Article Text

Abstract
Background Colony-stimulating factor 1 receptor (CSF1R), a classic tyrosine kinase receptor, has been identified as a proto-oncogene in multiple cancers. The CSF1/CSF1R axis is essential for the survival and differentiation of M2-phenotype tumor-associated macrophages (M2 TAMs). However, we found here that the CSF1R expression was abnormally down-regulated in colorectal cancer (CRC), and its biological functions and underlying mechanisms have become elusive in CRC progression.
Methods The expression of class III receptor tyrosine kinases in CRC and normal intestinal mucosa was accessed using The Cancer Genome Atlas and Gene Expression Omnibus datasets and was further validated by our tested cohort. CSF1R was reconstructed in CRC cells to identify its biological functions in vitro and in vivo. We compared CSF1R expression and methylation differences between CRC cells and macrophages. Furthermore, a co-culture system was used to mimic a competitive mechanism between CSF1R-overexpressed CRC cells and M2-like macrophages. We utilized a CSF1R inhibitor PLX3397 to ablate M2 TAMs and evaluated its efficacy on CRC treatment in animal models.
Results We found here that the CSF1R is silenced in CRC, and the reintroduced expression of the receptor in CRC cells can be cleaved by caspases and constrain tumor growth in vitro and in vivo, functioning as a tumor suppressor gene. We further identified CSF1R as a novel dependence receptor, which has the potential to act as either a tumor suppressor gene or an oncogene, depending on its activated state. In CRC tumors, CSF1R expression is enriched in TAMs, and its expression is associated with poor prognosis in patients ith CRC. In a co-culture system, CRC cells expressing CSF1R compete with M2-like macrophages for CSF1R ligands, resulting in a decrease in CSF1R activation and cell proliferation in macrophages. Blocking CSF1R by PLX3397 could deplete M2 TAMs and augments CD8+ T cell infiltration, effectively inhibiting tumor growth and metastasis and improving responses to chemotherapy and immunotherapy.
Conclusion Our findings revealed that CSF1R is a novel identified dependence receptor silenced in CRC. The silence abalienates its ligands to stimulate CSF1R expressed on M2 TAMs, which is an appealing therapeutic target for M2 TAM depletion and CRC treatment.
- gastrointestinal neoplasms
- tumor microenvironment
- receptors, immunologic
- macrophages
- immunotherapy
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Colony-stimulating factor 1 receptor (CSF1R), a member of the type III tyrosine kinase receptor, has been extensively characterized as an essential oncogene in tumors.
WHAT THIS STUDY ADDS
CSF1R is a novel identified dependence receptor. Ligand-free CSF1R promotes colorectal cancer (CRC) cell apoptosis while binding to its ligands facilitates proliferation.
The FMS-intronic regulatory element (FIRE) enhancer region of CSF1R is hypermethylated in CRC cells and hypomethylated in macrophages.
PLX3397, a small molecule inhibitor targeting CSF1R, significantly reduces M2-phenotype tumor-associated macrophage (M2 TAM) infiltration and increases CD8+ T cell infiltration in CRC tumor tissue.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
CSF1R is an appealing target for M2 TAM depletion to debate malignant CRC.
PLX3397 effectively restrains CRC tumor growth and liver metastasis by inhibiting M2 TAMs in vivo, which is expected to be implicated in combinatorial therapies to treat CRC.
