Article Text

Abstract
Background Chimeric antigen receptor (CAR) T cell therapy targeting B cell maturation antigen (BCMA) on multiple myeloma (MM) produces fast but not long-lasting responses. Reasons for treatment failure are poorly understood. CARs simultaneously targeting two antigens may represent an alternative. Here, we (1) designed and characterized novel A proliferation inducing ligand (APRIL) based dual-antigen targeting CARs, and (2) investigated mechanisms of resistance to CAR T cells with three different BCMA-binding moieties (APRIL, single-chain-variable-fragment, heavy-chain-only).
Methods Three new APRIL-CARs were designed and characterized. Human APRIL-CAR T cells were evaluated for their cytotoxic function in vitro and in vivo, for their polyfunctionality, immune synapse formation, memory, exhaustion phenotype and tonic signaling activity. To investigate resistance mechanisms, we analyzed BCMA levels and cellular localization and quantified CAR T cell–target cell interactions by live microscopy. Impact on pathway activation and tumor cell proliferation was assessed in vitro and in vivo.
Results APRIL-CAR T cells in a trimeric ligand binding conformation conferred fast but not sustained antitumor responses in vivo in mouse xenograft models. In vitro trimer-BBζ CAR T cells were more polyfunctional and formed stronger immune synapses than monomer-BBζ CAR T cells. After CAR T cell–myeloma cell contact, BCMA was rapidly downmodulated on target cells with all evaluated binding moieties. CAR T cells acquired BCMA by trogocytosis, and BCMA on MM cells was rapidly internalized. Since BCMA can be re-expressed during progression and persisting CAR T cells may not protect patients from relapse, we investigated whether non-functional CAR T cells play a role in tumor progression. While CAR T cell–MM cell interactions activated BCMA pathway, we did not find enhanced tumor growth in vitro or in vivo.
Conclusion Antitumor responses with APRIL-CAR T cells were fast but not sustained. Rapid BCMA downmodulation occurred independently of whether an APRIL or antibody-based binding moiety was used. BCMA internalization mostly contributed to this effect, but trogocytosis by CAR T cells was also observed. Our study sheds light on the mechanisms underlying CAR T cell failure in MM when targeting BCMA and can inform the development of improved treatment strategies.
- cell engineering
- immunotherapy
- receptors, chimeric antigen
- hematologic neoplasms
- T-lymphocytes
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Chimeric antigen receptor (CAR) T cell therapy for multiple myeloma (MM) mostly targets B cell maturation antigen (BCMA) as a single antigen. While very good responses are achieved, durability is limited, and most patients experience relapse. A proliferation inducing ligand (APRIL)-CARs have been designed for dual antigen targeting and overcoming BCMA loss, but APRIL-CAR T cell therapy using a monomeric APRIL binding moiety was not successful in a clinical trial. Trimeric APRIL provides a better binding to both target antigens than monomeric APRIL when incorporated into a CAR. While trimeric APRIL CAR T cells produced fast responses in preclinical models, durability of response was not assessed.
What this study adds
This study identified the molecular determinants in monomeric and trimeric APRIL CAR designs that are associated with CAR T cell performance. BBζ APRIL CAR T cells with a trimeric binding moiety formed stronger immune synapses than monomeric BBζ CARs. APRIL CAR T cells produced fast antitumor responses in vivo in mouse xenograft models, but responses were not sustained. Contact of CAR T cells with tumor cells led to rapid downmodulation of BCMA on target cells and occurred with all three binding moieties evaluated. BCMA downmodulation was mostly due to internalization by myeloma cells, but also trogocytosis by CAR T cells. Non-functional CAR T cells activated BCMA pathway through loose interactions but did not promote tumor progression.
How this study might affect research, practice or policy
This study sheds light on the mechanisms underlying CAR T cell failure in MM when targeting BCMA and can inform the development of improved treatment strategies for the future.
Introduction
Chimeric antigen receptor (CAR) T cell immunotherapy for multiple myeloma (MM) is currently becoming standard of care in advanced MM.1 2 Most approaches currently focus on targeting B cell maturation antigen (BCMA) as a single target because BCMA is critical for maintaining tumor cell phenotypes and expression is limited to the B and plasma cell lineage.3–8 While fast and deep antitumor responses are consistently reported across clinical trials, the duration of response is quite limited and most patients experience relapse and progression.9 10 The reasons for treatment failure are not well understood, but likely involve several mechanisms: (1) antigen escape under immune pressure, (2) insufficient function or exhaustion of circulating CAR T cells, (3) limited CAR T cell migration and persistence, and (4) immune suppression in the MM bone marrow microenvironment. Finally, it is not known whether non-productive binding of persisting exhausted or non-functional CAR T cells to MM target cells could stimulate BCMA pathway activity and contribute to protumorigenic events in residual tumor cells fueling rapid tumor progression. Complete antigen loss has only been described in two patients so far and does not seem to be a frequent event.9 11 12
A proliferation inducing ligand (APRIL) is the natural high affinity ligand for two receptors expressed on MM, BCMA and transmembrane activator and CAML interactor (TACI). APRIL-based CARs can overcome BCMA antigen escape in preclinical models,7 13 but an early phase clinical trial with a CAR using an APRIL monomer as a ligand binding moiety did not confirm efficacy in patients.14 Preclinically, a trimeric APRIL configuration yielded better polyfunctionality and short term antitumor function of CAR T cells than monomeric APRIL,13 but information on long term in vivo tumor control, sustainability of responses and possible mechanisms of resistance is lacking.
Here, we used molecular modeling and developed and characterized three distinct new APRIL CARs, one monomer and two trimer based. We deeply characterized their properties when expressed in human T cells in functional assays in vitro and in vivo. We identified the molecular determinants in monomeric and trimeric APRIL CAR designs that are associated with CAR T cell performance. APRIL CAR T cells with a trimeric binding moiety formed stronger immune synapses and were more polyfunctional than the monomer-based CAR with the BBζ endodomain. In vivo, trimer BBζ CAR T cell therapy produced fast antitumor responses, but tumors recurred in the follow-up.
Second, we investigated mechanisms of CAR T cell failure in preclinical MM model systems by comparing a clinically used single chain variable fragment (scFv) based CAR (11D5-3),3 15 a human heavy-chain-only (FHVH33) based CAR under clinical development,16 and our newly designed APRIL CARs. After contact of CAR T cells with myeloma cells, we found rapid BCMA downmodulation on target cells independently of the targeting moiety used. Both CAR T cells and MM cells were actively involved in this process with trogocytosis by CAR T cells and BCMA internalization in MM cells.
Hence, our study provides a better understanding of the function of APRIL CAR T cells and critical insights into determinants of CAR T cell therapy failure in MM when targeting BCMA.
Methods
A detailed description of all methods is provided in the online supplemental materials document, including molecular modeling, immunophenotyping and cell sorting, Image Stream analysis, enzyme-linked immunosorbent assay (ELISA), Mesoscale Discovery (MSD) assay, proliferation assay, mouse models, immunofluorescent staining, confocal imaging and light sheet live microscopy.
Supplemental material
Cell lines
MM.1S cells were purchased from the American Type Culture Collection. K562 (chronic myeloid leukemia) and NCI-H929 (MM) were purchased from the German Cell Culture Collection. KG-1a (acute myeloid leukemia) cells were a gift from Dr. Stephen Gottschalk, Center for Cell and Gene Therapy, Baylor College of Medicine, Houston, USA. All lines were maintained according to the suppliers’ instructions. Cell lines were authenticated in January 2022 by short tandem repeat (STR) profiling (Microsynth).
Generation of retroviral vectors
Plasmids encoding for human truncated APRIL (115–250 a.a, Uniprot O75888, excluding the heparan sulfate proteoglycan binding sites of APRIL),17 18 full-length BCMA (Uniprot Q02223), full-length TACI (Uniprot O14836), 11D5-3 single chain variable fragment (scFv)3 and the human heavy-chain-only FHVH3316 were codon-optimized and synthesized by GeneArt (Thermo Fisher Scientific). Retroviral constructs encoding APRIL CARs, 11D5-3 and FHVH33 CARs, BCMA, TACI and BCMA-Emerald were generated using the In-Fusion HD Cloning Kit (Takara, #638933) and expressed in the pSFG retroviral backbone.
