglec-6) is a novel target for immunotherapy that our laboratory discovered in the context of chronic lymphocytic leukemia (CLL), and recent reports have confirmed its absence from most healthy cells and tissues and have shown its expression in other cancers.
WHAT THIS STUDY ADDS
In the present study, additional patient-derived anti-Siglec-6 monoclonal antibodies (mAbs) were discovered which bind to the N-terminal lectin domain with higher affinity. Employing these anti-Siglec-6 clones as T cell-recruiting bispecific antibodies (T-biAbs) generated highly potent and selective therapeutic candidates.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The patient-derived mAbs reported here are highly valuable reagents for basic and applied cancer immunotherapy. More specifically, we present Siglec-6 targeting immunotherapeutic interventions of translational value that warrant further study in CLL and other cancers. Lastly, in the rapidly growing field of T-biAbs, the functional comparison of single-chain variable fragment–Fc and dual-affinity retargeting–Fc formats illustrates important fundamentals in cytolytic synapse engineering, especially with membrane–distal epitopes.
Background
Chronic lymphocytic leukemia (CLL) has the highest incidence of all adult leukemias1 and is defined by the expansion of malignant, mature B cells (CD19+/CD20+/CD5+). Current treatments that effectively manage CLL include anti-CD20 monoclonal antibodies (mAbs) and small molecules such as Bruton’s tyrosine kinase inhibitors (BTKis), which inhibit B-cell receptor signaling that drives the disease.2 However, these therapies are not curative, and all have known mechanisms of relapse.3–6 They also lack specificity for leukemic B cells and thus perpetuate the immune dysfunction in patients with CLL, making them more susceptible to infections, including SARS-CoV-2, which was found to be fatal to over one-third of patients with CLL.7 Vaccine-induced immunity offers little protection for these patients, as one study of the BNT162b2 mRNA vaccine found that only 16% of patients undergoing CLL treatment produced a detectable antibody response after vaccination, and 0% who had received mAbs against CD20, a universal B-cell marker, developed anti-SARS-CoV-2 antibodies.8 Therefore, there is a salient need for CLL-specific therapies that eliminate the leukemia and restore natural immune function.
Allogeneic hematopoietic stem cell transplantation (alloHSCT), a high-risk procedure that is ruled out for most of the CLL patient population, has yielded long remissions or cures in approximately half the patients who have undergone this treatment.9 While most of the alloHSCT-induced graft-versus-leukemia response is T cell-mediated, there is evidence of leukemia-targeting antibodies in the sera of treated patients, and these antibodies may be the key for innovative CLL-specific therapies.10 Using a phage display library of an alloHSCT antibody repertoire of a cured patient with CLL, we identified one of these antibodies, JML-1, which binds sialic acid-binding immunoglobulin-like lectin-6 (Siglec-6, also known as OBP1, CD33L1, and CD327).11 12
Siglec-6 belongs to the family of CD33-related siglecs that share an N-terminal sialic acid-binding domain (V-type Ig-like domain), followed by a variable number of C2-type Ig-like domains, a transmembrane domain, and an intracellular domain that typically bears immunoreceptor tyrosine-based inhibitory motifs (ITIMs). Siglec-6 is a promising target for antibody-based therapy due to its overexpression on CLL cells and coincidental absence on most healthy cell types, with exceptions limited to placental trophoblasts, mast cells, and a portion of activated B cells.13–15 However, our initial studies using the anti-Siglec-6 mAb JML-1 in IgG1 format demonstrated a lack of anti-CLL effector functions, which led us to consider alternative mechanisms or antibody formats to translate JML-1 into a potential therapeutic. It is now recognized that patients with CLL treated with BTKis exhibit increased persistence and activation of T cells, supporting the use of immunotherapies that employ T cells in targeting the latent leukemia that persists during BTKi treatment.16–19 Given the success of the clinically approved CD19×CD3 T cell-recruiting bispecific antibody (T-biAb) blinatumomab in acute lymphocytic leukemia and the mechanistic potency of this modality, we reasoned that T-biAbs targeting Siglec-6 might be an effective CLL immunotherapy, without the need for transplantation and other high-risk cell therapies.20 21 Blinatumomab is not approved for CLL, and it has numerous drawbacks including the elimination of all healthy B cells, the induction of cytokine release syndrome (CRS), and a short circulatory half-life due to its small size. In contrast, an anti-Siglec-6 T-biAb may work well for patients with CLL as it will have increased target cell specificity, may result in less systemic cytokine release due to lower target expression, and have a longer circulatory half-life if it is fused to an Fc domain.
In the current study, we reprobed the post-alloHSCT antibody library and identified additional clones targeting Siglec-6, with higher affinity than the original JML-1 clone. The anti-Siglec-6 antibodies were engineered into various T-biAb formats which successfully lysed Siglec-6+ cell lines and primary CLL cells. Corroborating their potency and specificity, a Siglec-6+ fraction of primary B cells from healthy donors (HDs) was also eradicated. The dual-affinity retargeting (DART) format, which creates a shorter cytolytic synapse between target and effector cell than single-chain variable fragment (scFv)-based T-biAbs, outperformed the scFv at killing target cells and activating T cells. In a mouse model of human CLL, we demonstrated that anti-Siglec-6 DART–Fc inhibits tumor growth and extends survival, comparable to a CD19-targeting control T-biAb. Therefore, Siglec-6 appears to be a promising target for T-biAbs, a potent immunotherapy that can successfully eliminate leukemic cells, with less on-target-off-tissue toxicity than current therapies.
Materials and methods
See online supplemental materials for cell lines, expression, and purification of antigens and antibodies, human Fab library selection, ELISA, surface plasmon resonance (SPR), hydrogen–deuterium exchange mass spectrometry (HDX-MS), homology modeling of Siglec-6, flow cytometry, T-cell expansion, and statistics.
Clinical samples
Cryopreserved CLL peripheral blood mononuclear cells (PBMC) were derived from treatment-naïve patients enrolled in an observational study at the National Institutes of Health (NIH) Clinical Center (www.clinicaltrials.gov; NCT00923507). Cryopreserved BTKi-treated samples were obtained from patients with CLL enrolled in phase II clinical trials at the NIH Clinical Center investigating single-agent ibrutinib (NCT01500733) or acalabrutinib (NCT02337829).
Nuclear factor of activated T cells (NFAT) activation assays
MEC1 or MEC1-002 cells (2×105 cells) were seeded in 96-well round-bottom plates, and T-biAb or Dulbecco’s phosphate-buffered saline (DPBS; vehicle) was added followed by a 30 min incubation at 37°C. Jurkat cells engineered to express luciferase under the transcriptional control of NFAT (Jurkat-Lucia NFAT, InvivoGen) were added at an effector-to-target (E:T) ratio of 1:1 (2×105 cells). After overnight culture, 20 µL of supernatant was combined with 50 µL of QUANTI-Luc substrate, and luminescence was recorded after 15 min using a SpectraMax M5 plate reader. The positive control for NFAT activation consisted of Jurkat-Lucia NFAT cells plated alone with 0.5 mg/mL concanavalin A. Percent NFAT activation was calculated as follows:

In vitro cytotoxicity assays
Cell line cytotoxicity experiments were modeled off a previously published luciferase assay format.22 MEC1-fLuc, MEC1-002 fLuc, or MEC1-fLuc-hS6 cells w seeded at 2.5×104 cells per well along with T-biAb or DPBS (vehicle) followed by a 30 min incubation at 37°C. T cells expanded from HD PBMC23 were added at a 1:1 ratio and cells were cocultured for 16–24 hours. Luciferin (Biosynth Carbosynth) was added to 0.15 mg/mL, and luminescence was recorded after 15 min as described previously. Luminescent signal from T-cell and T-biAb-treated wells was normalized to the positive viability control consisting of cells only. Specific lysis was calculated as follows:

Ex vivo activity assays
T-biAb activity on cryopreserved CLL PBMC was evaluated as previously described.17 Cell viability of the CLL population (CD3−/CD5+/CD20+) was assessed at various time points with the LIVE/DEAD fixable violet stain (Thermo Fisher) by flow cytometry. Patient samples in which the vehicle-treated cells did not meet the viability threshold (>20%) at a given time point were eliminated from analysis. T-cell populations were quantified with commercial mAbs to CD4, CD8, CD25, and CD69 (see online supplemental materials).