Introduction
The receptor tyrosine kinases (RTKs) are transmembrane receptors whose critical roles have been extensively characterized during cancer development.1 2 The RTKs are constituted by twenty sub-families containing 58 known members, and most of them are considered to be oncogenic receptors, implicating in various malignant cells displaying ‘oncogenic addiction’ to these receptors, such as ErbB1/2/4, VEGFR, ALK, PDGFR A, FGFR, NTRK, and FLT3, and therefore have been developed as representative therapeutic targets in targeted therapies.1 3 Somewhat surprisingly, other researchers and us have previously found that NTRK3 and RET, two receptors that belong to RTKs, behave very unconventionally and act as tumor suppressors in colorectal cancer (CRC).4–6 In parallel, other RTKs, including c-Kit, MET, and ALK, function as tumor suppressors in at least specific conditions rather than acting solely as oncogenes.7–10 Although this evidence seems paradoxical, previous independent studies have pointed straightly at a model that those types of receptors have been dubbed dependence receptors according to their ability to elicit two opposite biological functions: these active receptors exert their classic tumor-promoting function in a condition that their ligands are available. In contrast, in the withdrawal or absence of ligands, the receptors would rather trigger cell apoptosis when disengaged from their corresponding ligands.10–12
Out of RTKs, a set of dependence receptors, such as DCC, NGFR, Plexin D1, UNC5D, and Neogenin/RGM, also carry the functional traits of dependence receptors.10 13–17 Mechanistically, those dependence receptors share a molecular hallmark that an inactive state of the receptors can commonly initiate apoptosis, a programmed cell death, by activating caspases. Although the underlying molecular mechanisms of these inactive dependence receptors to induce apoptosis remain obscure, it has been hypothesized that their caspase-cleaved sites are exposed when those inactivated receptors stay at a monomer form, consequently leading to the activation of apoptotic cascades. Indeed, there is a common characteristic that all these receptors contain addiction or dependence domains and caspase cleavage sites in their intracellular segment, which are required for apoptosis induction.10–12 These functions and mechanisms confer these receptors to acquire tumor-suppressor activity. Therefore, such a trait has been assumed that tumor cells depressing these dependence receptors present a selective advantage for adaption to an abnormal tumor microenvironment with limited and constant ligands, especially where ligands are commonly rare in invasive or metastatic sites.10–12
Colony-stimulating factor 1 receptor (CSF1R), a member of type III tyrosine kinase receptor, has been intensively identified as an oncogene for multiple tumor types, such as CRC, peripheral T-cell lymphomas, and glioma.18–23 However, we found that CSF1R and its preferred ligand CSF1 are abnormally down-regulated in CRC and other tumors. Such a notable alteration that is unusual aroused our imagination that CSF1R may not solely act as a traditional oncogene in CRC.
Materials and methods
Human samples
The CRC tumors and their paracancerous tissues were collected from 39 donor patients under strict compliance with ethical guidelines authorized by the Institutional Review Board of Sun Yat-sen University (2021ZSLYEC-366). The pathology archives in the Sixth Affiliated Hospital of Sun Yat-sen University kindly provided formalin-fixed and paraffin-embedded sections of CRC specimens and non-cancer tissues. Two fresh CRC tumor tissues were obtained from patients ith CRC by surgical excision in the Six Affiliated Hospital of Sun Yat-sen University. Informed consent was attained from all patients included in this study.
Methylation-specific PCR
Relative quantitative methylation-specific PCR was performed in a QuantStudio 7 Flex PCR system (Thermo Fisher). Detailed procedures have been described in our previous publications.4 5 24 The primers and probes targeting CSF1R and the internal control AluC4 are presented in online supplemental table 1.
Supplemental material
Animal models
Mice (C57BL/6, BALB/c, and immunodeficient BALB/c-nude) were purchased from the Vital River (Beijing, China) and bred under specific pathogen-free conditions in the Animal Facility of the Sixth Affiliated Hospital of Sun Yat-sen University. All mouse experiments were randomized (randomized block) and blinded, following the policies of the Sun Yat-sen University Institutional Animal Care and Use Committee (IACUC-2020122803). Cancer cells were subcutaneously injected into mice. Mice were pre-administrated with CSF1R monoclonal antibody (mAb) or IgG2a (20 mg/kg) before modeling and maintained the administration to the end-point. In pharmacodynamic experiments for PLX3397, PLX3397 (30 and 60 mg/kg) was orally administrated three times a week, and 5-fluorouracil (5-FU, 60 mg/kg, i.p.) was set as a positive control. To investigate the potential synergic effect of PLX3397 on immunotherapy, we treated the C57BL/6J mice bearing MC38 tumors with PLX3397 (60 mg/kg, p.o., once every 2 days) in the presence or absence of mouse anti-PD1 mAb (10 mg/kg, i.p., once every 3 days) and anti-CTLA-4 mAb (10 mg/kg, i.p., once every 3 days), and mice with mouse IgG (10 mg/kg, i.p., once every 3 days) administration were set as isotype control. Tumor diameters were monitored every 2–3 days, and tumor volumes were calculated using a previously reported formula.24 The mice-bearing luciferase-expressed tumors were scanned with an In Vivo Imaging System (IVIS, Caliper Life Sciences). Carbon dioxide inhalation was applied for mouse euthanasia, and then xenografts were resected.