Peripheral blood mononuclear cells from healthy human donors
Buffy coats from deidentified healthy human volunteer blood donors were obtained from the Center of Interregional Blood Transfusion SRK Bern (Bern, Switzerland).
Generation of CAR T cells and transgenic cell lines
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by density gradient centrifugation (Lymphoprep, StemCell #07851). PBMCs were activated on plates coated with anti-CD3 (1 mg/mL, Biolegend, #317347, clone: OKT3) and anti-CD28 (1 mg/mL, Biolegend, #302934, clone: CD28.2) antibodies in T cell medium (RPMI containing 10% FBS, 2 mM L-Glutamine, 1% Penicilin-Streptomycin) with IL-15 and IL-7 (Miltenyi Biotec, 10 ng/mL each, #130-095-362 and #130-095-765, respectively) (online supplemental table 1). The day before transduction, a non-tissue culture treated 24-well plate (Grener Bio one, #662102) was coated with retronectin (Takara Bio, #T100B) in PBS (7 µg/mL, 1 mL per well), and incubated overnight at 4°C. Three days after activation, retronectin was removed and the plate was blocked with T cell medium for 15 min at 37°C. Then, media was removed, and retroviral supernatant was centrifuged onto the retronectin coated plate at 2000 g for 1 hour at 32°C. Retroviral supernatant was gently removed and activated T cells were added at 0.15×106 cells/mL. The plate was centrifuged at 1000 g for 10 min at 21°C. Cells were then incubated at 37 °C/ 5% CO2 for 3 days. After 48 to 72 hours of transduction, T cells were harvested and further expanded in T cell medium containing IL-7 and IL-15. In selected experiments, CAR T cells were positively selected with a CD271 selection kit (EasySep, #17849) to enrich for transduced T cells. The same protocol was used for the generation of transgenic tumor cell lines. After expansion in the appropriate medium, transduction efficiency was checked by flow cytometry for BCMA and/or TACI, or GFP expression, and transgenic cells were purified by fluorescence activated cell sorting (FACS).
Supplementary video
Sequential coculture assay
Target cell lines expressing or not GFP-FFLuc were cocultured with T cells in replicate wells at an effector to target (E:T) ratio of 1:1 (200,000 cells each in a 48-well plate). After 3–4 days, residual tumor cells and T cells were quantified by FACS combining antibody staining and absolute counting beads (CountBright Beads, Life Technologies #C36950). Fresh tumor cells were added back to replicate wells when >85% of tumor cells were killed at the analysis timepoint. Otherwise, the killing was considered incomplete and T cells were not rechallenged with fresh tumor cells.
Mouse xenograft experiments
All animal studies were conducted in accordance with a protocol approved by the Veterinary Authority of the Swiss Canton of Vaud and performed in accordance with Swiss ethical guidelines (authorization VD3390). NOD-SCID-γc-/- (NSG) mice were bred and maintained at the animal facility of the University of Lausanne. Animals were 7–11 weeks old at the start of the experiments, both males and females were used, and experimental groups were randomized based on animal weight at start of experiment. Specific information for each model used is provided in online supplemental materials.
Statistical analysis
Descriptive statistics were used to summarize data. For continuous variables, normality distribution was analyzed, and comparisons were made by t-test. When normality testing failed, pair-wise comparison of conditions was performed using Mann-Whitney U-test. Area under the curve comparisons were analyzed with unpaired t-test and Welch’s correction (T cell expansion in vitro), or unpaired Mann-Whitney test (non-parametric t-test) (in vivo bioluminescent imaging). Sequential co-cultures and survival of mice were analyzed by Kaplan-Meier method and significance assessed with log-rank (Mantel-Cox) test. Multiple comparisons (eg, BCMA cell surface levels, analysis of sBCMA, T cell phenotypes) were performed by repeated measures one-way analysis of variance (ANOVA) with Tukey’s multiple comparison. Analyses were performed in GraphPad Prism V. 8.1.2 or higher. Differences in T cell polyfunctionality was analyzed by dependent t-test for paired samples using Python scipy.stats package (V.1.5.4), where cells co-expressing three or more evaluated markers simultaneously were considered to be polyfunctional. P values <0.05 were considered significant, and the levels of significance are indicated in the figures and legends.
Results
APRIL CARs recapitulate physiological target recognition of BCMA and TACI and can be efficiently expressed in human T cells
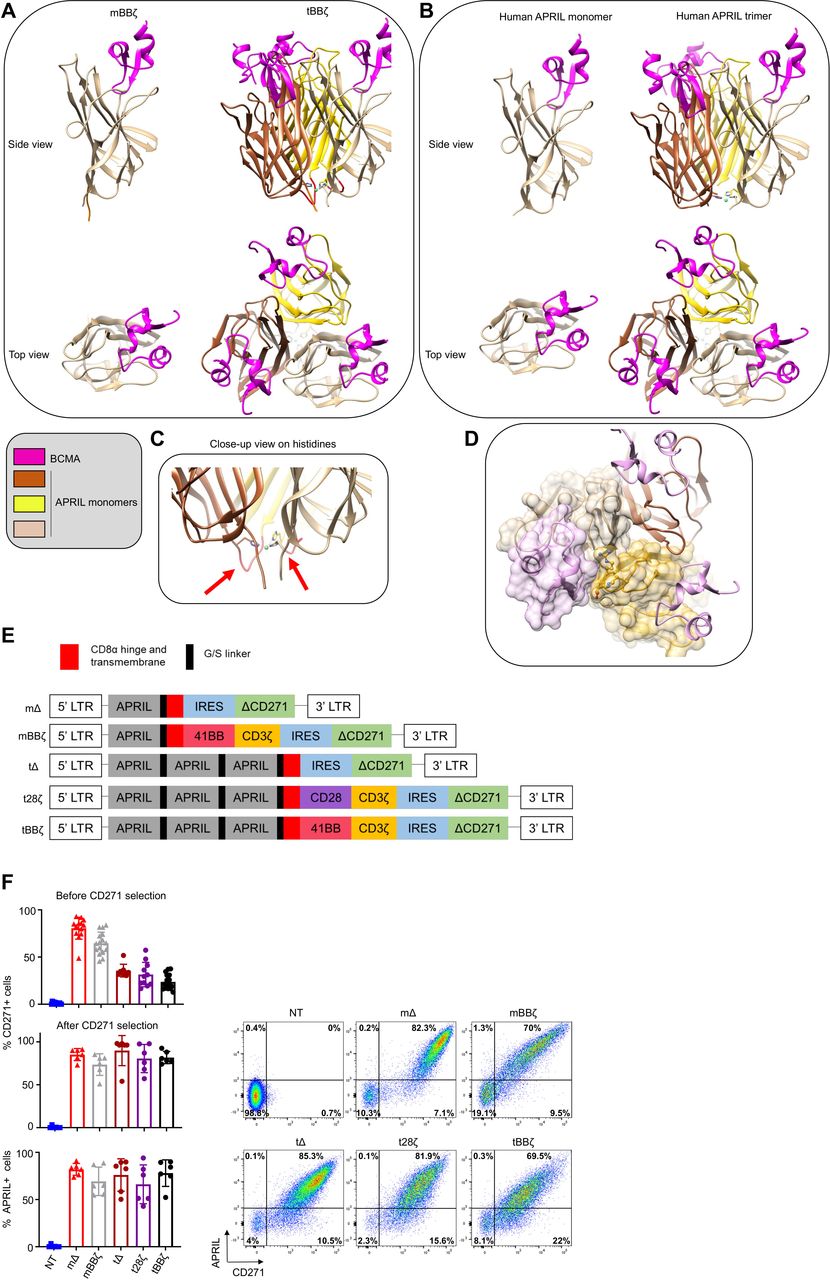
To investigate the optimal configuration of APRIL CARs to target MM, we performed molecular modeling of monomer (m) and trimer (t) APRIL CARs in contact with their natural target receptors BCMA and TACI and compared their binding interface to soluble human APRIL (figure 1A,B for BCMA and online supplemental figure 1A,B for TACI). A potentially important role for the histidine residues in position 115 of APRIL is illustrated in the trimer model, as the histidines form close contacts with each other and stabilize the trimer structure around a metal ion (figure 1C). From a structural point of view, the tCAR better recapitulates the physiological target recognition than the mCAR because optimal monomer binding requires binding support of some residues from a second monomer (figure 1D). Absence of a second monomer results in a loss of interaction surface area close to 200 Å2 (online supplemental text 1). Moreover, exposing to solvent some non-polar residues normally buried inside binding interfaces could weaken the stability of the mCAR (online supplemental text 2 and figure 1C). Because our modeling suggested a more physiological binding interface for tCARs than for mCARs, we designed the glycine-serine linker between APRIL monomers with a length that accommodates optimal trimer folding. We used a five amino acid linker with the sequence GGGGS between each truncated APRIL monomer and between the last APRIL monomer and the CD8α hinge and transmembrane sequence. APRIL mCAR and tCAR retroviral vector constructs were generated, including non-signaling control Δ and second generation 28ζ and BBζ CARs (figure 1E). Our tBBζ CAR construct is significantly different from the previously reported TriPRIL CAR (online supplemental figure 2).13 Primary human activated T cells were efficiently transduced with all our constructs, expanded, and CARs detected on the cell surface (figure 1F).