Supplemental material
Multispectral fluorescence imaging
MEC1-hS6 cells were stained with CellTrace Far Red dye (Thermo Fisher), followed by incubation with 5 µg/mL T-biAb for 30 min at 37°C. Excess T-biAb was washed off, and Jurkat T cells, stained with CellTrace CFSE dye, were added and cocultured (E:T=1:10) for 30 min at 37°C to allow synapse formation to occur. Conjugates were washed, fixed with Cytofix/Cytoperm solution (BD Biosciences), stained with Cy3-conjugated goat antihuman Fc (Jackson ImmunoResearch) and 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher), and imaged (~50,000 per sample) using an Amnis ImageStreamx Mk II instrument (Luminex) at ×40 magnification. IDEAS V.6.3 software (Luminex) was used for analysis. Images were gated to identify individual DAPI+ Jurkat and DAPI+ MEC1-hS6 cells, then a mask was created for the overlap between Jurkat and MEC1-hS6 cells. Next, data were filtered on the size of the overlap mask to eliminate cells that were clearly on top of each other or too far apart for a 1:1 conjugate, and an additional gate was added to require Cy3 (T-biAb) staining at the synapse. Synapse formation was quantified as the number of heterodoublet synapse+ conjugates relative to the total number of DAPI+ Jurkat cells. An additional mask was created using the union of DAPI and cell stains, and internuclear distance was calculated between the centroids of the DAPI–cell masks for each cell type using the distance theorem.
Xenograft mouse model
Female NOD-scid IL2Rγnull (NSG) mice (JAX #005557, 6–7 weeks old) were inoculated with 2×106 MEC1-fLuc-hS6 cells intravenously (tail vein). One day prior to each treatment, mice were randomized and preconditioned with 0.25 mL human serum by intraperitoneal injection. Mice were treated on day 7 with 3×106 human T cells and were given T-biAb (0.5 or 0.05 mg/kg, diluted in DPBS, five mice per group) two times per week for 2 weeks, all intravenously (tail vein). On day 13, the mice were given an additional 1×106 T cells. The mice were imaged with a Lago X instrument using templated imaging protocols and consistent timing after luciferin injection, and data were analyzed using Aura software (Spectral Instruments), similar to previous reports.24
Results
Discovery of new anti-Siglec-6 mAbs
The anti-Siglec-6 mAb JML-1 was discovered in a target-agnostic selection of a post-alloHSCT immune repertoire against primary CLL cells.11 12 To identify additional anti-Siglec-6 mAbs in the post-alloHSCT Fab-phage library and validate Siglec-6 as an immunogenic target for the humoral immune response to CLL, the library was reselected against recombinant human Siglec-6 (hS6) with either C-terminal or N-terminal human IgG1–Fc fusion (hS6-Fc). The Fab-phage panning on immobilized antigen successfully enriched polyclonal Fabs that bound hS6 in ELISA (online supplemental figure S1). Most clones shared the same heavy chain complementarity determining region 3 (HCDR3) as JML-1, except for RC-1 (figure 1A). HCDR1 and HCDR2 were also different for RC-1, and the RC-2 clone shared these new features while maintaining the JML-1 HCDR3, which warranted further investigation. To determine the binding kinetics of the selected Fabs, SPR analysis was employed using immobilized hS6-Fc and soluble Fab. A Kd of 0.49 nM was determined for RC-1, approximately sixfold lower than JML-1’s Kd of 3.15 nM, while the RC-2 clone had a Kd of 2.76 nM (figure 1A and online supplemental figure S2A–C). Both RC-1 and RC-2 maintained specificity for hS6 and did not bind related human siglec family proteins nor the closely related rhesus macaque (Macaca mulatta) Siglec-6 (figure 1B). Given the unique HCDR3 and high affinity of RC-1, along with the evidence that polyclonal phage bound hS6 in the presence of JML-1 Fab (online supplemental figure S1), we hypothesized RC-1 may bind a distinct epitope from JML-1. To this end, U937 (hS6+) cells were blocked with a titration of JML-1 Fab and subsequently stained with biotinylated RC-1 Fab (RC-1-bio). In the absence of JML-1, RC-1-bio bound strongly. However, when blocking with equimolar or excess JML-1, RC-1-bio binding was abolished, suggesting that the two clones bind identical or overlapping epitopes on hS6 (figure 1C). SPR corroborated this result as the combination of RC-1 and JML-1 did not increase binding in comparison to JML-1 alone (online supplemental figure S2D). To map the epitope targeted by these clones, the hS6 V-type (V) and C2-type I (C2i) Ig-like domains, previously shown to be required for JML-1 binding,25 were expressed as tandem domain and analyzed by HDX-MS (online supplemental figure S3A and table S1).26 Deuterium uptake in the presence or absence of RC-1 Fab was compared with identified residues involved in the epitope, yet one-third of the residues were not observed in the mass spectrometry (MS) data (figure 1D and online supplemental figure S3B,C). To assess the possibility of these missing residues forming the epitope, the sequence of hS6 was aligned with human CD33 (Siglec-3) and rhesus Siglec-6, which are both highly homologous to hS6 but are not bound by the Fabs. Chimeric hS6×CD33 mutants were rationally designed by individually replacing peptides that were implicated by or absent from the HDX-MS data and unique to hS6 (figure 1E and online supplemental figure S3D). In an ELISA, none of these four peptide replacement mutants were bound by the human Fabs, though three were bound by a commercial mouse mAb (figure 1F). Only the chimera that retained the hS6 V domain, but replaced the C2i domain, was bound by JML-1, RC-1, and RC-2. Taken together, these data suggest that the human Fabs bind to a conformational epitope in the membrane distal portion of the hS6 V domain.

