Article Text

Abstract
Background Despite the survival benefits observed with immune checkpoint blockade (ICB) treatment—programmed cell death-1/programmed cell death ligand-1 (PD-1/PD-L1), many patients with cancer have not benefited from these agents because of impaired antigen presentation and other resistance mechanisms. To overcome resistance to checkpoint therapy, we designed bispecific antibodies (BsAbs) targeting CD89 and tumor antigens. We demonstrated their immunomodulatory efficacy as a separate treatment strategy or combined with immune checkpoint inhibitors.
Methods We have previously generated a heterodimeric one-arm IgG1 Fc-based bispecific antibody. For animal efficacy studies, murine tumors in a humanized transgenic mice model were used to determine the effects of CD89-bispecific antibodies on antigen presentation and immune cell recruitment. The efficacy of the CD89 bispecific antibody against tumors resistant to pembrolizumab was evaluated in double-transgenic mice.
Results BsAbs targeting CD89 on tumor-associated macrophages (TAMs) increased the ratio of M1:M2 and activated the antigen presentation, leading to increased cytotoxic T cell-mediated tumor regression. CD89-BsAbs also potentiated a combinational antitumor effect with PD-1/PD-L1 inhibitors and overcame the ICB resistance by augmenting cytotoxic T-cell infiltration and reshaping tumor immune microenvironment. In an hCD89/hPD-1 double transgenic mouse model engrafted with pembrolizumab-resistant B16-HER2 tumor cells, the HER2-CD89 bispecific antibody potently inhibited tumor growth.
Conclusions CD89 BsAbs targeting tumor antigens and TAMs controlled tumor growth in animal models by improving antigen presentation and T-cell infiltration. Our results suggest a general strategy for overcoming resistance to ICB.
- Immunotherapy
- Macrophages
- Tumor Microenvironment
Data availability statement
No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Although immune checkpoint blockade (ICB) therapy has provided benefits for many patients with cancer, the majority of the patients remain resistant to programmed cell death-1 (PD-1) blockade therapy. Recent research has suggested that targeting tumor-associated macrophages (TAMs) may offer a promising therapeutic strategy and enhance the efficacy of ICB-based therapies when used as co-therapies.
WHAT THIS STUDY ADDS
We propose that impaired innate sensing may limit the complete T-cell activation. As TAMs play a key role in tumor microenvironment and local activation of TAMs restores the M1:M2 ratio and antigen presentation, we generated CD89 bispecific antibodies to activate macrophages. Our data suggest that this novel bispecific antibody improves tumor regression and antigen presentation, and significant tumor regression was observed in combination with PD-1.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
CD89-targeting therapies, such as the bispecific antibodies reported in this study, may hold promise for improving clinical outcomes in patients with advanced cancers when used alone or in combination with ICB therapies.
Background
Over the past several years, antibodies targeting the co-inhibitory immune checkpoints have demonstrated marked clinical outcomes in patients. Since the Food and Drug Administration first approved the anti-cytotoxic T-lymphocytes-associated protein 4 (CTLA-4) antibody ipilimumab for melanoma, followed by two programmed cell death-1 (PD-1) antibodies (pembrolizumab and nivolumab), increasing numbers of immune checkpoint blockade (ICB) agents have entered clinical trials in 2014.1 2 Although ICB agents are promising, clinical data also show that immune checkpoint antibodies benefit only a small percentage of patients, the objective response rate was approximately 10%–20% in most cancer types.3
Recent studies have stated the potential of targeting tumor-associated macrophages (TAMs) as a therapeutic strategy to activate antigen presentation.4 ,5–7 Macrophages are a major subpopulation of tumor-infiltrating immune cells in the tumor microenvironment (TME) and are widespread in all tumor types. TAMs play an important role in tumor progression and recruit inflammatory cells by secreting inflammatory cytokines.8 In recent years, researchers have found that TAMs exert significant anticancer effects after activation. Thus, TAMs have increasingly been recognized as attractive targets in cancer therapy.9 TAMs play an important role in PD-1/CTLA-4 therapy.10 11
CD89 is a transmembrane receptor of IgA and is expressed in myeloid lineage cells, including monocytes, several macrophage subsets, neutrophils, and eosinophils.12 13 Cross-linking of CD89 induces antigen presentation, phagocytosis, antibody-dependent cellular cytotoxicity (ADCC), and inflammatory mediators.14–16 Therefore, we hypothesized that activating CD89 could improve the phagocytosis and antigen-presenting ability of TAMs, leading to the tumor-specific T cells infiltration.
We generated novel bispecific antibodies (BsAbs) against CD89 and tumor antigens. Activation of CD89 signaling resulted in increased antigen presentation, M1/M2 ratios, and lymphocyte infiltration in the tumor tissues of CD89-Tg mice. Activation of antigen-presenting TAMs by treatment with CD89 BsAbs enhanced the PD-1 blockade efficacy and overcame pembrolizumab resistance in an hCD89/hPD-1 dual-Tg mouse model. Our results indicate that CD89-blocking agents, by improving and recovering the immune functions of TAMs, could serve as a connecting bridge between innate and adaptive immunity.
Methods
Animals
CD89 Tg mice were generated as previously described.17 Human PD-1 mice were generated by designing a vector containing human PDCD1 gene and the homologous arms (3.3 kb) on both 5′ and 3′ ends and replacing the murine Pdcd1 gene using homologous recombination. Both lines of Tg mice were generated on a C57BL6 background. Double-Tg mice expressing human CD89 and PD-1 (Tg hCD89/hPD-1) were generated by crossing the Tg hCD89 and hPD-1 mice. Mouse genotyping test was performed using PCR with the mouse tail DNA. The following transgene-specific primers: (CD89) 5′-GACGCAGAACTTGATCCGCA-3′ and 5′-GGATTCGTTGACGAGGACCC-3′; (hPD-1) 5′-CCAGGCCAGGGATAGCAGC-3′ and 5′-GGCCTACCCGCTTCCATTGCTC-3′.
The wild type (WT) mice were used as controls. The Animal Ethics Committee at Tongji University approved all animal research (No. TJLAC-018–032) and were performed according to the Animal Protection Ordinance and institutional animal guidelines. All mice were maintained in pathogen-free environments under the Experimental Animal Center of Tongji University. An appropriate attempt was made to reduce animal suffering.