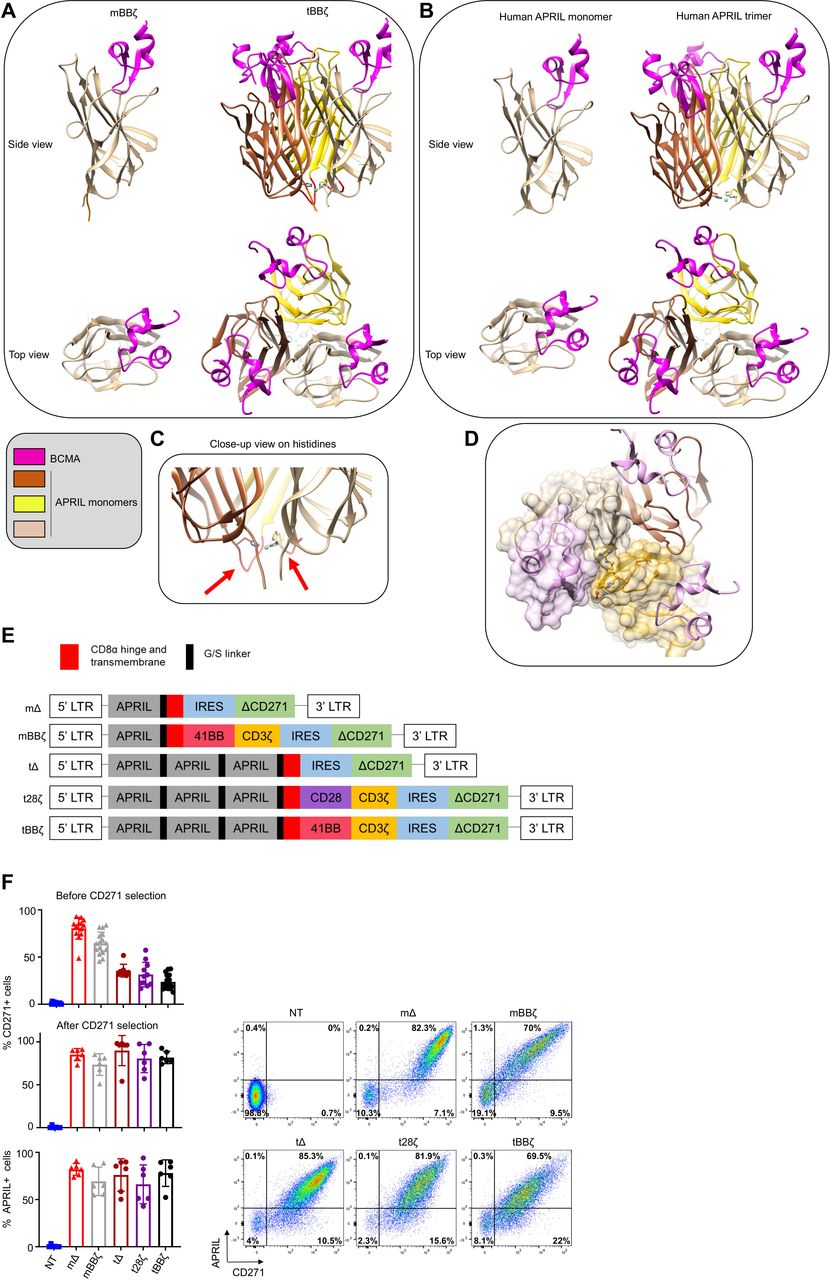
Molecular design and expression of APRIL CARs on human T cells. (A–D) Computational models of monomeric (m) and trimeric (t) APRIL ligand binding domains in CAR format. (A) left: mBBζ CAR, right: tBBζ CAR, compared with (B) left: human soluble APRIL monomer, right: human soluble APRIL trimer, interacting with BCMA (pink). (C) Close-up view on histidine residues contributing to the trimer folding, red arrows point to G/S linkers of optimal length. (D) Interaction between the APRIL-trimer (each monomer is colored differently in tan, dark brown and goldenrod), and three copies of BCMA (light pink). The surface of one BCMA is shown in transparent pink that is mainly binding one APRIL monomer (surface colored in transparent tan), additional interactions occur with His203, Asp205, Arg206, Tyr208 and His241 residues (in ball and stick representation) of a second APRIL monomer (surface colored in transparent goldenrod). (E) CAR construct schemes: mΔ, mBBζ, tΔ, t28ζ, tBBζ expressed in the pSFG retroviral vector. (F) Left: Bar charts represent summary of transduction efficiencies of APRIL CARs in human T cells before or after CD271 selection, based on CD271 marker gene or APRIL cell surface staining (n=8–16 donors; mean±SD). Right: representative FACS plots from one donor, after CD271 selection. APRIL, a proliferation inducing ligand; BCMA, B cell maturation antigen; CARs, chimeric antigen receptor; FACS, fluorescence activated cell sorting.
APRIL CAR T cells are cytotoxic in vitro and in vivo
To probe the cytotoxic activity of the different CAR constructs, we selected several MM cell lines and assessed cell surface BCMA and TACI expression by flow cytometry (figure 2A). NCI-H929 expressed BCMA only, while MM.1S expressed low levels of both BCMA and TACI. The myeloid cell lines KG-1a and K562 were negative for both antigens. We engineered K562 cells to express either antigen alone or in combination and confirmed their cell surface expression levels (figure 2A). To assess antitumor function and expansion of APRIL CAR T cells in vitro, we performed a sequential co-culture stress-test. T cells were challenged with fresh tumor cells up to five times, and residual tumor and T cells were quantified 3–4 days after each tumor challenge (figure 2B,C). Both mCAR and tCAR T cells produced significant antitumor activity against NCI-H929 and MM.1S cells. The mBBζ CAR T cells were the most potent in this assay with the best in vitro T cell expansion capacity (figure 2C). Similar results were obtained when performing the sequential co-culture assay with the engineered K562 targets (online supplemental figure 3). Analysis of the T cell differentiation and activation/exhaustion phenotype revealed that CD4+ t28ζ CAR T cells differentiated faster to an effector memory (TEM) state compared with mBBζ and tBBζ CAR T cells even at baseline in the absence of target antigen, while CD8+ mBBζ CAR T cells were more enriched in central memory (TCM) phenotype cells compared with control (online supplemental figure 4A). In addition, LAG3 levels were significantly higher on all except tBBζ CD4+ CAR T cells compared with ΔCARs and on mBBζ CD8+ CAR T cells at baseline. PD1 and TIM3 levels were comparable among groups (online supplemental figure 4B). APRIL CAR T cells did not spontaneously secrete significant amounts of IL-2 and did not proliferate in the absence of target antigen (online supplemental figure 5). Overall, these observations indicate absent to low tonic signaling activity of APRIL CARs which was least evident with the tBBζ CAR. Lastly, we evaluated the in vivo antitumor function of APRIL CAR T cells in a mouse xenograft model with NCI-H929 cells (figure 2D). mBBζ, t28ζ and tBBζ CAR T cells significantly delayed tumor growth (mΔ vs mBBζ: p=0.02, mΔ vs t28ζ: p=0.008, mΔ vs tBBζ: p=0.008, n=5) (figure 2E–G), but only tBBζ CAR T cells significantly enhanced overall survival of mice (mΔ vs tBBζ: p=0.003, mBBζ vs tBBζ: p=0.01, n=5) (figure 2H). The second CAR T cell infusion on Day 20 did not significantly delay disease progression with any of the CARs evaluated.