Siglec-6-targeting antibody clones selected from a post-alloHSCT library exhibit specificity and shared epitope. (A) Comparison of post-alloHSCT antibody clones including VH and VL amino acid sequence identities, IMGT-defined CDR3 alignments, and affinity data from SPR analysis of Fab binding to Siglec-6–Fc fusion protein. (B) Heatmap of ELISA data (triplicate) from directly coating human or rhesus (85% amino acid sequence identity within the ectodomain) Siglec–Fc fusion proteins, incubating with Fabs, and detecting with antihuman Fab or antihuman Fc (positive control). The post-alloHSCT Fabs are all specific for human Siglec-6. (C) Flow cytometry histograms (left) and quantification (right) demonstrating that JML-1 Fab can block the binding of biotinylated RC-1 (RC-1-bio) Fab (40 nM) by competing for Siglec-6 on the surface of U937 cells. Statistics (n=3) were calculated using Student’s t-test. ****p<0.0001. (D) Homology model of Siglec-6 (V and C2i domains, 27–236; see online supplemental material), colored according to the differential deuterium uptake in the presence of RC-1 Fab as determined by HDX-MS. Negative differential D2O values represent decreased solvent exposure in the presence of Fab. Black regions were not observed, and gray regions indicate no significant change in deuterium uptake. (E) Homology model of Siglec-6 illustrating with shades of yellow and orange the peptides that were individually substituted with corresponding CD33 sequences, and green indicating the C2i domain that was replaced entirely for epitope mapping. (F) Epitope mapping ELISA with Siglec-6×CD33 chimeric Fc-fusion proteins indicating the dependence of the mutated regions for binding with human Fabs but not for the commercial mouse mAb or sheep pAb to human Siglec-6 (hS6). alloHSCT, allogeneic hematopoietic stem cell transplantation; C2i, C2-type I; CDR3, complementarity determining region 3; HDX-MS, hydrogen–deuterium exchange mass spectrometry; mAb, monoclonal antibody; MFI, mean fluorescence intensity; pAb, polyclonal antibody; SPR, surface plasmon resonance; VH, variable heavy; VL, light chain domain.
Targeting Siglec-6 with T-biAbs
In previous experiments, JML-1 in IgG1 format failed to induce apoptosis or mediate antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity of primary CLL cells ex vivo, likely due to the low copy number of Siglec-6 on primary CLL cells, which varied from 843 to 6035 (n=6) on the initial treatment-naïve cohort we assessed (online supplemental figure S4A and table S2). We turned our attention to T cell-activating therapies because they have the potency to eliminate cells with very low target copy number,27 and T-biAbs have shown promise in CLL for other targets.17 28 Furthermore, both Kovalovsky et al and Jetani et al recently demonstrated the absence of Siglec-6 on healthy tissues including HSCs and most healthy B cells, indicating Siglec-6 targeting T-biAbs would have less risk of on-target-off-tissue toxicity and may help reverse immune dysfunction in patients with CLL.25 29 However, it has been recently reported that Siglec-6 is expressed on T cells in the peripheral blood of patients with bladder cancer.30 Since little is known regarding Siglec-6 expression on T cells in the context of CLL, Siglec-6 expression was assessed on the CD3+ population of CLL-derived PBMC (online supplemental figure S4B). No specific binding of anti-Siglec-6 mAbs to T cells was detectable when compared with isotype controls, confirming that the target is specific to the CLL B cells. To validate that the two lead anti-Siglec-6 post-alloHSCT mAbs bind to CLL cells from this patient cohort, JML-1 and RC-1 were used to stain whole PBMC from patients with CLL or HDs (online supplemental figure S4C). Both JML-1 and RC-1 stained CLL-derived PBMC more than the HD cells. Despite the Siglec-6 expression observed on primary CLL cells, the only Siglec-6+ CLL cell line that we have found is MEC1-002, a MEC1 subline that was sorted for JML-1 binding and expanded12 (expression quantified in online supplemental figure S4D). However, since MEC1-002 cells overexpress both FCRL4 and Siglec-6, relative to the parental MEC1 cells,12 both of which functionally inhibit B-cell receptor signaling,15 resulting in slower growth in vitro and in vivo,25 we transduced MEC1 to stably express Siglec-6 (without FCRL4) and fLuc to generate the new clonal cell line, MEC1-hS6, which we employed in our proof-of-concept experiments.
Siglec-6×CD3 T-biAbs were first generated in the IgG-like scFv–Fc format (figure 2A). The anti-CD3 T cell-engaging scFv arm (V9) of these T-biAbs was paired to Siglec-6-targeting or control (NT) scFv–Fc using knob-into-hole mutations.31 JML-1/V9 scFv–Fc and RC-1/V9 scFv–Fc were generated and validated for specificity against Siglec-6−, Siglec-6+, and CD3+ cells (figure 2B and online supplemental figure S5A–C). To determine whether these T-biAbs elicit T-cell signaling in the presence of Siglec-6-expressing cells, a Jurkat T-cell line with an NFAT-dependent luciferase reporter was cocultured with T-biAbs and MEC1-002 (fLuc−) cells at a ratio of 1:1. When the anti-Siglec-6 T-biAbs were added in the presence of MEC1-002 cells, NFAT activation occurred (figure 2C). Both RC-1 and JML-1 clones potently activated NFAT signaling, whereas the isotype NT control scFv–Fc did not. To assess their cytolytic activity, T-biAbs were added to cocultures of HD T cells and fLuc-expressing target cells. Specific lysis of MEC1-hS6 was comparable for CD19/V9 (EC50=2.4 pM), JML-1/V9 (EC50=2.4 pM), and RC-1/V9 (EC50=3.0 pM) scFv–Fc, and T cells upregulated CD69 and CD25, key markers of T-cell activation (figure 2D and online supplemental figure S6A). Importantly, there was no appreciable lysis of the Siglec-6− MEC1 cells. In these cytotoxicity experiments, both Siglec-6 targeting T-biAbs induced interferon gamma (IFN-γ) secretion, but it was significantly higher for RC-1/V9 scFv–Fc compared with JML-1/V9 scFv–Fc or CD19/V9 scFv–Fc (figure 2E). Interleukin (IL)-2 levels were similar between the RC-1 clone and the anti-CD19 control, yet the JML-1 clone induced lower levels of IL-2 secretion. Tumor necrosis factor alpha (TNF-α) secretion was higher for JML-1 and CD19 than for the NT control, while RC-1 was not statistically different from the JML-1 or the NT control (figure 2E). Analogous cytokine profiles were induced in the presence of MEC1-002 cells, which express lower levels of Siglec-6 (online supplemental figure S6B). These data suggest that the higher Siglec-6 affinity of RC-1 may result in stronger activation of T cells (higher IFN-γ and IL-2 secretion) than the JML-1 clone. However, the relatively low IL-2 secretion in the presence of MEC1-002 cells indicated that the scFv–Fc T-biAbs may not succeed in fully activating and expanding T cells in vivo, so alternative T-biAb formats were considered.

scFv–Fc T-biAbs mediate specific T-cell activation and CLL lysis. (A) T-biAbs were designed with scFv domains fused to human IgG1 Fc with knob-into-hole mutations to facilitate heterodimerization of Siglec-6 and CD3 binding arms. (B) Siglec-6− CLL cell line MEC1 (left), Siglec-6+ MEC1-002 (middle), and human T cells (right) were stained with 10 µg/mL of scFv–Fc T-biAb and an antihuman Fc secondary antibody to validate bispecificity for Siglec-6 (or CD19 as positive control) and CD3. (C) A Jurkat-Lucia NFAT reporter cell line was cultured overnight with scFv–Fc T-biAbs and MEC1 (left) or MEC1-002 (right) cells to determine T-cell activation in the absence or presence of target expression. (D) MEC1 (left) and MEC1-Siglec-6 (hS6) transgenic (right) cell lines were cocultured with human T cells and a titration of the indicated T biAbs. Following overnight incubation, cell lysis was assessed by intracellular luciferase activity. (E) The levels of type I cytokines in the cell lysis assay supernatants were determined by ELISA, and data were normalized to the CD19/V9 positive control. Statistics were calculated using an unpaired t-test, n=3. *P<0.05, **P<0.01. CLL, chronic lymphocytic leukemia; IFN-γ, interferon gamma; IL, interleukin; ns, not significant; scFv, single-chain variable fragment; NFAT, nuclear factor of activated T cells; T-biAb, T cell-recruiting bispecific antibody; TNF-α, tumor necrosis factor alpha; VH, variable heavy chain domain; VL, variable light chain domain.