Cell lines
The MC38 cell line was obtained from Dr Maoquan Chu (Tongji University, Shanghai, China), and the other cell lines were obtained from the American Type Culture Collection (ATCC). MC38 and Lewis lung carcinoma (LLC) cells (CRL-1642) stably expressing human CD20 antigen (MC38-CD20, LLC-CD20) and B16F10 (ATCC, CRL-6475) cells expressing the human HER2 antigen (B16-HER2) were established using lentivirus transfection as previously described.17 Cell culture medium consists of Dulbecco’s Modified Eagle Medium (DMEM;Life Technologies, Gaithersburg, Maryland, USA) and extra 10% fetal bovine serum (FBS; Gibco) at 37°C and 5% CO2 (v/v).
Tumor studies
Various CD89 BsAbs were engineered by genetically linking the scFv fragment derived from rituximab (anti-CD20) and trastuzumab (anti-HER2) into a human IgG1 framework (online supplemental figure S1). The sequence of anti-CD89 antibody was derived from our laboratory. All BsAbs were produced via transiently transfected plasmids. Antibody purification and production are described as before.18
Supplemental material
Supplemental material

CD89 BsAbs with superior cytotoxicity in vivo (A) Effect of the CD20-CD89 BsAb on the growth of LLC-CD20 cells in CD89 Tg mice. Tumor cells (2.5×105) were injected subcutaneously into female mice (n=6–8/group). Antibodies were administered by intravenous injection two times a week. Animals were monitored for tumor growth. (B) Survival curves for mice treated with CD20-CD89 (n=5/group). (C) CD20-CD89 BsAb and anti-CD20 antibody on the growth of LLC-CD20 in WT mice (n=6–8/group). (D) Effect of the CD20-CD89 on the growth of MC38-CD20 cells in CD89 Tg mice (n=6–8/group). (E, F) Tumor growth (E) and survival curves (F) for mice treated with HER2-CD89. (G, H) Enhanced phagocytosis mediated by CD89 BsAbs. CellTracker Green stained tumor cells mixed with CD89 BsAbs were co-incubated with CellTracker Deep red stained TAMs for 4 hours. Phagocytosis of tumor cells by TAMs was analyzed using flow cytometry and data represent per cent of phagocytosis (green positive cells within red positive TAMs)±SD of three independent experiments. *p<0.05, ***p<0.001 and ****p<0.0001, as determined by two-way analysis of variance. BsAbs, bispecific antibodies; LLC, Lewis lung carcinoma; TAM, tumor-associated macrophages; WT, wild type.
Six-to-eight weeks old female hCD89 or hCD89/hPD-1 Tg mouse were subcutaneously (s.c.) injected with 2.5×105 tumor cells (LLC-CD20, MC38-CD20, B16-HER2) and assigned to each treatment group (6–10 mice) with 100–150 mm3 tumor volume. Then, control antibodies (anti-CD19 monoclonal antibody (mAb) established by our laboratory), rituximab, trastuzumab, and CD89 BsAbs were injected intravenously, alone or in combination with mouse PD-1 antibody (BP0146, RMP1-14, Bio X Cell) and/or programmed cell death ligand-1 (PD-L1) antibody (atezolizumab), two times a week (10 mg/kg). Every other day, tumor sizes were measured and calculated as length×width2×0.5. In some studies, CD89 Tg mice were s.c. injected with the tumor cells and injected intravenously on the same day with CD89 BsAbs (10 mg/kg), alone or in combination with PD-1 antibody (10 mg/kg), two times a week.
Cell depletion and interferon blockade studies
Macrophages and CD8+ T-cell depletion in mice bearing tumor cells was accomplished using liposomal clodronate (Clophosome (neutral); FormuMax Scientific, Palo Alto, California, USA) or rat anti-mouse CD8 antibody (clone 169.4; Bio X Cell). Two-hundred microliters of clodronate or control liposomes (FormuMax Scientific) were injected intraperitoneally (i.p.) starting 2 days before antibody injection and continued every 4 days for the entire experiment. Macrophage depletion was determined by flow cytometry of peritoneal lavage and blood specimens. Depletion of CD8+ T cells was carried out by i.p. injecting 400 µg anti-CD8 antibody at the same time as CD89 BsAb treatment.
For the interferon (IFN) blocking test, 200 µg anti-IFNAR1 antibody (BE0241, Bio X Cell) was injected into the mice every 3 days (i.p.) from day 0 until the tumor volume reached 150‒200 mm3, and then 100 µg of anti-IFNAR1 antibody was injected (intratumoral) every 3 days. To block IFN-γ, 1 day before the treatment of CD89 BsAb, 500 µg anti-IFN-γ (BE0055, Bio X Cell) antibody was i.p. injected into mice, and were then injected three times every 3 days.
Analysis of tumor-infiltrating immune cells
The hCD89 Tg mice bearing LLC-CD20 or B16-HER2 tumors were treated with CD89 BsAbs alone or in combination with anti-PD-1/PD-L1 antibodies. Tumors were collected and dissociated using gentle MACS Dissociator (Miltenyi, Germany) according to the standard protocol. Flow cytometry was used to analyze tumor infiltrated myeloid cells. Single cell suspensions (106 cells/100 µL) were preincubated with rat anti-mouse CD16/CD32 antibody (Fc block, clone 2.4G; BD Biosciences, Heidelberg, Germany) and then tinted with one of the following fluorescently labeled antibodies at 4°C for 30 min: anti-CD45-AF700 (clone 30-F11), anti-CD8-PerCP-Cy5.5 (clone 53–6.7), anti-CD3-APC (clone 145–2 C11), anti-CD25-APC-Cy7 (clone PC61), anti-CD4-PE-Cy7 (clone RM4-5), anti-CD8-PE Texas Red (clone 5H10), anti-Foxp3-APC (clone FJK-16s), anti-CD11b-PE-TR (M1/70.15), anti-F4/80-PerCP-Cy5.5 (clone BM8), anti-Ly6C-PE-Cy7 (clone AL-21), anti-Ly6G-APC (clone 1A8), anti-CD206-PE (clone 19.2), anti-MHC class II-eFluor 450 (clone M5/114.15.2), anti-CD89-FITC (clone A59), anti-CD40-FITC (clone 3/23), or anti-CD80-FITC (16–10 A1). Labeled antibodies were purchased from eBioscience, BioLegend or Miltenyi Biotec. The ratio of M1/M2 was calculated for CD45+F4/80+CD89+ macrophages. For intracellular IFN-γ and tumor necrosis factor (TNF)-α staining, Fixation/Permeabilization Solution Kit (BD Biosciences) was used. Cells were fixed and permeabilized, and incubated with fluorescently labeled antibodies binding IFN-γ (clone XMG1.2) and TNF-α (clone 6401.1111) (BD Biosciences). Based on forward and side scatter and the fixable viability dye eFluor 506, dead cells and doublets were eliminated. Using the BD FACSVerse system, multicolor fluorescence-activated cell sorting (FACS) analysis was carried out. The FlowJo software (V.10.0; Tree Star, San Carlos, California, USA) was used for all data analysis.