In the liver metastatic model, CT26-Luc cells were injected into the spleen following PLX3397 (60 mg/kg, p.o.) and 5-FU (60 mg/kg, i.p.) administration every 2–3 days. At the experimental endpoint, metastatic tumors were monitored using the IVIS. The number of liver metastatic tumors was quantified by ImageJ software. H&E staining was conducted to detect liver metastases.
For the orthotopic colon model, CT26-Luc cells and MC38-Luc cells were injected into the submucosa of the cecum. Tumors were dissected for macrophage sorting when sufficient luciferin signaling was captured in the IVIS.
Magnetic bead-assisted sorting assay
Mice orthotopic MC38 and CT26 tumors were used to separate TAMs. After removing dead cells, we used microbeads coating F4/80 antibody to sort out macrophages in orthotopic tumors. In another single-cell suspension, CD45+ immune cells were removed using CD45 antibody-coated microbeads. The separation was performed on an AutoMACs Separator (Miltenyi Biotec). The validation of cell purity was assessed by flow cytometry (Bio-Rad).
Processing of Microwell-Seq and available scRNA-seq data analysis
Two fresh tumor tissues resected from patients with CRC were used to perform the Microwell-Seq according to previously reported protocols.25 Briefly, CRC tissues were digested for single-cell preparation. Single-cell samples and barcoded beads were transferred to the microwell array for lysis. Single-cell RNA was converted to cDNA using reverse transcriptase, followed by cDNA amplification. The sequencing was performed on an Illumine Hiseq system.
The raw unique molecular identifier (UMI) count matrices were downloaded from Gene Expression Omnibus (GEO) datasets (GSE178318 and GSE161277), followed by converting into a Seurat object by the R package Seurat (V.4.1.0).26 After quality control, 25,122 genes across 38,039 single cells in GSE178318 and 21,025 genes across 3292 single cells in GSE161277 were retained for analysis. Next, we used the Find Variable Features function to identify the top 2000 highly variable features and employed the Find Clusters function to perform the clutter on 50 principles components with resolutions 0.5 or 1.0, and cell types were annotated using the recognized cell markers.27 28
Statistical tests
The statistical significance was assessed with a standard Student’s t-test, paired-sample t-test or one-way Analysis of variance with Tukey’s multiple comparison test. Kaplan-Meier survival curves comparison with the most significant split was performed using the log-rank (Mantel-Cox) test. Data are generally presented as the means±SD and in some cases that were otherwise stated in figure legends, displayed as means±SE of the mean (SEM). *p<0.05, **p<0.01, ***p<0.001 are considered statistically significant, and ‘ns’ indicates the statistic does not reach statistical significance (p>0.05). Suitable statistical analysis was performed using GraphPad Prism V.7 software.
Results
CSF1R and its preferred ligand CSF1 are downregulated in CRC
The available mRNA expression in The Cancer Genome Atlas (TCGA) datasets was employed to compare the class III RTKs (CSF1R, FLT3, KIT, PDGFRA, and PDGFRB) expression between CRC tumors and matched paracancerous normal tissues. As shown in figure 1A, most of the class III RTKs (CSF1R, FLT3, KIT, and PDGFRA) were significantly downregulated in CRC, except PDGFRB, which was upregulated as expected. Due to its typical class III RTK, which has been well identified as an important oncogene in several cancers, we chose the CSF1R gene as a representative for further investigation. Indeed, CRC tumors exhibited an abnormal CSF1R-silenced phenotype with a frequency of 81.6% (40/49) in the TCGA cohort (figure 1A), and this downregulation can also be verified in a dataset derived from the GEO (figure 1B). We then further carried out a validation study utilizing an independent tissue queue, showing an aberrant decrease of CSF1R expression in CRC tissues (figure 1C,D). The TCGA database showed that the silence existed in multiple cancer types, including bladder urothelial carcinoma, breast cancer, hepatocellular carcinoma, lung cancer, pancreatic cancer, prostate adenocarcinoma, stomach cancer, and uterine corpus endometrial carcinoma (online supplemental figure 1A). In addition, CSF1, a preferred ligand of the CSF1R, showed dramatically reduced expression in the tissues of patients with CRC (online supplemental figure 1B). The expression of IL34, an alternative CSF1R ligand,29 was invariable in tumor tissues, but its expressive abundance in CRC was lower than CSF1 (mean 2.34 vs 4.42) (online supplemental figure 1C). These results indicate that the CSF1-CSF1R axis is inactivated in cancer cells.