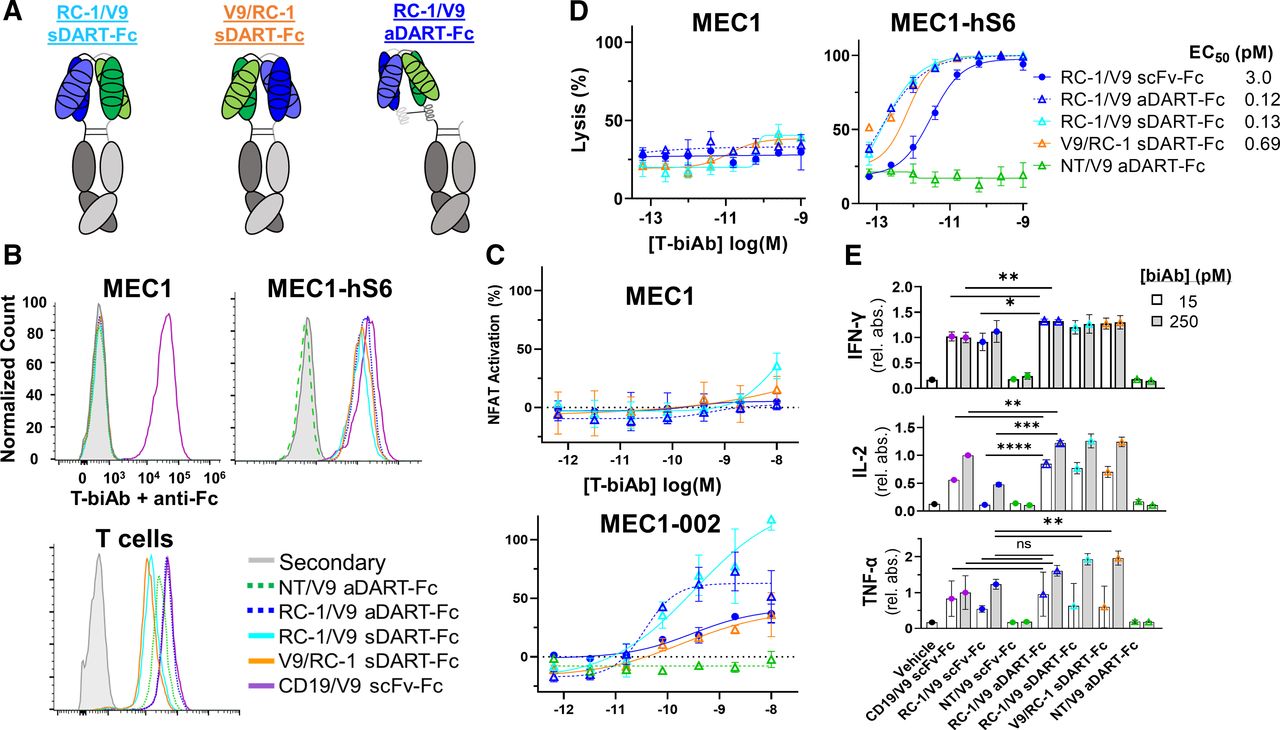
It is well-established that T-biAbs that bind to membrane-proximal epitopes on target cells have greater efficacy than those that bind to membrane–distal epitopes. The theory is that shorter cytolytic synapses—those closer in size to the endogenous T-cell receptor (TCR)–major histocompatibility complex (MHC)-peptide interaction—result in the exclusion of large immunoinhibitory phosphatases from the cytolytic synapse, reducing TCR signaling and cell lysis potential.23 32 Since the epitopes of all three anti-Siglec-6 mAbs from our post-alloHSCT library are in the outermost V domain (the third Ig-like domain from the surface), we were faced with a significant challenge. We hypothesized the synapse length could be shortened by using the compact and rigid diabody format known as DART.33 For our DART–Fc engineering studies, we chose to pursue RC-1 since it has higher affinity for Siglec-6 and elicited higher IL-2 and IFN-γ secretion than JML-1 in the scFv–Fc format. Two DART formats were employed: a symmetrical dual-affinity retargeting (sDART) architecture to form a dimer and another with an asymmetrical dual-affinity retargeting (aDART) architecture to form a trimer (figure 3A).34 35 Two sDART–Fc constructs were assembled, and they were named according to the order of VL and VH for each clone, RC-1/V9 sDART–Fc and V9/RC-1 sDART–Fc, while only one aDART–Fc construct, RC-1/V9 aDART–Fc, was made (online supplemental figure S7A–D). DARTs were tested for specificity using flow cytometry and showed specificity only for Siglec-6+ and CD3+ cell types (figure 3B). All DART–Fc T-biAbs demonstrated the ability to activate Jurkat-Lucia NFAT cells, with RC-1/V9 aDART–Fc appearing to be the most potent against Siglec-6+ cells without having background NFAT activation in the presence of Siglec-6− cells, which was observed with both sDART–Fc formats (figure 3C). The background NFAT activation may be attributable to the sDART–Fc format’s tendency toward aggregation (online supplemental figure S7A). RC-1/V9 aDART–Fc was also the most potent in terms of directed T-cell lysis of CLL cell lines, with an EC50 of 0.12 pM, a 25-fold increase in potency compared with the RC-1/V9 scFv–Fc (figure 3D). In these cytotoxicity assays, the fraction of T cells with both CD69 and CD25 activation markers was larger for the DARTs at 5 pM (~67%) than it was for the RC-1 clone in the scFv–Fc format (27.3%) (online supplemental figure S6C). Against the MEC1-hS6 cell line, the RC-1/V9 aDART–Fc resulted in higher levels of IFN-γ and IL-2 than the RC-1/V9 scFv–Fc, yet only the sDART–Fc formats yielded higher levels of TNF-α than the scFv–Fc, potentially indicating higher risk of CRS36 (figure 3E). Against MEC1-002 cells, IL-2 levels were higher with the aDART–Fc than the scFv–Fc, but IFN-γ and TNF-α levels were not significantly higher (online supplemental figure S6B). The aDART–Fc also outperformed both sDART–Fc constructs in terms of IFN-γ and IL-2 induction in the presence of these cells with low Siglec-6 copy number (online supplemental figure S6B). From these data, it was clear that RC-1/V9 aDART–Fc was the DART–Fc candidate with the most therapeutic potential, so we chose to investigate it further. To better predict responses in patients with CLL with lower Siglec-6 expression and lower E:T ratios, lysis assays were conducted against MEC1-002 (Siglec-6 antibody-binding capacity (ABC) = 1452) and MEC1-hS6 (Siglec-6 ABC=1 89 390) cell lines, titrating both the T-biAb and the effector cell number (online supplemental figure S8A–C). Specific lysis of MEC1-002 cells occurred down through the low E:T ratio of 1:30, and although the efficacy (maximum lysis) of both Siglec-6 and CD19-targeting T-biAbs decreased at low E:T ratios, the potency of RC-1/V9 aDART–Fc (EC50=1.1 pM) was still greater than CD19/V9 scFv–Fc (EC50=2.9 pM) (online supplemental figure S8D,E). These data strongly support RC-1/V9 aDART–Fc for potently and specifically killing target cells and activating T cells even with relatively few effector cells present and low target expression.