Antigen uptake investigation
BsAbs were administered into LLC-CD20 or B16-HER2 bearing mice (n=5/group). With the final BsAb injection, an antigen (OVA-Alexa Fluor 488) was given intravenously. Tumors were evacuated after 24 hours, and flow cytometry was used to analyze cells with OVA-Alexa Fluor 488 staining in the gated CD45+ population.
TAM phagocytosis
On day 15, TAMs were isolated in LLC-CD20 or B16-HER2 bearing Tg mice. Using a human tumor dissociation kit, tumors were subdivided into single-cell suspensions (Miltenyi Biotec). The cell suspensions were sorted using BD FACSAria II after being initially labeled with F4/80-PE and CD11b-FITC antibody (BD Bioscience). TAMs were further labeled with CellTracker Deep Red (Thermo Fisher Scientific, Waltham, Massachusetts, USA) after 24 hours, and were then treated for an additional 4 hours with CellTracker Green (Thermo Fisher Scientific)-dyed tumor cells mixed with CD89 BsAbs. Using flow cytometry, the phagocytosis of tumor cells by TAMs was analyzed.
ELISA experiments
Single-cell tumor preparations were incubated with FcR-blocking reagent (BD Biosciences) and magnetic microbeads conjugated to anti-CD8 antibodies (Miltenyi Biotech MACS Microbeads) at 4°C. As instructed by the manufacturer, CD8+ cells attached to the magnetic beads were removed out of the cell suspension. Total protein concentrations were measured using a BCA Protein Assay Kit after whole tumors or CD8+ T cells were lysed in radioimmunoprecipitation assay buffer (Pierce, Bonn, Germany). SimpleStep ELISA Kits were used to quantify IFN-γ, TNF-α, and granzyme B in a total of 500 g of total protein lysate from cells (Abcam, USA).
Quantitative reverse transcriptase-PCR experiments
TRIzol was used to extract total RNA from samples (Invitrogen, Carlsbad, California, USA). ProtoScript-II First Strand cDNA Synthesis Kit (NEB, Hitchin, UK) was used to obtain complementary DNA from 500 ng RNA. PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) was used for quantitative PCR in accordance with the manufacturer guidelines. The relative amounts of messenger RNA (mRNA) expression were normalized to GAPDH’s. Murine primers were designed against the following mRNA targets: II6, Nos2, Tnfa, β2m, H2-Aa, Tgfb1, ArgI, Ccl2, Idno, Ccl3, Ccl5, Cxcl9, Cxcl10, Prf1, Gzmb, Ifng, Ctla-4, Pdl-1, and Gapdh using gene-specific primers (online supplemental figure S3).