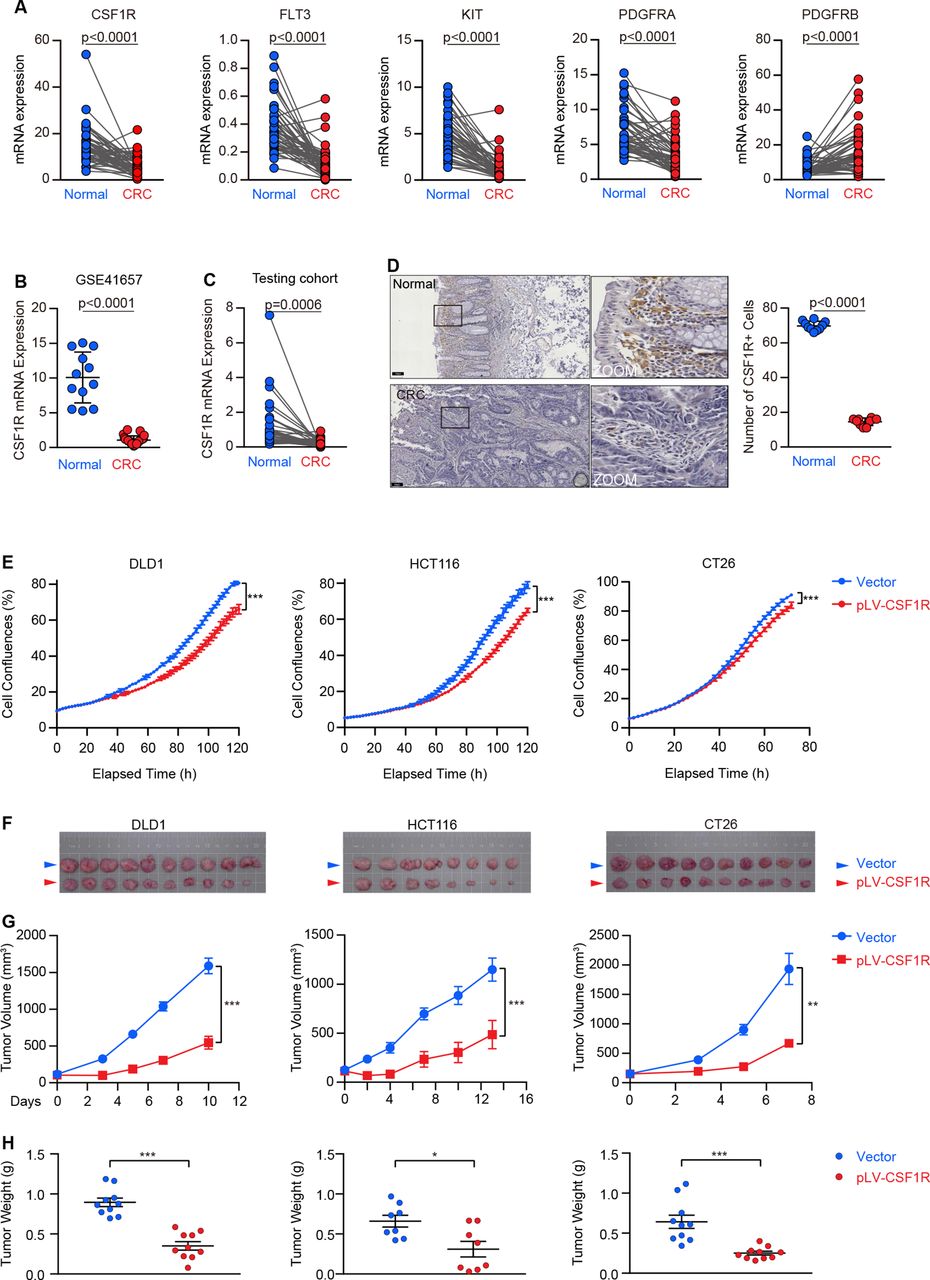
CSF1R is downregulated in colorectal cancer (CRC), and its reconstitution inhibits tumor growth in vitro and in vivo. (A) Paired comparison of receptor tyrosine kinases (RTKs) (CSF1R, FLT3, KIT, PDGFRA, and PDGFRB) mRNA expression in CRC (n=50 pairs) versus adjacent normal tissues using the TCGA datasets. (B) CSF1R mRNA expression in CRC (n=25) and normal tissues (n=12) was analyzed using an available GEO cohort. (C) Paired differential expression analysis of colon adenocarcinomas versus matched adjacent normal colon epitheliums (n=39 pairs) in our testing cohort. (D) Representative CSF1R IHC images of CRC (n=10) and normal (n=10) tissues. (Scale bars, 100 µm). The representative regions in boxes are enlarged, and 10 regions of each sample were randomly selected for quantitatively counting CSF1R positive cells. (E) The real-time cell confluence of DLD1, HCT116, and CT26 cells with or without CSF1R overexpression. Data are means±SD. (F–H) Bright-field images (F), tumor volumes (G), and tumor weights (H) of subcutaneous xenografts were established using DLD1 (n=10), HCT116 (n=8), and CT26 (n=10) cells with CSF1R or empty vector stable transfection. Data are presented as means±SEM. The values at the end-point were statistically analyzed (E, G). *p<0.05, **p<0.01, ***p<0.001 by two-sided unpaired Student’s t-test. Data are expressed as mean±SD. P values were calculated by a two-sided paired (A, C,) or unpaired (B, D, H) Student’s t-test. GEO, Gene Expression Omnibus; IHC, immunohistochemistry; TCGA, The Cancer Genome Atlas.
CSF1R is a novel dependence receptor
Given the abnormal downregulation, we hypothesized that CSF1R has a potential tumor-suppressive effect responsible for its silence. We; therefore, stably reconstructed CSF1R expression on three CRC cell lines (CT26, DLD1 and HCT116) to assess its biological function in CRC (online supplemental figure 2A,B). The stable reconstitution of CSF1R substantially slowed cell growth in all of these cell lines, as tested by the in vitro proliferation experiment (figure 1E). In mouse xenograft models, the clear mitigating growth curves and lightening tumor burdens in CSF1R-reconstructed tumors demonstrated that CSF1R served as a tumor suppressor in vivo (figure 1F–H).