In vitro and in vivo antitumor function of APRIL CAR T cells. (A) BCMA and TACI expression on cell lines relative to isotype control staining. (B) Scheme of rechallenge co-culture stress test, effector to target ratio E:T 1:1. (C) Kaplan-Meier curves in the top row depict probability of tumor control in the rechallenge co-culture stress test, n=6 donors, log-rank test (table top right). Line graphs show T cell expansion in co-cultures, n=6, mean±SD, unpaired t-test with Welch’s correction on AUC (table bottom right). (D) Scheme of the in vivo experimental timeline. 1 of 2 representative experiments shown, n=5 mice per group per experiment. (E) Tumor growth measured by BLI, individual mouse pictures (color scale min 1×105, max 1×107 p/s/cm2/sr). Dorsal view identifies tumor growth in the spine and skull. (F) Individual line graph of tumor growth for each mouse per group. BLI flux (p/s) normalized to signal intensity measured on day 10. (G) BLI flux summary n=5, mean±SD. Unpaired Mann-Whitney-U test (non-parametric t-test) on AUC. (H) Survival of mice as defined by humane end-point criteria. Log-rank test. (C, G, H) Definition of significance levels: ns=not significant, *p<0.05, **p<0.01, ***p<0.001. APRIL, a proliferation inducing ligand; AUC, area under the curve; CAR, chimeric antigen receptor; BLI, bioluminescent imaging.
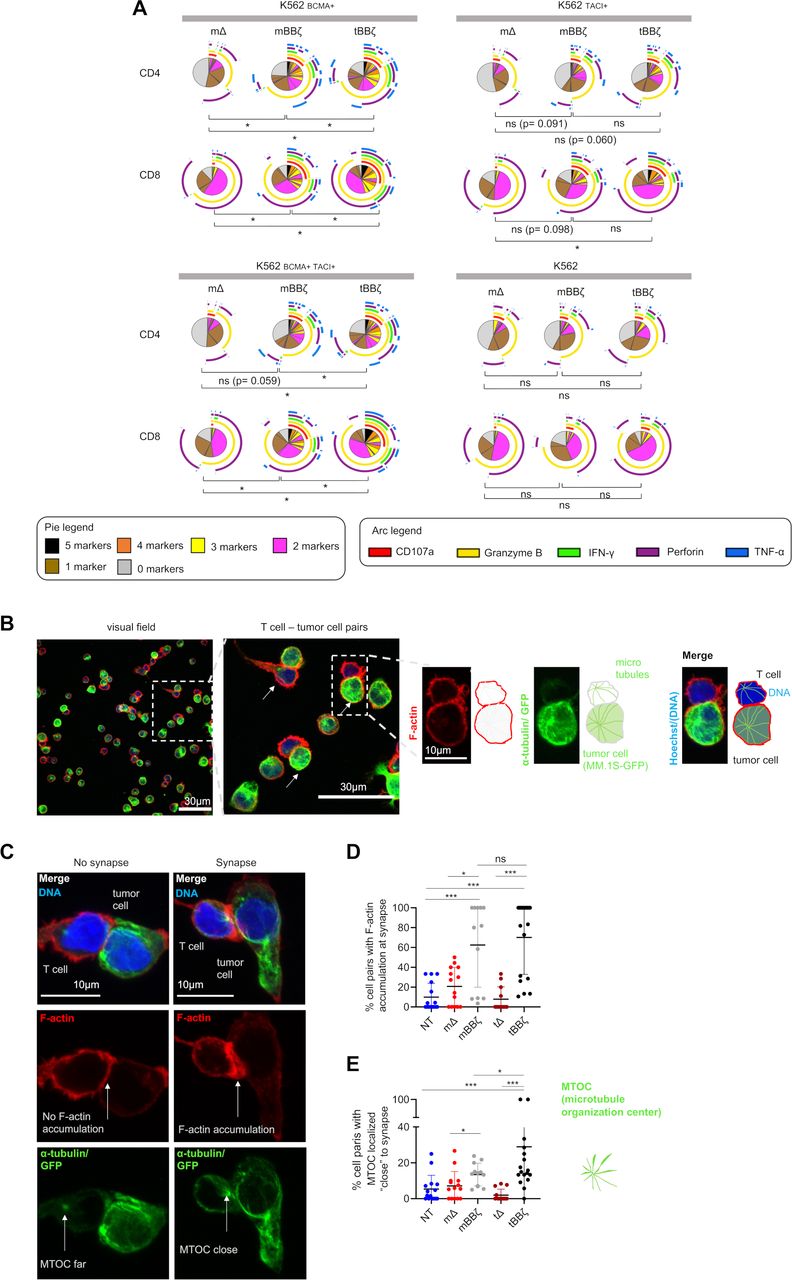
tBBζ CAR T cells are more polyfunctional and form stronger immune synapses than mBBζ CAR T cells
By intracellular cytokine staining (figure 3A) and analyses of the immune synapse (figure 3B–E), we assessed CAR T cell polyfunctionality and the strength of T cell–target cell interaction. In both CD4+ and CD8+ CAR T cells, polyfunctionality was enhanced when comparing mBBζ to tBBζ CARs (figure 3A and online supplemental figure 6) despite considerable donor variability. To further investigate the differences between mBBζ and tBBζ CARs, we performed confocal microscopy and analyzed features of the engineered immune synapse (figure 3B). While actin accumulation at the T cell–target cell interface was comparable between mBBζ and tBBζ CAR T cells, the microtubule organization center (MTOC) localized significantly closer to the synapse in tBBζ compared with mBBζ CAR T cells (figure 3C–E), suggesting the formation of stronger immune synapses with the tBBζ CAR.