Supplemental material
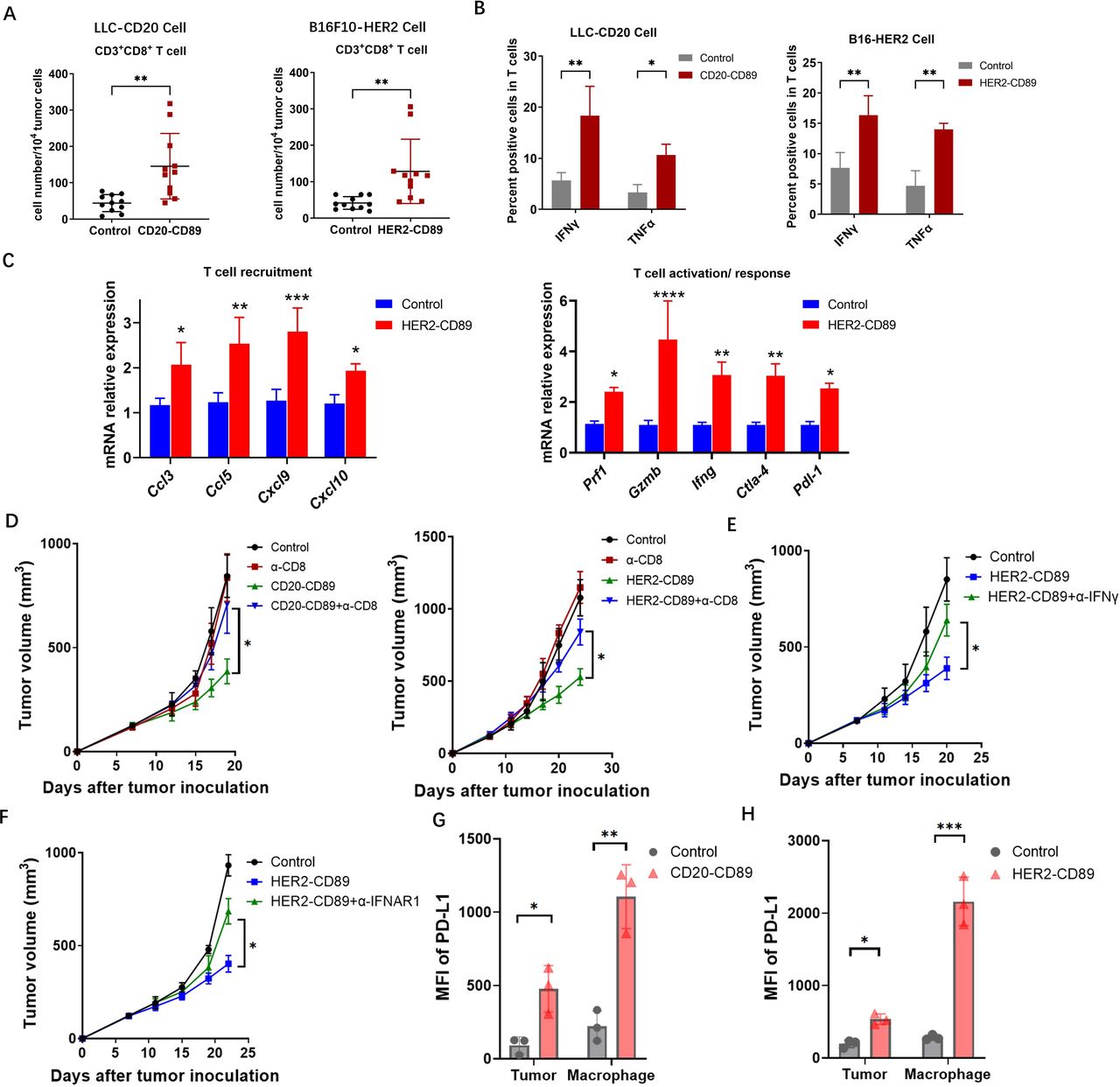
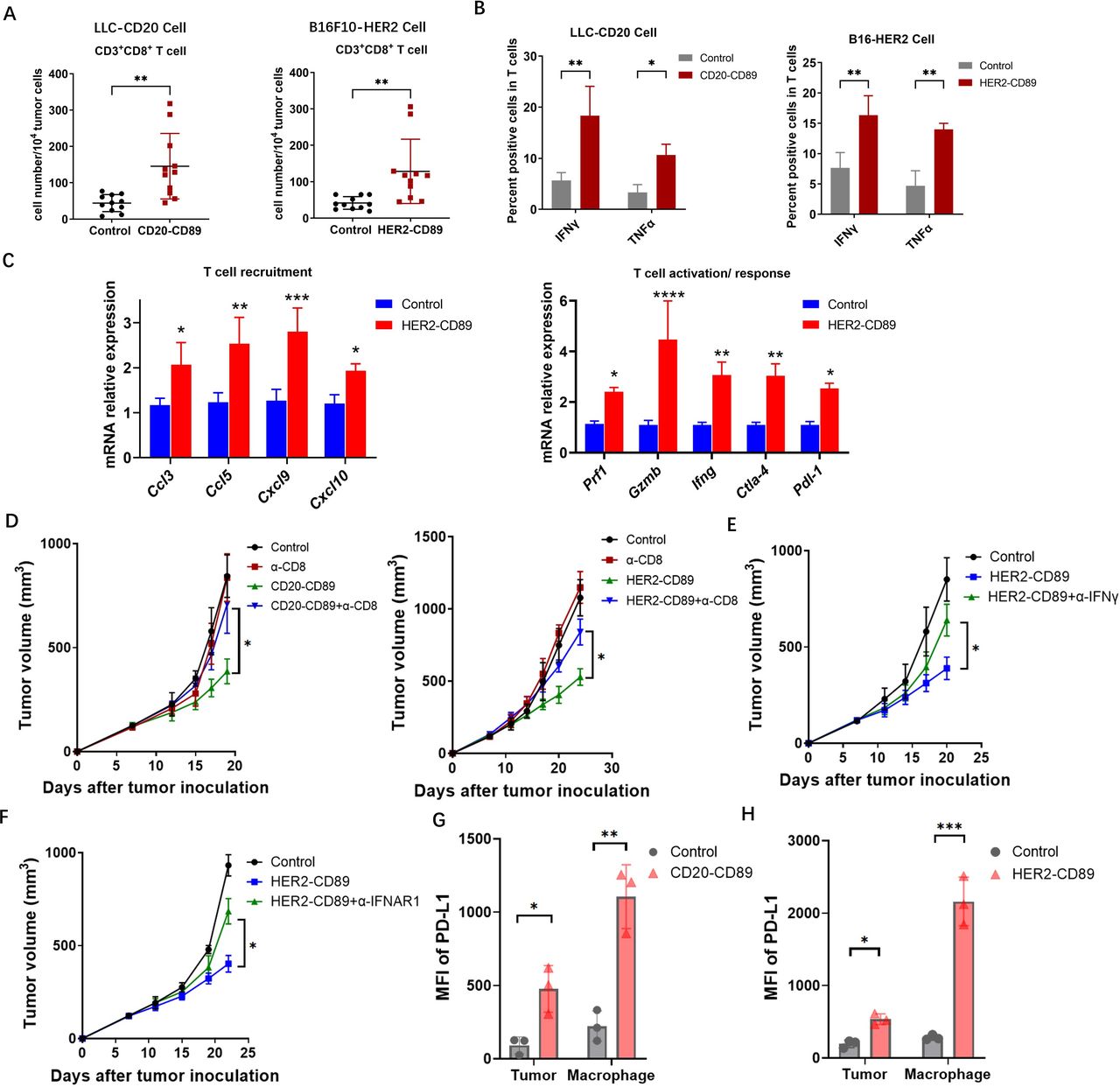
CD89 BsAb enhanced activated CD8+ T cells in TME (A) Tumors were harvested on day 21 after the treatment of BsAbs. Tumor-infiltrating CD8+ T cells were gated on CD45+CD3+CD8+ populations. Each subset is represented in a scatter plot. (B) IFN-γ and TNF-α expression in T cells isolated from tumors in CD20-CD89, HER2-CD89 and negative control-treated mice. n=3 biological replicates. (C) Messenger RNA expression levels of genes involved in T-cell immune responses in HER2-CD89 and negative control-treated mice. n=3 biological replicates. (D) Effect of CD8+ T-cell depletion on tumor growth. Anti-CD8 mAb or mouse IgG control were injected on days 7. Tumor-bearing mice (n=6–8/group) were treated with CD89 BsAbs therapy. Tumor volumes are plotted as the mean+SEM. (E, F) Effect of IFN depletion on tumor growth. Anti-IFN-γ mAb (E) or anti-IFNR1 mAb (F) were injected on days 7, and tumor-bearing mice (n=6–8/group) were treated with CD89 BsAbs therapy. Tumor volumes are plotted as the mean+SEM. T cells isolated from tumors in mice treated as above, and IFN-γ expression were detected. n=3 biological replicates. (G, H) PD-L1 expression on tumor cells and macrophages after the treatment with CD20-CD89 (G) or HER2-CD89 (H). P values were determined by two-way analysis of variance (D–F). BsAbs, bispecific antibodies; IFN, interferon; mAb, monoclonal antibody; MFI, mean fluorescence intensity; PD-L1, programmed cell death ligand-1; TME, tumor microenvironment; TNF, tumor necrosis factor.
Statistical analysis
GraphPad Prism V.9.0 was used to analyze and graph all data (GraphPad Software). Data are presented as the mean, SEM, or median and range (SEM). Unpaired, two-tailed Student’s t-tests and two-way analysis of variance were used to evaluate the data. Statistical significance was determined by p 0.05 (*p 0.05, **p 0.01, ***p 0.001, and ****p 0.0001). To guarantee that the results could be reproduced, experiments were conducted repeatedly.