DART–Fc T-biAbs elicit more potent activation and lysis than scFv–Fc. (A) Cartoon depiction of Siglec-6×CD3 DART–Fc constructs. In all three constructs, Fc dimerization was mediated by knob-into-hole mutations. Two sDART constructs were made, alternating the positioning of VL and VH for each clone. sDART–Fc T-biAbs are labeled in terms of the VL clone at the N-terminus of the hole–Fc chain, RC-1/V9 sDART–Fc and V9/RC-1 sDART–Fc. The aDART–Fc construct consists of three polypeptide chains and was made by inserting oppositely charged coiled-coil domains at the C-terminus of each VH domain to stabilize the interaction between the free VL–VH chain and the two chains with dimerized Fc domains. (B) Siglec-6− CLL cell line MEC1, Siglec-6+ MEC1-hS6, and human T cells were stained with 10 nM of DART–Fc T-biAb and an antihuman Fc secondary antibody to validate T-biAb specificity for Siglec-6 (or CD19 as positive control) and CD3. (C) A Jurkat-Lucia NFAT reporter cell line was cultured overnight with scFv–Fc T-biAbs and MEC1 or MEC1-002 CLL cell lines (E:T=1:1) to determine T-cell activation in the absence or presence of target expression. (D) MEC1 and MEC1-hS6 cell lines were cocultured with human T cells (E:T=1:1) and a titration of the indicated T-biAbs. Following overnight incubation, cell lysis was assessed by intracellular luciferase activity. (E) Cytokine levels in the culture media from the overnight cytotoxicity assay were determined by ELISA, and data were normalized to the CD19/V9 positive control at the high dose (250 pM). Statistics were calculated using an unpaired t-test, n=3. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. aDART, asymmetrical dual-affinity retargeting; DART, dual-affinity retargeting; E:T, effector-to-target; IFN-γ, interferon gamma; IL, interleukin; NFAT, nuclear factor of activated T cells; ns, not significant; scFv, single-chain variable fragment; sDART, symmetrical dual-affinity retargeting; T-biAb, T cell-recruiting bispecific antibody; TNF-α, tumor necrosis factor alpha.
Efficacy of T-biAbs against primary CLL cells ex vivo
To evaluate the clinical utility of Siglec-6 targeting T-biAbs, they were tested against samples of treatment-naïve patients with CLL ex vivo. PBMC from patients with CLL (n=15, online supplemental table S2), with T cell : CLL cell ratios ranging from 1:15 to 1:200, were cultured with either scFv–Fc or aDART–Fc T-biAbs. After 11 days in culture, the only Siglec-6 T-biAb that resulted in statistically significant lysis of CLL cells was the RC-1/V9 aDART–Fc (median=24.9%); both the JML-1/V9 (median=8.8%) and RC-1/V9 scFv–Fc (median=2.4%) exhibited low target cell lysis (figure 4A and online supplemental table S3). Thus, these data agree with the findings from experiments on cell lines, that the aDART–Fc is the most effective T-biAb format for antibody clones targeting a membrane distal epitope on Siglec-6. An expanded CLL cohort (n=38) was tested against RC-1/V9 aDART–Fc in ex vivo cultures, and cell lysis was not significant until day 10, suggesting that T-cell expansion may be a factor for Siglec-6×CD3 T-biAbs (figure 4B, online supplemental figure S9A). T-cell expansion was evaluated at day 9 of ex vivo culture, and this verified that both CD4+ and CD8+ T-cell subsets proliferated and upregulated activation markers in response to T-biAb treatment, indicating that activation and expansion are occurring by this time point (online supplemental figure S10A,B). To determine whether CLL lysis is dependent on T-cell number and/or quality, allogeneic PBMC from an HD were cultured with primary CLL cells at an E:T ratio of 1:1. T-biAb-mediated specific lysis was significant at day 7 of this coculture, and higher in comparison to an autologous culture run in parallel, suggesting that both low E:T ratio and T-cell dysfunction in treatment-naïve CLL samples37 play a role in limiting efficacy of Siglec-6×CD3 T-biAbs (figure 4C,D). As expected, RC-1/V9 aDART–Fc-mediated primary CLL cell lysis correlated with Siglec-6 expression at baseline (figure 4E). Interestingly, there was a correlation between Siglec-6 expression and E:T ratio at baseline, indicating a potential relationship between immune activation (exemplified by expansion of T cells) and expression of Siglec-6 in B cells (online supplemental figure S9B). When patients with CLL were grouped based on Siglec-6 expression, specific lysis was higher in the Siglec-6+ group when treated with RC-1/V9 aDART–Fc (online supplemental figure S9C). The increased lysis of Siglec-6+ CLL cells by RC-1/V9 aDART–Fc was dependent on Siglec-6 expression, and not E:T ratio, because specific lysis did not increase between Siglec-6+ and Siglec-6− primary CLL cell samples with the CD19/V9 scFv–Fc. Since most patients with CLL receive BTKis such as ibrutinib or acalabrutinib as a front-line treatment, we assessed Siglec-6 expression on a cohort of patients (n=7) before (pre-BTKi) and while undergoing (on-BTKi) treatment (online supplemental figure S11A–C). We found that Siglec-6 expression does not change on-BTKi, although the E:T ratio increased as anticipated. RC-1/V9 aDART–Fc cytotoxicity assays with BTKi-treated patients revealed significant levels of CLL cell lysis at day 3 and day 11 of ex vivo culture, suggesting that Siglec-6 directed therapies will maintain efficacy in combination with BTKi (online supplemental figure S11D,E, online supplemental table S4).