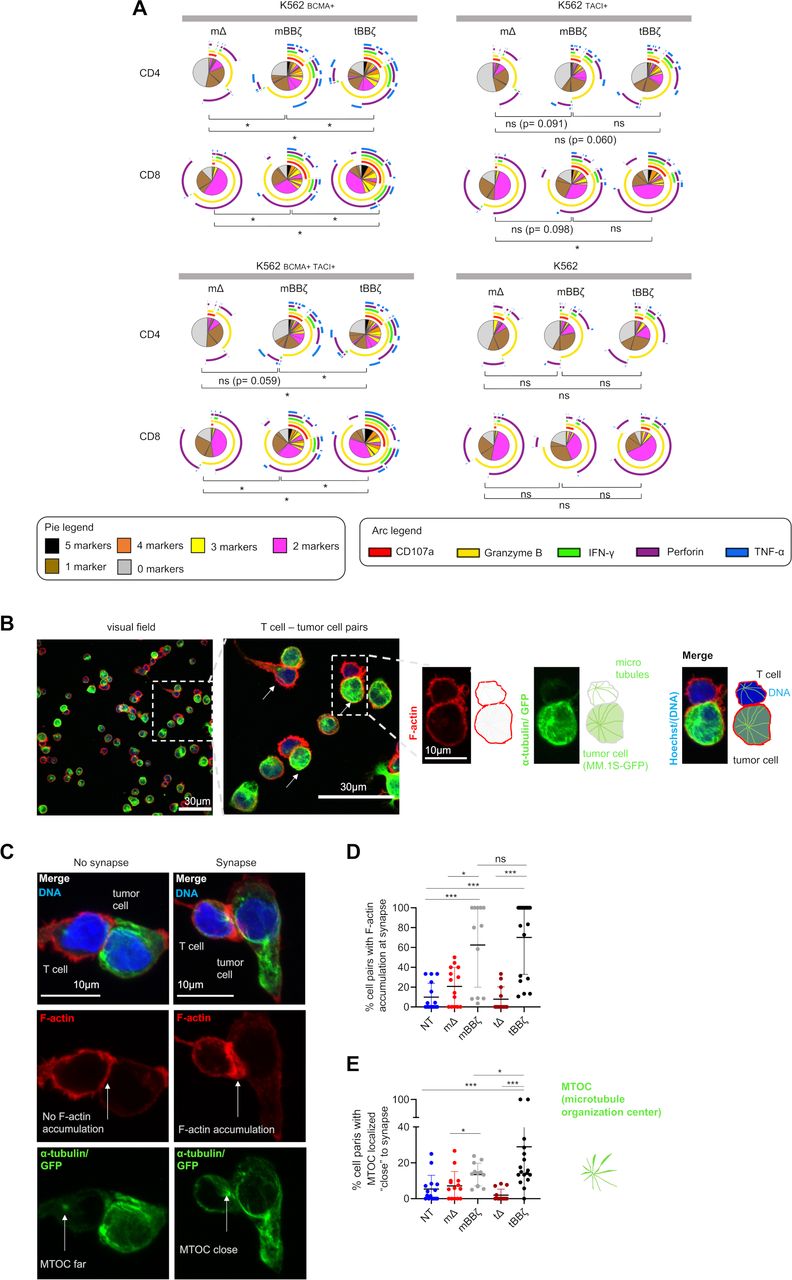
Analysis of polyfunctionality and immune synapses of APRIL CAR T cells. (A) Polyfunctionality by intracellular cytokine staining. Pies depict all different combinations of populations expressing 5, 4, 3, 2, 1 or 0 markers, and arcs around pies indicate the analyte detected. Pies represent mean of n=6 donors. Dependent t-test for paired samples was used to compare populations of CAR T cells expressing at least three markers. (B–E) Fixed cell confocal microscopy for GFP (green, tumor cell), α-tubulin (green, showing the microtubule organizing center (MTOC), centrosome), F-actin (red) and DNA (blue, Hoechst). (B) Left panel: Representative confocal microscopy field of view showing NT T cells and MM.1S-GFP-FFLuc tumor cells, scale bar 30 µm. Middle panel: enlarged inset of left panel, arrows marking T cell - tumor cell pairs, scale bar 30 µm, Inset: enlarged view of one T cell (upper cell)–tumor cell (lower cell) pair, scale bar 10 µm. Scheme on the right explains the immunofluorescence. (C) Left column: representative confocal microscopy images for T cell–tumor cell pair without features of an immunological synapse. Right column: representative images with features of an immunological synapse. First row: Merged images. Second row: F-actin, left: no F-actin accumulation at the cell-cell interface, right: F-actin accumulation at the synapse. Third row: alpha-tubulin and GFP. Arrows are pointing toward the centrosome, the MTOC. Scale bars 10 µm. (D) Quantification of % cell pairs with F-actin accumulation at the cell–cell interface. Each data point corresponds to one field of view, mean±SD, Mann-Whitney-U test. (E) Quantification of % cell pairs with the centrosome close to the cell–cell interface. Each data point corresponds to one field of view, mean±SD, Mann-Whitney-U test. (A, D, E) Definition of significance levels: ns=not significant, *p<0.05, ***p<0.001. APRIL, a proliferation inducing ligand; CAR, chimeric antigen receptor; GFP, green fluorescent protein.
CAR T cells mediate BCMA downmodulation on target cells by trogocytosis and internalization
We next investigated possible mechanisms of resistance when MM cells are exposed to CAR T cells, evaluating APRIL CARs and two different well-studied BCMA-targeting CARs (FHVH33 and 11D5-3) (figure 4A).3 9 16 19 First, we analyzed BCMA levels on target cells after contact with the different CAR T cells. MM.1S cells expressed low levels of BCMA that remained stable in the presence of mΔ and tΔ CAR T cells, but decreased significantly after contact with mBBζ, tBBζ as well as FHVH33Δ, 11D5-3Δ, FHVH33BBζ and 11D5-3BBζ CAR T cells (figure 4B). BCMA reduction on target cells was not due to an increased BCMA shedding as soluble BCMA (sBCMA) levels in the culture supernatants were also reduced in the presence of mBBζ, tBBζ, FHVH33Δ and 11D5-3Δ CAR T cells (figure 4C). However, we detected an increase of BCMA on the cell surface of mBBζ and tBBζ CAR T cells compared with mΔ and tΔ (figure 4D), a process known as trogocytosis.20 NCI-H929 cells expressed high levels of BCMA on the cell surface that rapidly decreased in the presence of FHVH33Δ and 11D5-3Δ CAR T cells, but BCMA levels were restored after 24 hours with all ΔCARs (figure 4E). Similar to MM.1S cells, NCI-H929 cells did not downmodulate cell surface BCMA levels after contact with mΔ and tΔ CAR T cells. However, we detected significant BCMA reduction on residual tumor cells within 6 and 24 hours of co-culture with all BBζ CAR T cells and MM.1S cells, but only at 24 hours with NCI-H929 cells (figure 4B,E, and online supplemental figure 7A). sBCMA levels were decreased with both cell lines (figure 4C,F). We also detected BCMA uptake from NCI-H929 cells by mBBζ and tBBζ CAR T cells consistent with trogocytosis (figure 4G and online supplemental figure 7B). In addition, FHVH33Δ and 11D5-3Δ CAR T cells had high levels of BCMA on their cell surface after co-culture with NCI-H929 (figure 4G and online supplemental figure 7B) that were higher than in MM.1S co-cultures (figure 4D). This may be explained by the overall much higher levels of BCMA on NCI-H929 compared with MM.1S. Since treatment with γ-secretase inhibitors (GSI) has been shown by others to be a potential strategy to increase BCMA cell surface levels on target cells by reduction of BCMA shedding,21 we investigated addition of the GSI LY3039478 during co-culture. When LY3039478 was added at initiation of co-culture, LY3039478 treatment could not prevent BCMA downmodulation in both MM.1S and NCI-H929 cells (figure 4H,I, and online supplemental figure 7C). These results led us to explore BCMA internalization as a potential explanation for reduced BCMA cell surface levels during co-culture. We encoded emerald-tagged BCMA into a retroviral vector (figure 4J) and generated MM.1S- and K562-BCMA-Emerald transgenic cell lines. Co-localization of BCMA-Emerald with cell surface BCMA staining was analyzed by Image Stream after co-culture with T cells. Contact of 11D5-3Δ CAR T cells with MM.1S cells led to a significant reduction of BCMA co-localization on the tumor cell membrane (NT vs 11D5-3Δ: 66.5±6.3% vs 35.0±16.3%, n=5, mean±SD, p=0.005), with a consequent increase in the fraction of cells with no co-localization (NT vs 11D5-3Δ: 14.2±4.0% vs 44.5±14.1%, n=5, mean±SD, p=0.01) (figure 4K,L, and online supplemental figure 8A). Both K562-BCMA-Emerald and MM.1S-BCMA-Emerald cells co-cultured with 11D5-3Δ CAR T cells significantly downmodulated cell surface BCMA levels, while emerald was only slightly reduced (online supplemental figures 8B,C,F and G). 11D5-3Δ CAR T cells gained emerald on their surface (online supplemental figures 8D,E,H and I) consistent with trogocytosis. After co-culture with APRIL CAR T cells, we found that only tBBζ but not tΔ CAR T cells induced BCMA internalization in MM.1S-BCMA-Emerald cells (figure 4M,N and online supplemental figure 9A) or in K562-BCMA-Emerald cells (online supplemental figure 9B). Very low but significant levels of emerald were also detected on tΔ or tBBζ APRIL CAR T cells consistent with trogocytosis (online supplemental figures 9C,D and 10). Altogether, these data suggest that BCMA downmodulation on MM cells is mediated by a combination of trogocytosis and BCMA internalization after CAR T cell exposure with internalization accounting for most of the effect.