Results
Antitumor activity of CD89-BsAbs
To co-target tumor cells and CD89 on innate immune effector cells, IgG1 Fc-based bispecific immunoengagers targeting CD20 or Her2 on tumor cells and CD89 on innate immune effectors were developed and named CD20-CD89 and HER2-CD89, respectively (online supplemental figure S1). The CD89 BsAb contains a fully human IgG1 Fc and a knobs-in-to-holes strategy, as previously described.18
Next, we assessed the antitumor effects of mouse effector cells in vivo. Owing to the lack of homologous CD89 expression in mice, we generated a Tg mouse strain expressing human CD89. Because we focused on CD89-mediated functions in macrophages, an authentic murine CD14 promoter was used, as described in our previous report.17 18 Mouse LLC cells or MC38 cells transduced with the hu-CD20 gene (LLC-CD20, MC38-CD20) and mouse B16F10 cells expressing human Her2 (B16-HER2) were injected into CD89 Tg mice to facilitate tumor growth. The mice were then treated with either the control (cIgG) or BsAbs. CD89 BsAbs exhibited notable antitumor activity and improved overall survival in the LLC-CD20 Tg mouse model (figure 1A and B). In WT mice implanted with LLC-CD20 cells, as shown in figure 1C, there was no response to CD89 BsAb therapy because of the lack of CD89 expression in macrophages. CD89 BsAbs also resulted in significant tumor regression in the MC38-CD20 model (figure 1D). Furthermore, the effects of HER2-CD89 BsAbs were observed in Tg mice implanted with B16-HER2 cells (figure 1E and F).
Subsequently, we investigated whether CD89 BsAbs could enhance phagocytosis by tumor cells. BsAbs were preincubated with Raji, Daudi, or BT474 or SK-BR-3 cells (labeled with CellTracker Green) before being co-cultured with TAMs (CD11b+F4/80+) obtained from tumor-bearing CD89 Tg mice (labeled with CellTracker Deep red). The proportion of phagocytosis increased in the CD89-BsAbs treated groups compared with that in the control groups, according to phagocytosis of macrophages with double positive (deep red and green) signals (figure 1G and H). Taken together, these results show that CD89 BsAbs significantly increased antitumor activity as a result of phagocytosis of tumor cells by CD89+ TAMs.
Increased antigen uptake by TAMs and improved M1/M2 ratio in the TME
Immune cells were isolated to further understand how CD89 BsAbs affected TAMs. TAMs were described as CD45+CD11b+F4/80+ subset. We further differentiated TAMs into the M1-like (MHC-II+CD206−) and M2-like (MHC-II–CD206+) macrophage phenotypes. In accordance with the more potent inhibition of tumor growth, FACS data demonstrated that CD89 BsAb therapy decreased CD206 expression and that M1:M2 macrophage ratios (the ratio of M1/M2) were increased in the BsAb-treated tumors (figure 2A). In agreement with this, we identified mRNA expression in TAMs, suggesting upregulation of M1 function-related genes (Il6, Nos2, Tnfa, β2m, and H2-Aa) and downregulation of M2 function-related genes (Tgfb, Arg1, Ccl2, and Idno) after CD89 BsAb treatment (figure 2B).

CD89 BsAb enhanced M1/M2 ratio in the tumor microenvironment and increased antigen uptake of TAMs (A) LLC-CD20 or B16-HER2 cell bearing mice were treated with CD89 BsAb and tumors were harvested on day 15. Tumor-infiltrating cells were analyzed by flow cytometry. MHC-II expression was used to identify M1 macrophages, and CD206 expression identified M2 macrophages. The M1/M2 ratios were calculated as the per cent of the M1 (MHC-II+CD206−) population divided by that of the M2 (MHC-II–CD206+) population among CD45+F4/80+CD89+ cells. The M1/M2 ratios of TAMs are represented in scatter plots. Each dot represents an individual animal, and the bars indicate means±SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. (B) mRNA expression of selected M1 and M2 markers in LLC-CD20 and B16-HER2 tumors as determined by RT-PCR. n=3 biological replicates. (C, D) CD40, CD80, and MHC-II expression in TAMs. (E) Analysis of antigen uptake by TAMs in vivo. Antigen (OVA–Alexa Fluor 488) was intratumoral injected with the last BsAbs injection. Twenty-four hours later, tumors were harvested and OVA–Alexa Fluor 488–positive cells in the gated CD45+ population were evaluated by flow cytometry. (F) Effect of macrophage depletion on tumor growth. HER2-B16 tumor bearing mice were treated with intravenously with HER2-CD89, 200 µL of Clophosome or control liposome was administered intraperitoneally on day 5 and every 4 days thereafter. Data are reported as means+SEM. P values were determined by two-way analysis of variance .(G)Interferon-γ expression in T cells isolated from tumors in mice treated as above were shown. n=5 biological replicates. BsAbs, bispecific antibodies; LLC, Lewis lung carcinoma; mRNA, messenger RNA; RT-PCR, reverse transcription-PCR. TAM, tumor associated macrophages.
To confirm that CD89 BsAb injection promoted antigen presentation, we used flow cytometry to assess antigen uptake and expression of maturation markers in TAMs isolated from tumors following CD20-CD89 therapy. Examination of the expression levels of costimulatory molecules revealed that the proportions of CD40+, CD80+, and MHC-II+ populations increased in TAMs (figure 2C and D). Mice bearing LLC-CD20 that had given CD20-CD89 treatment were administered Alexa Fluor 488-conjugated OVA intravenously in order to analyze antigen uptake by TAMs in further detail. Tumors were isolated 24 hours later, CD11b+F4/80+ macrophages linked with antigens (Positive Alexa Fluor 488 cells) were identified by flow cytometry. When compared with the control treatment, CD89 BsAb therapy significantly improved the amount of antigen-positive macrophages in tumors (figure 2E). These data indicated that BsAbs act directly on TAMs and enhance antigen presentation after anti-CD89-mediated phagocytosis by macrophages. Next, we deleted macrophages from the CD89 Tg mouse tumor models using liposomal clodronate. The therapeutic efficacy of HER2-CD89 was substantially lost in the absence of macrophages, resulting in reduced IFN-expression in T cells (figure 2F and G).