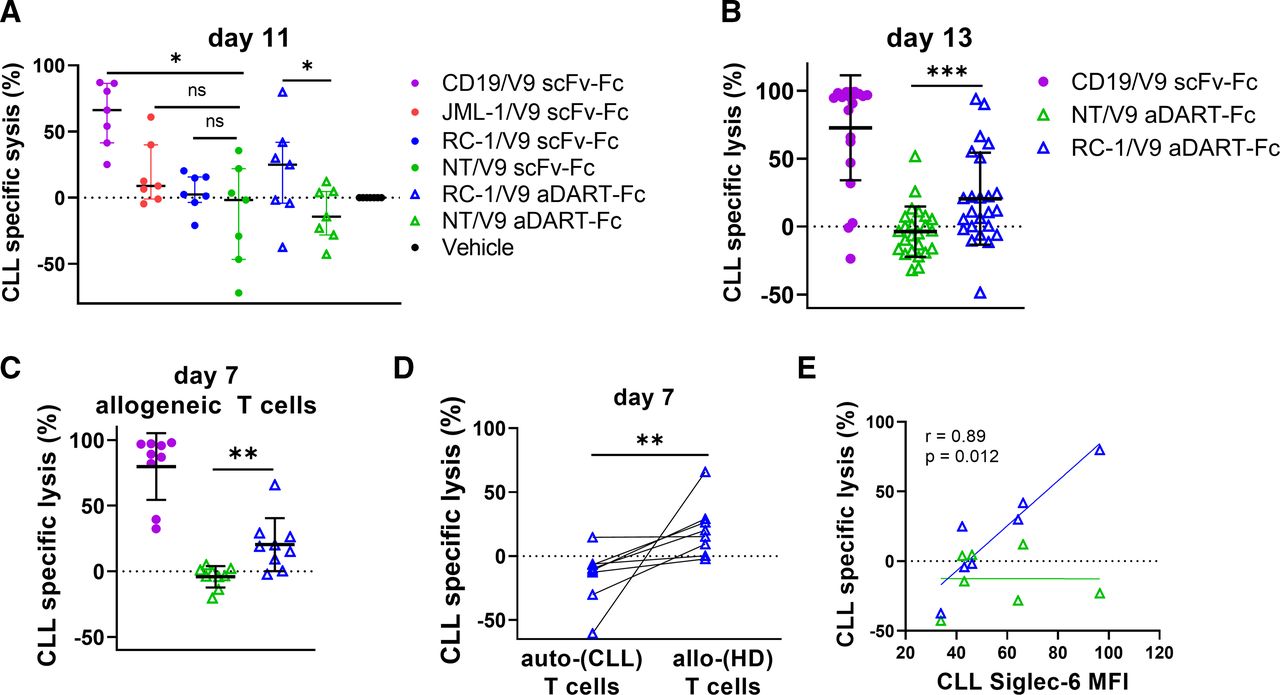
RC-1/V9 aDART–Fc mediates lysis of primary CLL B cells. (A) Treatment-naive CLL PBMC (n=7) were cultured with 6 nM T-biAbs for 11 days and analyzed by flow cytometry to determine specific lysis of CLL cells by autologous T cells at endogenous E:T ratios. The activity of each T-biAb was compared with the corresponding NT control of the same construct. (B) CLL PBMC were cultured (as in A) but for 13 days on a larger set of patients (n=24). (C) To assess lysis by allogeneic (allo-) T cells, HD PBMC were cocultured with CLL PBMC (n=9) at an E:T ratio of 1:1, and specific lysis was assessed on day 7. (D) Comparing T-biAb-mediated specific lysis of CLL cells from individual patients (n=8) by autologous (auto-) or allo-T cells after 7 days of ex vivo culture. (E) Cell lysis at day 11 correlated with Siglec-6 expression levels as determined by Spearman’s correlation analysis. All pairwise comparisons were made using the Wilcoxon matched-pair signed-rank test. *P<0.05, **P<0.01, ***P<0.001. aDART, asymmetrical dual-affinity retargeting; CLL, chronic lymphocytic leukemia; HD, healthy donor; MFI, mean fluorescence intensity; ns, not significant; E:T, effector-to-target; NT, non-targeting; T-biAb, T cell-recruiting bispecific antibody.
Although T-biAbs targeting CD19 were more effective than those targeting Siglec-6, CD19 is a marker found on all healthy B cells, while Siglec-6 expression is restricted to a subset of activated B cells. In ex vivo cultures of HD PBMC (using conditions identical to the CLL cytotoxicity assays), the CD19/V9 scFv–Fc killed a larger fraction of healthy B cells at the day 11 time point compared with the RC-1/V9 aDART–Fc and the non-targeting NT/V9 aDART–Fc (online supplemental figure S12A). Notably, there was no significant difference between the latter two, despite the fact that the E:T ratio in these HD PBMC was up to 1000-fold higher than in the CLL PBMC (online supplemental figure S12A,B, and table S5). Since a subset of HD B cells express Siglec-6 (median=23.3%), some lysis was still observed from RC-1/V9 aDART–Fc. As expected, the fraction of viable healthy B cells expressing Siglec-6 was lower in the RC-1/V9 aDART–Fc treatment group than in the NT/V9 aDART–Fc or vehicle control groups, suggesting the Siglec-6+ fraction of healthy B cells were selectively depleted (online supplemental figure S12C). Since the overall percent lysis was higher than expected based on the initial size of the Siglec-6+ fraction of B cells, we wondered whether the culture conditions facilitated the outgrowth of activated B cells (which encompasses the Siglec-6+ fraction of healthy B cells). The fraction of B cells expressing Siglec-6 increased slightly over the course of these experiments in both the vehicle and NT/V9 aDART–Fc treatment groups, yet the difference was not significant (online supplemental figure S12D). These data collectively indicate that Siglec-6-targeting T-biAbs eliminate Siglec-6-expressing CLL and healthy B cells but spare the bulk of healthy B cells.
Investigating the basis for differential activity of T-biAb formats
To determine whether T-biAb format affected the affinity to Siglec-6 or CD3 on the cell surface, T-biAbs were titrated and used to stain cells for flow cytometry (figure 5A). In terms of Siglec-6 binding, RC-1/V9 aDART–Fc (apparent Kd=0.79 nM) fell between the scFv–Fcs JML-1/V9 (apparent Kd=0.61 nM) and RC-1/V9 (apparent Kd=1.1 nM). Against T cells, CD3 binding appeared to be more potent by a factor of two for the scFv–Fc than for the aDART–Fc. To quantify the affinities more accurately, we used SPR, which revealed that the affinity to both Siglec-6 and CD3 was slightly higher for the RC-1/V9 scFv–Fc than the RC-1/V9 aDART–Fc (figure 5B). However, these differences were subtle, and both in the same direction, thus the format change did not reverse the target preference ratio. Therefore, affinity alone cannot account for the >10-fold higher potency of the DART–Fc over the scFv–Fc in lysis assays.
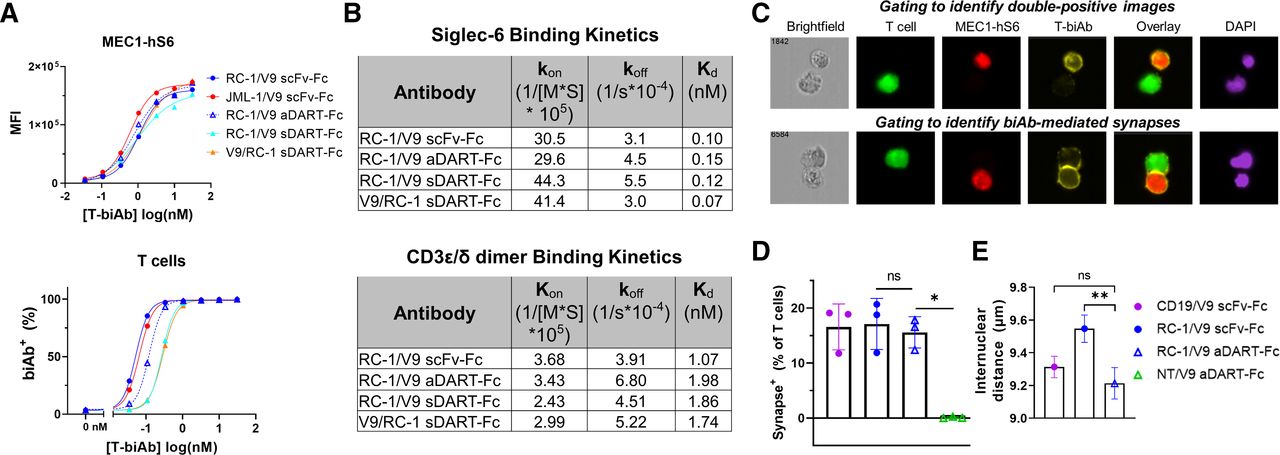
aDART–Fc binds with similar affinity but creates a shorter synapse than scFv–Fc. (A) MEC1-hS6 (top) or primary human T cells (bottom) were stained with a titration of T-biAbs, and binding was detected by the addition of an antihuman Fc secondary antibody. (B) RC-1/V9 T-biAbs were immobilized to a CM5 Biacore chip via the Fc domain, soluble Siglec-6 or CD3ε/δ dimer was injected, and SPR sensorgrams were used to determine kon, koff, and Kd. (C) FarRed-labeled MEC1-hS6 cells (1×106 cells) were bound with 50 nM T-biAb, washed, and cocultured with CFSE-labeled Jurkat cells (1×105 cells) for 1 hour at 37°C followed by fixation, permeabilization, and staining with Cy3-conjugated antihuman Fc and DAPI. Samples were processed on an Amnis ImageStreamx MK II (Luminex) instrument and analyzed by IDEAS V.6.3 software to identify CFSE+/FarRed+ images (top) and T-biAb+ synapses (bottom). (D) The percentage of T cells forming synapses was quantified for three independent experiments and data were compared using a paired t-test. (E) Internuclear distance was calculated by measuring the distance between synapse-positive cells in the DAPI channel. The mean and SE of the mean for approximately 1000 events per sample are plotted from one representative experiment and statistics were calculated using an unpaired t-test, *P<0.05, **P<0.01. aDART, asymmetrical dual-affinity retargeting; CFSE, carboxyfluorescein succinimidyl ester; DAPI, 4′,6-diamidino-2-phenylindole; scFv, single-chain variable fragment; SPR, surface plasmon resonance; T-biAb, T cell-recruiting bispecific antibody.