B cell maturation antigen (BCMA) downmodulation on MM cells by CAR T cell trogocytosis and BCMA internalization. (A) Schemes of FHVH33 and 11D5-3 CAR retroviral vector constructs. (B) BCMA cell surface levels on MM.1S cells co-cultured with CAR T cells (E:T 1:1) at indicated time points. (C) Soluble BCMA (sBCMA) levels in MM.1S co-culture supernatant (24 hours). (D) BCMA levels on CAR T cells co-cultured with MM.1S cells, normalized to NT (6 hours). (E) BCMA cell surface levels on NCI-H929 cells co-cultured with CAR T cells (E:T 1:1) at indicated time points. (F) sBCMA levels in NCI-H929 co-culture supernatant (24 hours). (G) BCMA levels on CAR T cells co-cultured with NCI-H929 cells, normalized to NT (6 hours). (H–I) Cell surface BCMA levels on MM cells co-cultured with CAR T cells (E:T 1:1) at indicated time points, in the presence of γ-secretase inhibitor, 0.1 µM Crenigacestat (LY3039478). n=5, mean±SD, one-way ANOVA with Tukey’s comparison. (J) Scheme of BCMA-Emerald retroviral vector with ΔCD271 as selectable marker. (K–N) MM.1S-BCMA-Emerald cells were co-cultured for 1 hour with NT or 11D5-3Δ CAR T cells (K–L) or with NT, tΔ or tBBζ CAR T cells (M–N). Co-localization of BCMA-Emerald and BCMA cell surface staining (PE) was analyzed by Image Stream. (K, M) Representative images and gating. (L, N) Summary graphs show percentage of no co-localization (no co-loc) or co-localization (co-loc) of BCMA-Emerald and cell surface BCMA (PE) in MM.1S-BCMA-Emerald cells. n=4–5, mean±SD, paired t-test. (B–I, L, N) Definition of significance levels: ns, not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (B, D, E, G, H, I) BCMA cell surface levels were determined by FACS. (C, F) sBCMA levels were determined by ELISA. CAR, chimeric antigen receptor; NT, non-transduced; ANOVA, analysis of variance.
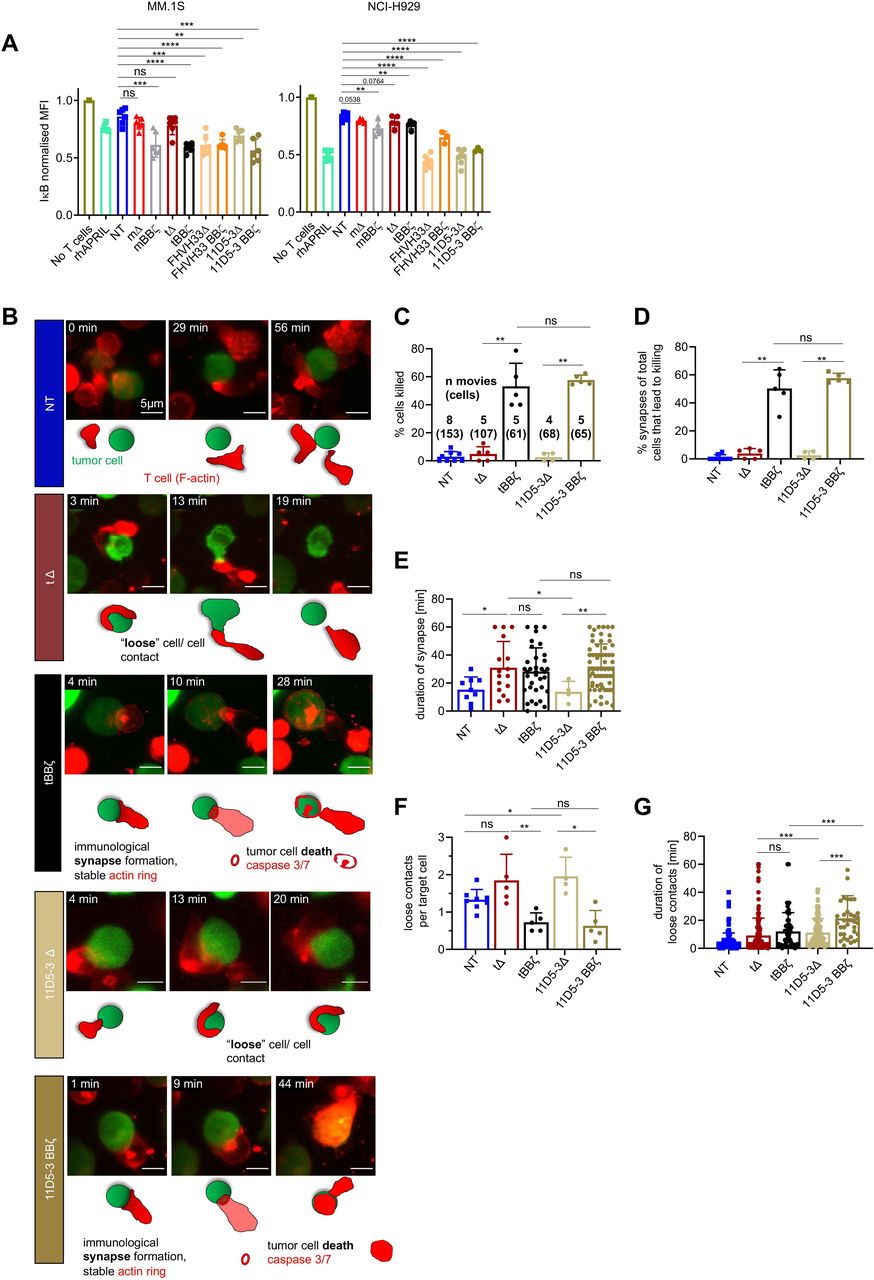
CAR T cell–tumor cell interactions trigger BCMA signaling in MM cells
Since disease relapse with BCMA positive disease was reported in clinical trials despite CAR T cell persistence,9 22 we next assessed if BCMA directed CAR T cells have the potential to activate BCMA signaling in MM cells. After short co-culture of CAR T cells with MM.1S and NCI-H929 cells, mΔ and tΔ CAR T cells induced only minimal NFκB activation while mBBζ, tBBζ, FHVH33Δ, FHVH33-BBζ, 11D5-3Δ and 11D5-3 BBζ CAR T cells all induced strong NFκB activation (figure 5A). To investigate if differences in T cell–tumor cell interactions could explain differences in NFkB pathway activation in MM.1S cells, we next investigated target cell killing and the quality of the interaction (immune synapse formation, loose interactions, and their durations) at the single cell level by live time-lapse light sheet microscopy (figure 5B). Live imaging revealed that both tBBζ and 11D5-3BBζ CAR T cells led to efficient target cell killing (figure 5C) via stable immune synapse formation (figure 5D,E). While non-signaling tΔ and 11D5-3Δ CAR T cells did not form synapses nor kill target cells, they formed significantly more loose contacts with target cells than tBBζ and 11D5-3BBζ CAR T cells, suggesting significant interactions that may lead to BCMA pathway activation (figure 5F). The duration of these loose contacts was longer with 11D5-3Δ compared with tΔ CAR T cells (figure 5G), potentially explaining why 11D5-3Δ CAR T cells, and not tΔ, activated BCMA pathway. These results suggest that significant CAR T cell - tumor cell interactions can take place in the absence of killing which led us to hypothesize that those interactions could lead to a survival advantage of residual BCMA positive tumor cells.