Increased CD8+ T cells with activity in the TME
We hypothesized that the observed strong control of tumor growth resulted from macrophage activation, combined with adaptive immunity localized to the TME. CD8+ cytotoxic T cells in tumors were investigated to evaluate whether the activation of TAMs by CD89 BsAb therapy results in the recruitment of tumor-specific T cells to the tumor regions. A substantial increase in CD8+ T cells was observed in the tumor-infiltrating immune cells in FACS analysis (figure 3A). IFN-γ+ and TNF-α+ CD8+ T cells were used to identify activated CD8+ T cells. T cells were collected from LLC-CD20 or B16-HER2 tumors treated with CD89 BsAbs. LLC-CD20 or B16-HER2 tumor-specific immune responses were confirmed by detecting enhanced IFN-γ and TNF-α expression in T cells using FACS (figure 3B). Consistently, when analyzing mRNA expression in tumor tissue specimens after CD89 BsAb treatment, we observed that HER2-CD89 therapy resulted in an upregulation of genes and proteins related to T-cell activation (figure 3C). The treatment with BsAb boosted the expression of genes involved in T-cell activation and recruitment.
To confirm that tumor-specific CD8+ T cells induced by CD89 BsAb therapy contributed to tumor growth suppression, CD8+ cells were depleted using an anti-CD8 mAb. In both tumor models, CD89 BsAb therapy’s suppressive effects were reversed by CD8+ cell depletion (figure 3D). Furthermore, IFN-γ-depleted and IFN-α-depleted CD89 Tg mouse tumor models were generated using anti-IFN-γ and anti-IFNAR1 antibodies and similar suppressive antitumor effects were observed, as shown in figure 3E and F. Together, these findings demonstrate that BsAb therapy-induced CD8+ T cells can inhibit tumor growth, and that this impact is dependent on the IFN signaling pathway.
Given their remarkable capacity to promote T-cell responses, CD89 BsAbs may cause tumor cells or APCs to overexpress PD-L1. Tumor tissues were obtained, and tumor cells (CD45–hCD20+ or CD45–hHER2+) and macrophages (CD45+CD11b+F4/80+) were examined in order to identify PD-L1 expression. PD-L1 was shown to be upregulated in both tumor cells and macrophages (figure 3G and H).
Combining CD89 BsAb with PD-1/PD-L1 inhibition prevented the growth of tumor
Based on these results, we postulated that PD-1/PD-L1 antibody resistance owing to limited T-cell infiltration and suppressive TAM cells may be overcome by combined CD89-targeting therapy with PD-1/PD-L1 antibodies. We injected MC38-CD20 cells into CD89 Tg mice and treated them with CD20-CD89 and anti-mPD-1 antibody (RMP1-14), alone or together (figure 4A). Comparing the CD20-CD89 or anti-mPD-1 antibody treatment alone to the combined treatment group, remarkable tumor regression was shown. Using an MC38-CD20 tumor model in which CD20-CD89 and/or anti-mouse PD-1 antibodies were simultaneously administered during MC38-CD20 cell implantation, we further evaluated the efficacy of the combination strategy. Both agents had only limited therapeutic efficacy, however the combination therapy significantly increased the antitumor effects and reduced tumor growth (figure 4B). Triple combination therapy was performed using CD20-CD89, anti-mPD-1, and the anti-mPD-L1 antibody atezolizumab (which is capable of binding mice and human PD-L1). We found that compared with dual therapy, the triple combination strategy remarkably delayed tumor growth (figure 4C).

Targeting CD89 combined with anti-programmed cell death-1 blockade (A, B) As shown in the schematic, CD89 Tg mice were treated with antibodies CD20-CD89 or mPD-1, alone or in combination (n=6–8 mice/ group). MC38-CD20 cells (2.5×105) were injected s.c. into CD89 Tg mice. Antibodies were administered by intravenous injection as shown in the schematic, when the tumor volumes reached 100–150 mm3 (A) or simultaneously injected during MC38-CD20 cell implantation (B). (C)Triple treatment comprised of CD20-CD89, anti-mPD-1, and anti-mPD-L1 was administered to mice (n=8 mice/group). (D, E) B16-HER2 cells (2.5×105) or LLC-CD20 (2.5×105) cells were injected s.c. into CD89 Tg mice (n=6–8 mice/group). Antibodies were administered by intravenous injection as shown in the schematic. (F) MC38-CD20 cells (2.5×105) were injected s.c. into CD89 Tg mice. Anti-mPD-1 was administered as shown in the schematic, CD20-CD89 was injected when the tumor volumes reached 400 mm3.Tumor sizes were monitored individually. Tumor volumes are plotted as the mean±SEM. P values were determined by two-way analysis of variance (A–F). LLC, Lewis lung carcinoma; s.c., subcutaneously.
Notably, tumor regression induced by the combination strategy was also observed in PD-1 resistant B16-HER2 tumors and LLC-CD20 tumors (figure 4D and E). In another MC38-CD20 tumor model, anti-mouse PD-1 antibody was injected according to the schematic and replaced with CD20-CD89 when the tumor volume reached 400 mm3. As shown in figure 4F, sequential therapy significantly controlled tumor growth in the late stage of treatment compared with PD-1 blockade alone.
Augmenting antitumor immunity through combination therapy
After treating pre-established MC38-CD20 flank tumors in immunocompetent CD89 Tg mice with CD20-CD89 and/or anti-mPD-1, the tumors were resected to determine changes in the immune microenvironment. FACS data having a greater proportion of CD8+ T cells were observed in the combination treatment group than in the CD20-CD89 or anti-mPD-1 alone groups. CD4+ helper T-cell infiltration did not change among the different groups, although the amount of regulatory T cells (Treg cells) infiltrating tumors decreased after the PD-1 blockade (figure 5A). Furthermore, analysis of tumor-infiltrating lymphocytes (TILs) using flow cytometry in the combination treatment group showed increased TAM infiltration and a higher M1/M2 ratio (figure 5B). Next, we confirmed that combination therapy promoted T cell-mediated cytotoxicity. When compared with those administered with a single agent, T cells from inhibitor-treated mice showed considerably higher levels of IFN-γ and granzyme B. (figure 5C). Together, these results indicate that combination therapy promotes high intratumoral T-cell infiltration, resulting in both Th1 and cytotoxic immune responses.