It has been widely established that CSF1R activation is carcinogenic in CRC and other tumor types.18–21 In the CSF1R-reconstructed experiments, we did not co-express or further add its ligands, creating a circumstance that the overexpressed CSF1R may be inactive. To determine the effect of CSF1R activation in CRC cells, we used CSF1 and IL34 to stimulate the receptor, and the downstream cellular CSF1R signaling cascades were examined. We confirmed that CSF1R was successfully stimulated after being treated with IL34 and CSF1, and its downstream Akt and Erk cascades were also activated in CSF1R-overexpressed DLD1 and HCT116 cells (figure 2A). CSF1 and IL34 markedly accelerated cell proliferation in CSF1R-overexpressed DLD1 and HCT116 cells in a concentration-dependent manner. Still, they could not equivalently hasten growth in empty vector-expressed DLD1 and HCT116 cells carrying low CSF1R expression (figure 2B). Data from the Cancer Cell Line Encyclopedia (CCLE) database showed that among CRC cell lines, CACO2 had a comparatively high endogenous CSF1R mRNA expression (online supplemental figure 2C). We confirmed that CACO2 cells treated with CSF1 and IL34 displayed a CSF1R-stimulated status (online supplemental figure 2D). CSF1 and IL34 potentially enhance cell proliferation in CACO2 cells, agreeing with ectopic CSF1R-expressed DLD1 and HCT116 cells (figure 2C), indicating that the endogenous CSF1R remain to be an oncogene when its ligands are available and activate the receptor. Collectively, these traits confer CSF1R as a dependence receptor that is silenced in CRC. Dependence receptors lacking their ligands prefer to be cleaved by caspases in the intracellular domain as a prerequisite for triggering apoptotic death in tumor cells. Here, we discovered a rise in proapoptotic caspase 3/7 activity in the CSF1R-overexpressed DLD1, HCT116, and CT26 cells, which could be reverted by CSF1 and IL34 (figure 2D). To explore whether CSF1R could also be a caspase substrate, we used a CSF1R C-terminal recognized antibody to detect its cleaved fragments. As shown in figure 2E, we observed that CSF1R was cleaved in the CSF1R-overexpressed DLD1 and HCT116 cells, generating C-terminal cleavage fragments with a molecular mass of 60–70 kDa, which could be more detectable when cells were treated with a proteasome inhibitor MG-132, and this cleavage was rescued by adding a pan-caspase inhibitor Q-VD-Oph.