NFkB pathway activation in multiple myeloma (MM) cells after interaction with chimeric antigen receptor (CAR) T cells. (A) Normalized intracellular IκBα MFI in MM cells co-cultured with CAR T cells for 30 min. MM.1S cells (left, E:T 1:1), NCI-H929 cells (right, E:T 10:1). n=3–6 donors, mean±SD, unpaired t-test. (B–G) Light sheet live imaging of immunological synapse formation, target cell killing and loose T cell–tumor cell interactions. (B) Representative stills from light sheet time-lapse movies. NT (dark blue), tΔ (dark red), tBBζ (black), 11D5-3Δ (beige), and 11D5-3BBζ (dark beige) CAR T cells with MM.1S-GFP-FFLuc (green) target cells. T cell F-actin (SPY-actin live dye (red)), and cell death (biotracker caspase 3/7 stain, red). One image per minute for 60 min. Scale bar 5 µm. (C) Percentage target cell killing per 60 min movie, mean±SD. Numbers of movies and numbers of single cells (in parentheses) analyzed from n=3 independent donors and experiments. (D) Percentage of observed immunological synapses between T cells and target cells (appearance of an F-actin ring stable for >2 min leading to target cell death measured by Biotracker caspase 3/7 red signal), mean±SD. (E) Contact time between T cell and target cell at each synapse in minutes, mean±SD. (F) Quantification of loose contacts per target cell, mean±SD. (G) Contact time between T cell and target cell in minutes for loose contacts, mean±SD. (A, C–G) Definition of significance levels: ns=not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Non-functional BCMA-directed CAR T cells do not promote tumor progression in vivo
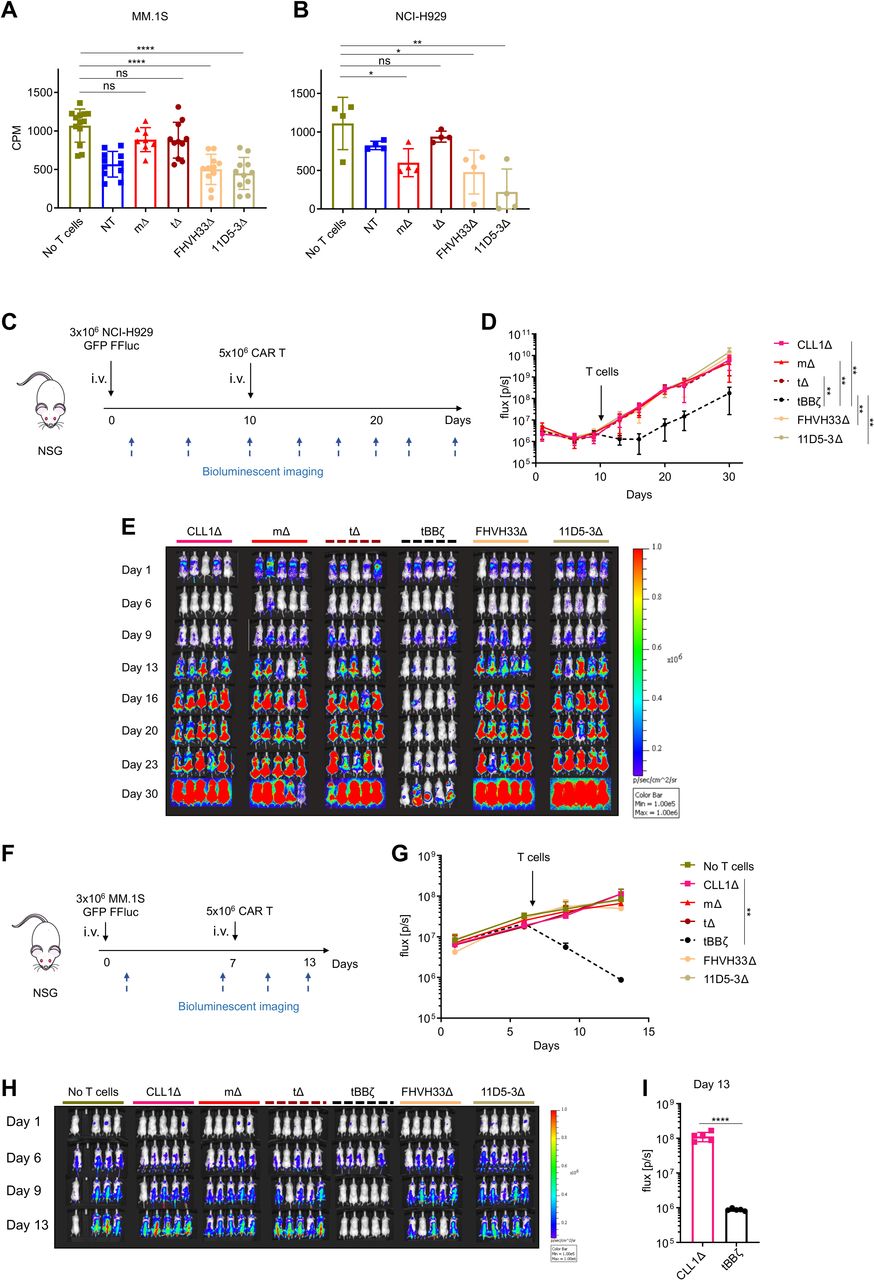
Soluble APRIL is well known to promote tumor progression by activation of BCMA signaling in MM cells.23–28 Thus, we next investigated if APRIL, FHVH33 or 11D5-3 ΔCAR T cells mediated MM cell proliferation. We showed that CAR T cells did not induce proliferation of MM.1S or NCI-H929 cells in vitro (figure 6A,B). Similarly, in vivo in two different mouse xenograft models, we found no tumor promoting capacity of APRIL, FHVH33 or 11D5-3 ΔCAR T cells, neither in mice systemically engrafted with NCI-H929 cells (figure 6C–E) nor in mice systemically engrafted with MM.1S cells (figure 6F–H). We confirmed fast and significant antitumor activity with tBBζ CAR T cells in both models (figure 6C–I), with early recurrence in the NCI-H929 model. The follow-up in the MM.1S model could not be extended beyond day 14 due to significant T cell receptor mediated alloreactivity against MM.1S cells in all treatment conditions evaluated. Thus, despite significant T cell–tumor cell interactions that activated BCMA signaling, non-functional APRIL, FHVH33 or 11D5-3 CAR T cells did not promote tumor growth in vivo.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Non-functional B cell maturation antigen (BCMA)-directed CAR T cells do not promote tumor growth. (A, B) H3 thymidine incorporation assay on chimeric antigen receptor (CAR) T cell co-culture with (A) MM.1S cells at E:T ratio 1:1 or (B) NCI-H929 cells at E:T ratio 5:1. Background T cell H3 thymidine incorporation was subtracted. n=4–11 donors, colored symbols depict mean of technical triplicates, bars mean±SD, unpaired t-test. (C) Scheme of the NCI-H929 mouse xenograft model. 1 representative experiment of 2. n=5 mice per group per experiment. (D) BLI summary and quantification of total flux (p/s), mean±SD, unpaired Mann-Whitney-U test on area under the curve (AUC). (E) Tumor growth measured by BLI, individual mouse pictures (color scale min 1×105, max 1×106 p/s/cm2/sr). Dorsal view identifies tumor growth in the spine and skull. (F) Scheme of the MM.1S mouse xenograft model. 1 representative experiment of 2. n=4–5 mice per group per experiment. (G) BLI summary and quantification of total flux (p/s), mean±SD, unpaired Mann-Whitney-U test on AUC. (H) Tumor growth measured by BLI, individual mouse pictures (color scale min 1×105, max 1×106 p/s/cm2/sr). Dorsal view identifies tumor growth in the spine and skull. (I) Bar graph shows comparison of BLI signal intensity at day 13 between CLL1Δ and tBBζ CAR T cell treated mice. (A, B, D, G, I) Definition of significance levels: ns, not significant, *p<0.05, **p<0.01, ****p<0.0001. BLI, bioluminescent imaging.
Discussion
We show that a trimeric configuration of truncated APRIL can be exploited as efficient binding moiety in CARs leading to dual antigen recognition on MM. Polyfunctionality and immune synapse formation were superior in BBζ CAR T cells with our trimer-derived compared with monomer-derived APRIL. The tBBζ construct was most potent in vivo in two different MM mouse xenograft models and led to fast antitumor responses, but responses were not sustained, and tumors recurred in the NCI-H929 model. We identified BCMA downmodulation as a common escape mechanism for APRIL, scFv and FHVH-CAR T cells that was attributed in part to trogocytosis and mostly to BCMA internalization. Despite activation of survival pathways in residual tumor cells and significant T cell–tumor cell interactions via loose contacts, we demonstrate that APRIL, scFv, and FHVH-CAR T cells cannot promote tumor cell proliferation or tumor growth in vitro and in vivo.
Truncated APRIL for the generation of natural ligand-based CARs targeting MM has previously attracted attention due to APRIL’s natural high affinity for BCMA and TACI expressed on MM, allowing for dual antigen targeting.7 13 In addition, both BCMA and TACI provide survival and proliferation signals to MM cells and are expressed on putative MM stem or progenitor cells.6 25 28 While the APRIL monomer based CD28-OX40ζ CAR showed promising preclinical activity,7 the subsequent clinical trial was terminated early due to insufficient efficacy in patients.29 A trimeric APRIL configuration as CAR binding moiety linked to a CD8α hinge and transmembrane and a BBζ endodomain (TriPRIL) resulted in better polyfunctionality compared with a monomeric APRIL BBζ CAR.13 Both groups reported in their preclinical models that APRIL CARs have the capacity to overcome BCMA negative antigen escape. Fast antitumor responses were seen in mice-bearing BCMA-TACI+ myeloma xenografts, but follow-up was short in both papers, ranging from 9 to a maximum of 21 days, and the sustainability of those responses was not investigated. A clinical trial assessing safety and efficacy of TriPRIL CAR T cells has recently started.30 In our study, we evaluated alternative APRIL CAR configurations that are distinct from the previously reported ones,7 13 and found that a histidine residue in position 115 of APRIL is potentially favoring the proper trimer folding and stabilization of the trimeric binding module. Linker length is also critical for the stability of the fold. Based on our models, we chose a linker for our tBBζ CAR that is shorter than the one used in the TriPRIL CAR. We show that immune synapse formation with our tBBζ CAR is enhanced compared with mBBζ CAR T cells, correlating with polyfunctionality by intracellular cytokine staining. In vivo in the mouse xenograft models, tBBζ CAR T cells produced significantly better tumor control than mBBζ CAR T cells, even though tumors recurred in the follow-up in the NCI-H929 model. Overall, APRIL is an interesting natural dual antigen binder that can be used as a targeting moiety in CARs. Additional optimization is most likely required to deploy its full therapeutic potential.