Effects of combination therapy on antigen-specific T-cell responses in vivo (A)Tumors were harvested on day 21 and tumor-infiltrating cells were analyzed by flow cytometry. Tumor-infiltrating T cells were gated into CD45+CD3+CD8+, CD45+CD3+CD4+ and CD3+CD4+CD25+FoxP3+ populations. (B)Tumor-associated macrophages were identified as the CD45+F4/80+CD89+ subset. The M1/M2 ratios were calculated as indicated above. Each subset is represented with a scatter plot. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. (C) IFN-γ and granzyme B protein concentrations. (D) mRNA expression levels of immune response genes from whole tumors after antibody treatment. n=3 biological replicates. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 compared with negative control. IFN, interferon; mRNA, messenger RNA.
Immune response gene expression and antigen-presenting gene expression were enhanced by CD89 BsAbs and PD-1 inhibitors, whereas they inhibited immune suppressive gene expression. These indicators were further elevated by the combination of therapy (figure 5D). These results show that through a combination of adaptive and innate immune activation localized to the TME, CD89 BsAbs could be combined with T cell-targeted therapy to promote antitumor immune responses that induce sustained tumor regression in mouse cancer models.
CD89 BsAbs overcame resistance to pembrolizumab inhibition
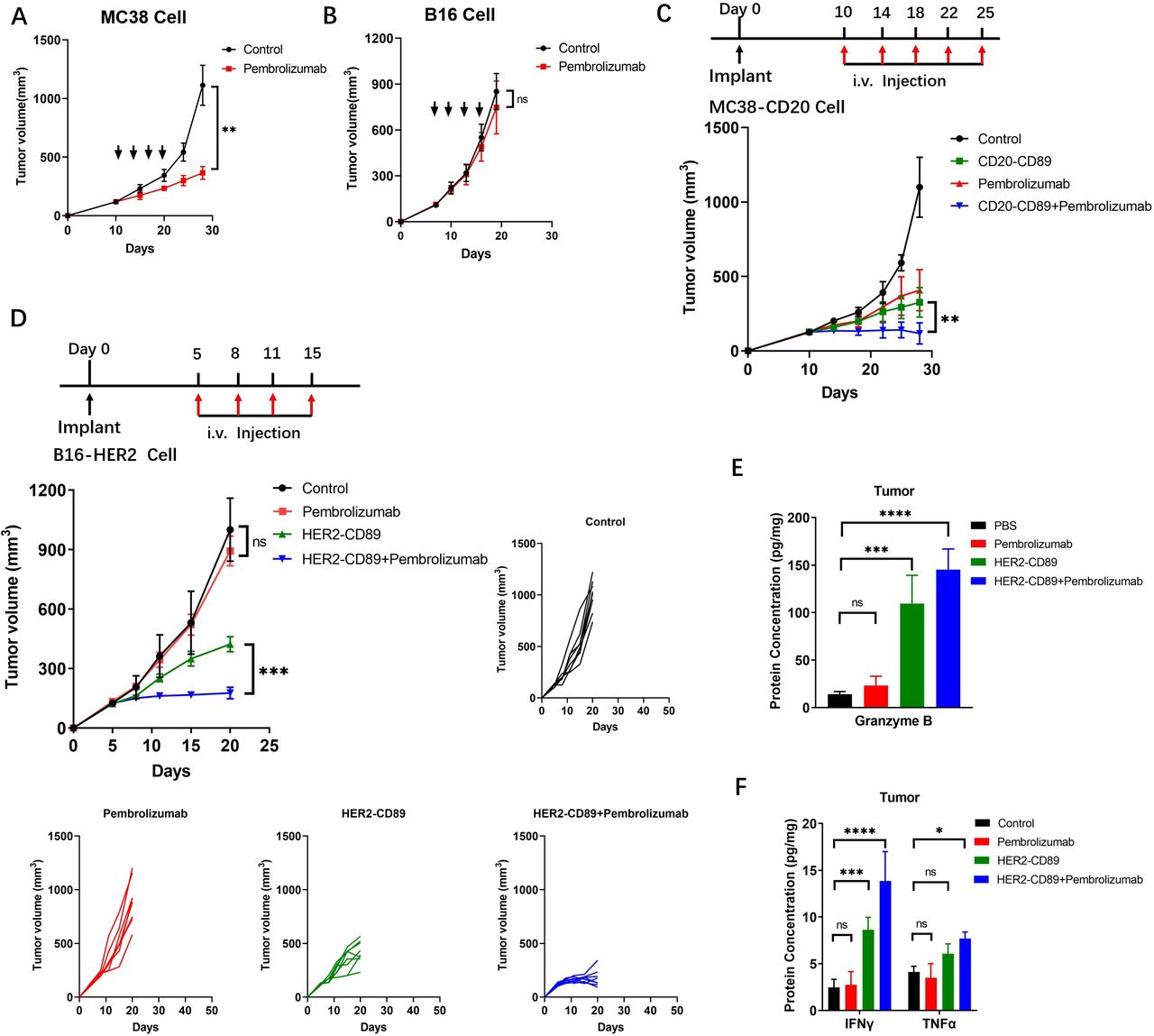
Because pembrolizumab only recognizes and inhibits human PD-1, we generated an hPD-1 Tg mouse strain by replacing both copies of the murine PD-1 gene with the full-length human PD-1 gene (online supplemental figure S2). MC38 (respondent to PD1/PD-L1 inhibitors) and B16F10 (non-respondent to PD1/PD-L1 inhibitors) tumor cells were implanted into hPD-1 Tg mice. Pembrolizumab conferred a therapeutic benefit to MC38 tumors but was ineffective against B16F10 tumors (figure 6A and B).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Resistance to checkpoint blockade therapy was overcome when combined with a CD89 bispecific antibodies (A, B) Effect of the pembrolizumab on the growth of MC38(A) or B16F10(B) cells in hPD-1 Tg mice. MC38 or B16F10 cells (2.5×105, 2.5×105) were injected subcutaneously into female mice (n=6–8/group). Antibodies were administered by intravenous injection. Animals were monitored for tumor growth. (C, D) Therapeutic regimen, mean tumor volumes and tumor volume changes of subcutaneous MC38-CD20 (C) and B16-HER2 (D) tumors after treatment with negative Control, HER2-CD89, alone or in combination with pembrolizumab in hCD89/hPD-1 Tg mice. Individual volumes of B16-HER2 -derived tumors treated with negative control or HER2-CD89 (alone or in combination with pembrolizumab) are shown. Tumor volumes are plotted as the mean+SEM. (E, F) Protein concentrations of granzyme B (E) and IFN-γ, TNF-α (F) measured in T cells isolated from B16-HER2 tumors after treatment (n=5 biological replicates); *p<0.05, ***p<0.001 and ****p<0.0001 as determined by two-way analysis of variance. IFN, interferon; PBS, phosphate buffered saline; TNF, tumor necrosis factor.