To quantify characteristics of the synapse, we used multispectral fluorescence imaging. To control for any subtle differences in binding orientation of different clones, we focused on the RC-1 clone and compared the scFv–Fc and aDART–Fc formats, since they showed 25-fold difference in cytotoxicity EC50 (figure 3D), and the aDART–Fc lacked the non-specific background attributed to sDART–Fc aggregation (figure 3C and online supplemental figure S7A). Target (MEC1-hS6) and effector (Jurkat T cells) cells were cocultured with T-biAb, fixed, and analyzed. Initial gating to identify double-positive images containing both cell types lacked the stringency to selectively identify synapses (figure 5C, top). However, additional gating to require overlap between target and effector cells, as well as colocalization of T-biAb at the synapse, clearly delineated images with true T-biAb-mediated synapses (figure 5C, bottom). The percentage of T cells forming synapses was similar for CD19/V9 scFv–Fc, RC-1/V9 scFv–Fc, and RC-1/V9 aDART–Fc, confirming that at a saturating concentration, all constructs are equally adept at forming synapses (figure 5D). To investigate our initial hypothesis, namely, that the DART–Fc would result in a shorter synapse, we measured the distance between the nuclei in each synapse+ cell pair, as a proxy for synapse length (figure 5E). This revealed that the RC-1/V9 aDART held cells closer together than the RC-1/V9 scFv–Fc. Accordingly, the improvement in efficacy of the DART–Fc may be attributed to the shorter or more rigid synapse formed when compared with a scFv–Fc T-biAb.
RC-1/V9 aDART–Fc activity in vivo
To characterize the function of the RC-1/V9 aDART–Fc in vivo, we generated a MEC1-fLuc-Siglec-6 xenograft mouse model in NSG mice as a systemic cell line-derived xenograft (CDX) mouse model of CLL. After the first treatment with low dose (0.05 mg/kg) or high dose (0.5 mg/kg) RC-1/V9 aDART–Fc, the leukemic burden was significantly reduced compared with the NT/V9 aDART–Fc (figure 6A,B). By day 21, the MEC1-hS6 cells were cleared in mice treated with Siglec-6-targeting and CD19-targeting T-biAbs. No signs of treatment-related toxicity or decrease in body weight were observed over the course of the study (figure 6C). By day 21 and later, the tumor burden decreased in some T cells only and NT/V9 aDART–Fc-treated mice, which was attributable to allogeneic killing—a common phenomenon in CDX models.25 Despite this non-specific T-cell activity, the 0.05 mg/kg dose of RC-1/V9 aDART–Fc significantly extended the overall survival compared with the negative controls, similar to the CD19/V9 scFv–Fc treatment (figure 6D). Surprisingly, no dose–response was observed between the 0.05 mg/kg and 0.5 mg/kg doses of RC-1/V9 aDART–Fc. The only relapse observed was in the high-dose group, suggesting that 0.5 mg/kg may be above the optimal activity window for the three-component binding model, resulting in a hook effect.38 A follow-up pharmacokinetic study revealed that the half-life of the aDART–Fc in NSG mice is 7.9 days after conditioning with human serum and intravenous injection. Therefore, RC-1/V9 aDART–Fc would have accumulated over the course of the study with dosing two times per week, reaching 100 nM in the blood, ~800-fold higher than the EC50 for lysis in vitro (online supplemental table S6). Collectively, these results demonstrate that a Siglec-6×CD3 T-biAb in aDART–Fc format is highly potent at clearing CLL in vivo, has long circulatory half-life, and warrants further preclinical studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RC-1/V9 aDART–Fc clears CLL cells in a CDX model. NSG mice were inoculated intravenously with 2×106 MEC1-fLuc-hS6 cells and on day 7 were treated with 3×106 T cells intravenously, followed by treatment two times per week with T-biAb (0.5 mg/kg or 0.05 mg/kg) for 2 weeks. On day 13, mice received a second injection of only 1×106 T cells. (A) Bioluminescence images and (B) quantification (one line per mouse) showed complete clearance of CLL cells in mice treated with T cells and CD19/V9 or RC-1/V9 T-biAbs, but not the NT/V9 T-biAb. Statistics were calculated comparing both doses of RC-1/V9 with NT/V9 using an unpaired t-test with Welch’s correction, n=5 mice per group (**p<0.01, ***p<0.001, # = image saturation). (C) Weight loss was not observed in any animals over the course of treatment. (D) Kaplan-Meier survival analysis showed that RC-1/V9 (0.05 mg/kg) and CD19/V9 (0.5 mg/kg) both improved survival over the NT/V9 control (0.5 mg/kg). Statistics were calculated using the Mantel-Cox log-rank analysis, HR=9.95 (*p<0.05). aDART, asymmetrical dual-affinity retargeting; ns, not significant; NT, non-targeting; scFv, single-chain variable fragment; T-biAb, T cell-recruiting bispecific antibody.
Discussion
Siglec-6 is an emerging target for cancer immunotherapy that offers increased specificity over conventional CLL targets which are universal B-cell markers. We identified high-affinity, patient-derived anti-Siglec-6 mAbs that hold translational potential, given that they arose in a human allogeneic setting and may have contributed to curing CLL in this patient. We mapped the epitope of these mAbs to the N-terminal lectin domain, raising the possibility there may be functional consequences of interfering with siglec–glycan interactions.