CAR T cell therapy in MM has so far mostly focused on targeting BCMA as a single antigen. While fast and deep responses have been consistently reported in several clinical trials, most patients do not experience long-term sustained remissions.10 22 31–35 Mechanisms of resistance to BCMA CAR T cell therapy have not been entirely elucidated yet.36 Inspired from the CD19 CAR T cell experience, tumor escape by target antigen loss was already investigated. Indeed, rapid BCMA downmodulation on residual plasma cells has been reported in several clinical trials of BCMA-CAR T cell therapy, but the mechanism is not known. Further, it is unclear if the BCMA antigen density on persisting myeloma cells is below the antigen sensitivity of the respective CAR T cells.22 31 In most patients, malignant plasma cells will re-express BCMA to higher levels on their cell surface during disease progression, suggesting that BCMA downmodulation is a transient effect. Only two cases of definitive loss by genetic deletion have been described to date,9 11 12 probably because BCMA provides crucial signals for maintaining and expanding the malignant myeloma cell population.28 Our data now indicate that BCMA downmodulation consistently occurs in malignant plasma cells after CAR T cell exposure, and we identified two mechanisms that contribute to this effect. First, CAR T cells can extract BCMA from their target cells and present it on the T cell surface. This phenomenon is known as trogocytosis and can potentially lead to CAR T cell exhaustion and fratricide, and thus limit CAR T cell function, expansion and persistence in vivo.20 Second, plasma cells internalize BCMA after CAR T cell exposure, leading to a rapid reduction of BCMA cell surface levels. Similar internalization has been observed after exposure to an antibody-drug conjugate targeting BCMA.32 Interestingly, several clinical trials indicate that a significant proportion of patients experienced disease progression despite peripheral blood CAR T cell persistence, suggesting a dysfunctional state of the persisting CAR T cells.9 22 Thus, other mechanisms of therapy resistance may include the limited fitness or exhaustion of persisting CAR T cells, limited trafficking of CAR T cells to the bone marrow, local immune suppression of CAR T cells in the tumor microenvironment, or BCMA pathway stimulation by continuous but insufficient CAR T cell–tumor cell interactions. In preclinical models, we investigated whether persisting non-functional mAPRIL, tAPRIL, 11D5-3, and FHVH33-CAR T cells could stimulate the BCMA pathway and through this interaction promote tumor growth. Even though NFkB signaling was activated after loose T cell–tumor cell contacts with all CARs investigated except with mΔ and tΔ, this stimulation did not translate into measurable MM cell proliferation in vitro or faster tumor progression in vivo in two mouse xenograft models. Thus, these findings are encouraging and suggest that BCMA targeted CAR T cells are not actively involved in tumor progression even if they persist in vivo in a non-functional or exhausted state. Even though soluble APRIL is a strong survival factor involved in MM progression and high levels in patients are associated with poor prognosis,26 37 truncated APRIL presented on non-functional CAR T cells did not promote tumor progression. A potential explanation could be that we used a truncated sequence devoid of the heparan sulfate proteoglycan binding (HSPG) site that is required to engage CD138 on plasma cells and critical for mediating the pro-proliferative and survival effects mediated by soluble APRIL.38
Due to the importance of BCMA in MM cell biology, BCMA remains a very attractive target for CAR T cell therapy. Strategies to upregulate BCMA levels on target cells before and early during the course of CAR T cell therapy are investigated clinically by the use of GSI.21 39 In our study, however, when adding GSI simultaneously with CAR T cells to the co-culture, we nevertheless found BCMA downmodulation to a similar extent as in the absence of GSI. A pretreatment phase with GSI seems to be critical to achieve the desired effect on increasing BCMA levels on target cells.21 Another avenue for improvement is the use of fully human binding sequences to reduce immunogenicity of CAR T cells, as we do with APRIL CAR T cells. In clinical trials using murine scFvs in CARs such as the 11D5-3 scFv, anti-drug antibodies (ADAs) were detected in a significant proportion of patients,9 33 and the presence of ADAs was a risk factor for relapse or progression after CAR T cell therapy.33 Further, efficacy may be increased with dual antigen targeting CARs that combine BCMA targeting with a second antigen that is involved in myelomgenesis but through a different pathway. Combinatorial targets include explorations of GPRC5D, SLAMF7, CD38, CD19, TACI and BAFF-R.40–47 Other future directions include combinatorial targeting with CAR T cells and microenvironment modulation or the exploration of enhanced manufacturing modalities that favor the long-term function and persistence of adoptively transferred T cells.
In summary, we show that trimeric APRIL CAR T cells can efficiently target both BCMA and TACI on MM, and that our tBBζ construct was most potent. However, the fast initial antitumor responses were not sustained in the NCI-H929 model that is mostly assessing the BCMA-targeted component of the response. We reveal BCMA downmodulation on tumor cells as an important mechanism of immune evasion after CAR T cell contact mediated by trogocytosis and BCMA internalization, independent of the binding moiety used. Importantly, when non-functional mAPRIL, tAPRIL, 11D5-3 or FHVH33 CAR T cells persisted in vivo, they were not able to promote tumor proliferation and growth in vivo. Our results shed light on the mechanisms underlying CAR T cell treatment failure targeting BCMA in MM and may help to devise more efficient therapeutic strategies in the future.
Supplemental material
Supplementary video
Supplementary video
Supplementary video
Supplementary video
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involved deidentified human volunteer blood donors who donated buffy coats for the isolation of peripheral blood mononuclear cells. Buffy coats were obtained from the Center of Interregional Blood Transfusion SRK Bern (Bern, Switzerland).
Acknowledgments
We are very grateful to Maude Varrin for technical assistance and lab management, to Dr Rosa Paolicelli, University of Lausanne, for granting us access to her light sheet microscope for live imaging, and to Francisco Sala de Oyanguren for expert technical assistance with the Image Stream analysis. We thank all Arber lab members for helpful and stimulating discussions and critical feedback on the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @imwolfb1, @GfellerD, @ArberCaroline
Presented at International Conference on Lymphocyte Engineering 31 March–2 April 2022 Munich, Germany https://doi.org/10.1089/hum.2022.29200.abstracts
Contributors NC designed research, performed experiments, analyzed and interpreted results, and wrote parts of the manuscript. BW designed research, performed microscopy studies, and wrote parts of the manuscript. GC performed statistical analysis of polyfunctionality assessment. DG supervised statistical analysis of polyfunctionality assessment. VZ designed research, performed molecular modeling, analyzed and interpreted results and wrote parts of the manuscript. CA designed research, supervised the entire study, analyzed and interpreted results, wrote the manuscript, is responsible for the overall content of the publication and acts as a guarantor. All authors reviewed and approved the final version of the manuscript.
Funding BW receives funding from the Fondation Dr Henri Dubois-Ferrière Dinu Lipatti. GC receives funding through the H2020 Marie Skłodowska-Curie Actions Individual Fellowship (H2020-MSCA-IF-2020, No. 101027973). CA receives funding from Swiss Cancer Research KFS-4542-08-2018-R, Stiftung für Krebsbekämpfung, and the Department of oncology UNIL CHUV, University of Lausanne and Lausanne University Hospital.
Competing interests CA holds patents and provisional patent applications in the field of engineered T cell therapies and receives royalties from Immatics. VZ is a consultant for Cellestia Biotech. All other authors declare no competing financial interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.