CSF1R activation by CSF1 and IL34 promotes colorectal cancer (CRC) cell proliferation and reduces proapoptotic caspase 3/7 activity. (A) The activation of the CSF1R (phospho-Tyr723) and its downstream AKT (phospho-Ser473) and ERK (phospho-Thr202/Tyr204) pathways in transfected DLD1 and HCT116 cells treated with CSF1 and IL34 was detected using Western blotting. (B, C) The proliferation of ectopic CSF1R-overexpressed DLD1 and HCT116 cells (B) and CACO2 cells (C) expressed endogenic CSF1R in the treatment with CSF1 or IL34 at various concentrations. A statistical comparison between cells with or without CSF1R overexpression (DLD1 and HCT116) was performed. The significant difference between the viability of CACO2 cells treated with CSF1 or IL34 was compared with control. P values were calculated using the unpaired Student’ t-test. (D) Caspase 3/7 activity was assessed in CSF1R-reconstructed DLD1, HCT116, and CT26 cells in the presence or absence of CSF1 and IL34. (E) The transfected DLD1 and HCT116 cells treated with MG132 (5 µM) for 2 hours or Q-VD-Oph (25 µM) for 24 hours, the cleaved CSF1R was detected using Western blotting. Significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, and ‘NS’ indicates no significance. ANOVA, analysis of variance.
CSF1R is enriched in TAMs and associated with prognosis in patients with CRC
In light of the general agreement that CSF1R is required and that its expression is increased progressively as macrophage mature, we hypothesized that CSF1R had a high propensity for expressing on tumor-associated macrophages (TAMs). Using information from the CCLE datasets, we compared the levels of CSF1R mRNA expression in cancer cell lines, revealing that it was most highly expressed in leukemia cell lines that contained mononuclear cancer cells, but its expression was impoverished in CRC cell lines and other solid tumor cells (online supplemental figure 3A). In a validation study, the unbalanced CSF1R expression between human or mouse monocytes and CRC cell lines in a panel of cell lines was depicted in figure 3A, where it can be observed that CSF1R expression was enriching in mouse and human monocytes, but low expression levels were observed in CRC cell lines. Next, we further compared different CSF1R expression levels between F4/80+, F4/80-, and CD45- cells separated from mouse orthotopic MC38 and CT26 tumor tissues using antibody-coated microbeads. In stark contrast to F4/80- and CD45- cells, F4/80+ cells displayed abundant CSF1R expression in MC38 and CT26 orthotopic tumors (figure 3B,C and online supplemental figure 3B). We obtained fresh tumors from two patients with CRC to perform a Microwell-seq, the results demonstrate a marked enrichment of the CSF1R expression in TAMs but not in other cell-type clusters, including cancer cells, B cells, T cells, endothelial cells, monocytes, and fibroblasts (figure 3D). We further accessed available scRNA-seq data with large sample sizes from the GEO database, both two independent datasets (GSE161277 and GSE178318) show that CSF1R is specifically expressed on TAMs at high levels, rather than other cell clusters (figure 3D, online supplemental figure 4), indicating that TAMs was the major cell type that expressed CSF1R in the CRC tumor microenvironment. Moreover, the Pearson linear regression analysis provides evidence for the substantial positive correlation between CSF1R expression and total marker of macrophage (CD11b, R2=0.7063) as well as M2-phenotype tumor-associated macrophages (M2 TAMs) makers (CD206, R2=0.6668; CD163, R2=0.4450) (figure 3E). In further analysis, we observed a relatively weak correlation between CSF1R expression and other immune cell markers, such as M1 TAM (CD86, CD32, and CD80), dendritic cells (CD11c), CD8+ T cells (CD8), natural killer cells (CD56), and B cells (CD19) (online supplemental figure 3C). Based on these findings, M2 TAMs, rather than malignant cells and other stroma or immune cells, have a large contribution rate to the CSF1R expression detected in CRC tissues; at least in part, the CSF1R expression reflects the level of infiltrated M2 TAMs in the tumor microenvironment. Thus, it is not difficult to understand why we observed a higher baseline CSF1R gene expression in CRC tumors that was associated with a significantly worse overall survival of patients in the TCGA cohort (figure 3F). Out of CRC, patients with high CSF1R-expressed cancers also displayed poor prognosis in other tumor types, implicating kidney cancer, stomach adenocarcinoma, hepatocellular carcinoma, and bladder urothelial carcinoma (online supplemental figure 5A). Similarly, patients with CRC with high expression of CSF1 and IL34 in tumor tissues also exhibited a poor prognosis (online supplemental figure 5B), suggesting that the CSF1/IL34-CSF1R axis in M2 TAMs contributes to CRC progression.