We engineered Siglec-6 clones into T-biAbs and demonstrated the potency of this modality for killing Siglec-6+ CLL cells while sparing the majority of healthy B cells, which are Siglec-6−. In vitro, T-biAbs directed the killing of target cells with subpicomolar potency and activated T cells to secrete type I cytokines. We found that Siglec-6 x CD3 scFv–Fc only weakly induced IL-2 secretion, which we attributed to the membrane–distal epitope location on Siglec-6. We were able to improve T-biAb activity via antibody engineering into the DART–Fc format, increasing lysis potency >10-fold and IL-2 levels >2-fold. Mechanistically, neither the binding kinetics nor the number of synapses formed favored the DART–Fc versus the scFv–Fc, as has been reported when comparing DARTs and BiTEs targeting CD19.35 Rather, we observed the formation of tighter synapses when using the same anti-Siglec-6 clone in aDART–Fc versus scFv–Fc format, and the reduction in synapse length is known to affect T-biAb function.32 39 One limitation of our work was using a flow cytometry-based system that lacks the resolution to measure the distance between opposing membranes in a synapse. As a correlate for synapse length, we measured internuclear distance of floating cell conjugates. The benefit of the approach was the throughput—it allowed us to capture enough events for a Gaussian distribution to reveal that RC-1 clone in the aDART–Fc format held cells closer together than in the scFv–Fc format, but not in comparison to the scFv–Fc targeting CD19, which has one fewer extracellular domain than Siglec-6. Thus, the DART–Fc, which constrains its paratopes by covalent linkage at each end, appears to reduce the effective synapse length by approximately one Ig-like domain. The aDART antibody engineering strategy employed here illustrates a blueprint for targeting membrane–distal epitopes with T-biAbs that can be applied to other targets.
The RC-1/V9 aDART–Fc mediated the killing of CLL cells ex vivo by autologous T cells at endogenous E:T ratios for patients with CLL (1:10 to 1:100), and lysis correlated with Siglec-6 expression, indicating that Siglec-6 targeted therapies will be most effective for the ~50% of patients that overexpress Siglec-6. The CD19/V9 scFv–Fc is more efficacious, as one might anticipate, given the higher expression of CD19. However, CD19-targeted therapy has not been approved in CLL, and CD19 is a universal B-cell marker, like CD20, so its targeting results in humoral immunodeficiency.8 We observed that only a fraction of HD B cells are killed ex vivo with RC-1/V9 aDART–Fc, since only ~23% of healthy B cells express Siglec-6, indicating that Siglec-6 targeting will not eliminate all B cells in vivo, as CD20-targeting mAbs do. Additional studies will be needed to assess Siglec-6 expression on healthy circulating and tissue-resident B cells and mast cells of patients with CLL and the effects of Siglec-6 targeting on these hematopoietic cell populations in vivo. Notably, Siglec-6 expression is maintained on CLL cells from BTKi-treated patients, unlike targets such as CD20, which can be downmodulated by BTKi.40 Importantly, treatment of CLL patients with a BTKi has been reported to enhance T-cell cytotoxicity in response to a CD19×CD3 T-biAb.17 18 Here, we have demonstrated efficacy of Siglec-6×CD3 T-biAbs in PBMC from patients treated with BTKi. Additional studies on combination treatments with other CLL therapeutics as well as checkpoint inhibitors may further increase responses.
Since little is known regarding the function of Siglec-6 in CLL, the possibility of downmodulation in response to targeted treatment remains a potential concern for therapeutic development that must be investigated further. However, Siglec-6 expression is reportedly higher in the proliferative fraction (CXCR4dimCD5bright),41 so downmodulation may reduce clonal expansion in vivo.
Concurrent to our T-biAb efforts, two groups developed chimeric antigen receptor (CAR)-T cells using our original anti-Siglec-6 clone, JML-1, for the treatment of CLL and acute myeloid leukemia (AML).25 29 Their findings corroborate ours and demonstrate a proof of concept for targeting Siglec-6-expressing cells with T cell-based therapies. However, our study introduces RC-1, a novel human anti-Siglec-6 clone with sixfold higher affinity than JML-1. We found that T-biAbs induce higher levels of IL-2 secretion using RC-1 versus JML-1. While the JML-1 CAR-T does not elicit IL-2 in the presence of cells with low target expression,25 29 the RC-1/V9 aDART–Fc results in high levels of IL-2 secretion, even with low target copy number. Additionally, our work expanded the number of patients with CLL tested and revealed higher Siglec-6 expression in patients who have undergone BTKi therapy. In vivo, the RC-1/V9 aDART–Fc eliminated a CLL cell line, extended survival at a low dose of 0.05 mg/kg, and demonstrated a half-life of over 1 week. In comparison to CAR-T cells, T-biAbs offer the advantage of being an off-the-shelf therapy for CLL, which will be accessible to more patients, at a lower cost, and with shorter time to treatment.
In conclusion, Siglec-6 is an excellent target for next-generation CLL treatments. Beyond CLL, there are additional indications that exhibit specific expression including AML,29 42 mast cell disorders (eg, mastocytosis),14 and prenatal complications (eg, gestational trophoblastic disease and pre-eclampsia).43 44 Pending further investigation into its immunoinhibitory signaling capacity, solid tumors could also benefit from blocking Siglec-6 on tumor-infiltrating mast cells.45
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human subjects. Cryopreserved chronic lymphocytic leukemia (CLL) PBMC were derived from treatment-naïve patients enrolled in an observational study at the National Institutes of Health (NIH) Clinical Center (www.clinicaltrials.gov; NCT00923507). Cryopreserved BTKi-treated samples were obtained from patients with CLL enrolled in phase II clinical trials at the NIH Clinical Center investigating single agent ibrutinib (NCT01500733) or acalabrutinib (NCT02337829). The studies were approved by the institutional review boards of the NIH Clinical Center (#08-H-0105, #12-H-0035, and #15-H-0016) and The Scripps Research Institute (#18-7179) and were in accordance with the Declaration of Helsinki. All patients gave written informed consent to participate in the study before taking part.All procedures were approved by the Institutional Animal Care and Use Committee (IACUC #15–026) of The Scripps Research Institute (Jupiter, Florida, USA) and were performed according to the NIH Guide for the Care and Use of Laboratory Animals.
Acknowledgments
We are grateful to Drs James C Paulson, Matthew E Pipkin, and Travis S Young for generously providing their expertise and feedback. We thank Alta Johnson for assistance with flow cytometry and fluorescence microscopy experiments. We also like to thank Dr Raquel Dias for protein modeling advice. Additionally, we are grateful to our collaborators who provided CLL cell lines, including Dr Natarajan Muthusamy, Jessica Nunes, and Dr Ian McNiece.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @CyrialDilutions
Contributors Conceptualization: CR and MGC; methodology: MGC, MM, JQ, HP and JC; investigation: MGC, MM, DE, JH and SJN; visualization: MGC, VVC and EMG; analysis: MGC, MM, EMG, TV, SJN, PRG, AW and CR; funding acquisition: CR, MGC, PRG and AW; writing (original text): MGC and VVC; writing (review and editing): all authors. CR is responsible for the overall content as the guarantor.
Funding This study received funding support from the following: National Cancer Institute (NCI), National Institutes of Health (NIH), through grant R21 CA229961 awarded to CR; NCI, NIH, through grant R01 CA174844 awarded to CR; National Center for Advancing Translational Sciences and NIH, through grant TL1 TR002551 awarded to MGC; National Heart, Lung, and Blood Institute, NIH Intramural Research Program, supporting AW, EMG, MM, DE, and JH.
Competing interests CR and MGC are inventors of a patent application for recombinant human antibodies targeting Siglec-6 and their applications.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.