To test the combined effect of CD89 BsAbs and pembrolizumab, hCD89/hPD-1 double-Tg mice containing both transgenes were generated. In MC38-CD20 bearing hCD89/hPD-1 Tg mice, the combination therapy showed improved antitumor effects and restrained tumor progression compared with the therapeutic efficacy of either single agent (figure 6C). We then examined whether HER2-CD89 renders B16-HER2 tumor cells sensitive to pembrolizumab blockade. Significantly delayed tumor growth was observed with HER2-CD89 and pembrolizumab combination treatment than with PD-1 blockade alone (figure 6D). This strong control of tumor growth coincided with increased granzyme B levels (figure 6E). After combination therapy, protein expression signatures related to effective antitumor immunity (IFN-γ and TNF-α responses) were highly enriched (figure 6F). Thus, targeting CD89 makes tumor cells more susceptible to T cell-mediated cytotoxicity and overcomes pembrolizumab resistance.
Discussion/conclusion
Tumor immunotherapy is a hot topic in cancer research. Regardless of the PD-1/PD-L1 and CTLA-4 antibodies that have already been introduced to the market, TIM3, LAG3, Siglec-15, and other inhibitor targets that are still in clinical research,19 tumor immunotherapy is mainly focused on improving the killing function of specific T cells against tumor cells. Although these methods can be used to directly kill tumor cells, they do not normalize the immune system in the TME. In the TME, TAMs are the most prevalent form of innate immune cell and can directly regulate cytotoxic T lymphocyte responses via their expression of molecules such as MHC-II or PD-L1, and by their production of inhibitory cytokines,20–22 Additionally, TAMs can indirectly limit T-cell responses by managing the immunological microenvironment by recruiting immunosuppressive populations (such as Treg cells) or inhibiting dendritic cells.23 24 Therefore, achieving immune normalization by targeting TAMs may emerge as a future trend in tumor immunotherapy.20
In this study, we used a BsAb targeting CD89 and a tumor antigen that induced remarkable immune cell-mediated antitumor responses. Since mice do not exhibit a CD89 homolog, there is still little in vivo evidence of CD89’s potential. A transgenic mouse model whose macrophages and monocytes specifically express the CD89 gene was generated in a previous study,18 which allowed us to observe macrophage activation by CD89 BsAbs in vivo. To further evaluate the antitumor efficacy of combination treatment with anti-PD-1 antibody, Tg mice expressing human PD-1 and double-Tg mice expressing human CD89 and PD-1 were generated. Using immunocompetent hCD89 and hCD89/hPD-1 double-Tg mouse models, we observed promoted T-cell activation and recruitment by antigen presentation through CD89 activation; A stronger antitumor response was obtained with combination treatment, which overcame resistance to checkpoint block therapy.
Although the understanding of the impact of ICB on tumors and immune cells continues to evolve,25 26 it is generally accepted that the failure of ICB therapy may be caused by (1) insufficient infiltration of intratumoral immune cells resulting from impaired antigen processing or presentation (such as B2M mutations),27 (2) insufficient tumor-specific T-cell activity caused by severe T-cell exhaustion,28 or (3) abundant suppressive immune cells (TAMs, Tregs, or myeloid- derived suppressor cells (MDSCs)) in the TME.29 Several studies have demonstrated a negative role for TAMs in regulating the inactivation of immune checkpoint inhibitors.10 11 30
Our findings demonstrate that CD89 BsAb leads to the activation of phagocytosis by TAMs, in turn leading to the release of inflammatory cytokines and the presentation of antigenic tumor peptides to T-cell priming. Increased lymphocyte activation at the tumor site was evidenced by elevated production of IFN-γ and TNF-α in the TILs. Thus, targeting CD89 represents a rational strategy for overcoming resistance to ICB therapy. The innate and adaptive immune systems being active via anti-CD89 and checkpoint blockade as combination therapy has been demonstrated to drive macrophage-mediated and T cell-mediated immunity against cancer in syngeneic mouse models.
Due to the importance of TAMs in controlling tumor immunity, treatment approaches that target macrophages have generated a lot of interest, such as targeting the CCL2–CCR2 and CSF1–CSF1R axes to deplete TAMs, CD40 agonists, and PI3Kγ inhibitors to alter protumoral TAM activities.31–34 In addition to these widely known strategies, targeting Fc receptors on TAMs enables them to enroll in ADCC and antibody dependent cellular phagocytosis (ADCP). CD16A bispecific antibodies targeting CD30 on tumor cells can improve the TMEs tumoricidal activity through ADCP, mediated by different macrophage subtypes, especially M2 macrophages. CD16A-specific immuno-engager are presently being assessed in several clinical trials for the treatment of CD30-positive malignancies.35 36 Triggering the IgA Fc receptor CD89 also provided promising results for multiple tumor-associated antigens. However, there have only been a few in vivo studies on CD89-targeting therapy, in part because mice do not express CD89.14 37 Using the murine CD14 promoter, which is an excellent marker for monocytes and macrophages, we generated a CD89 transgenic mouse model that is ideal for studying the antitumor function of macrophages. Considering that many various myeloid lineage cells express CD89, we believe that targeting CD89 could serve as a promising antitumor therapy in clinical settings.15 37–41
As a result, using Tg mouse models we demonstrate that substantial activation of CD89 signaling is essential for boosting the effectiveness of checkpoint inhibitors in cancer. When considered collectively, these promising developments provide a solid foundation for targeting macrophages for the further exploration of the possibilities of such therapeutics and for improving future cancer treatment strategies.
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
We would like to express our gratitude to all of the laboratory's staff for their assistance and constructive criticism.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LX and BL contributed equally.
Contributors JF, BL and LX conceived and designed the experiments. LX, BL, CP, ZZ, FT, YF, XX and YY performed experiments. KX provided analysis of in vivo data. HG provided critical reagents and advice. HG and LX interpreted data and wrote the manuscript. JF is the guarantor and controlled the decision to publish.
Funding This research was funded by funds from the Shanghai Science & Technology Basic Research Program (18JC1414400), the China Postdoctoral Science Foundation Grant (2018M640652, 2019M652466), and the Key Science and Technology Program of Henan Province (212102310872).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.