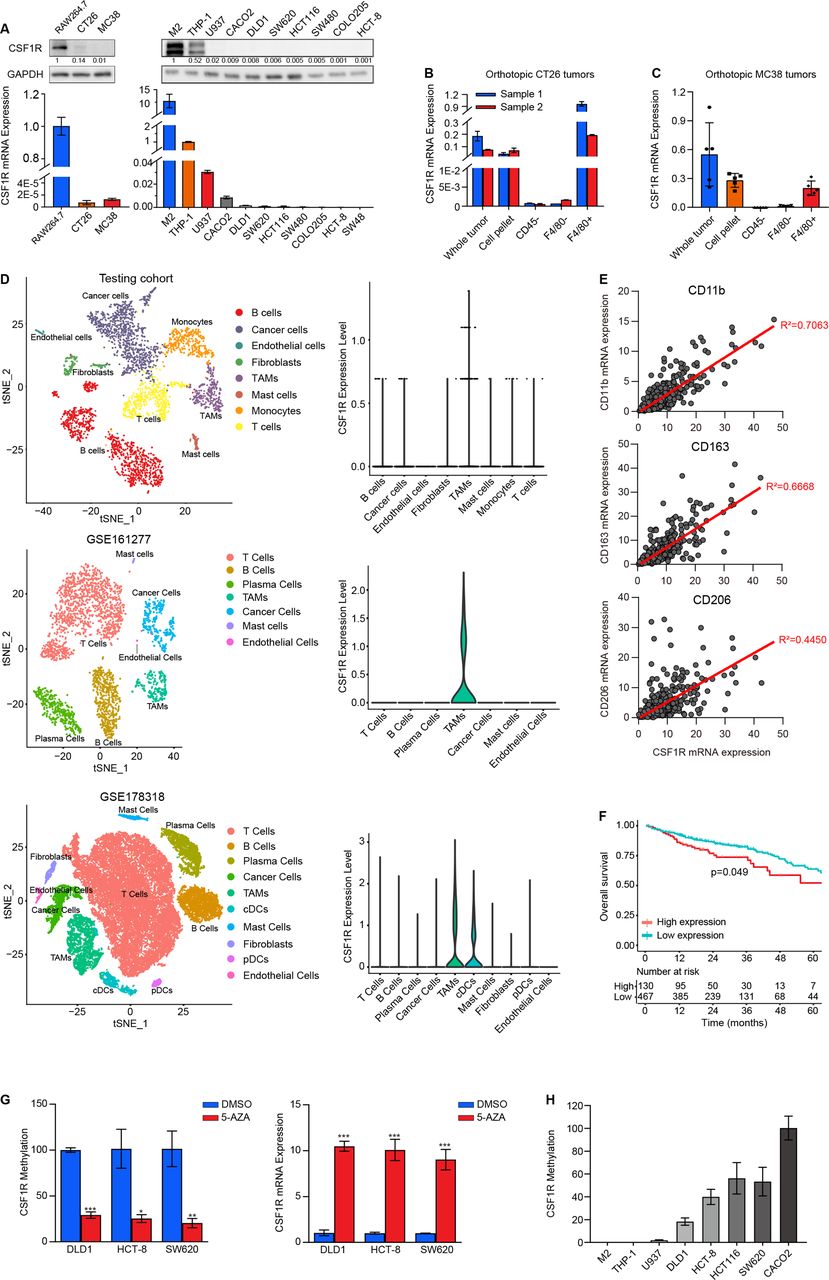
CSF1R expression is enriched in tumor-associated macrophages (TAMs) and correlates with prognosis. (A) The CSF1R protein and mRNA expression levels on a panel of cell lines containing mouse and human monocyte cells and CRC cells were detected using Western blotting and RT-PCR, respectively. (B, C) F4/80+, F4/80- and CD45- cells were separated from orthotopic CT26 (B) and MC38 (C) tumors, and their CSF1R mRNA expression levels were detected by RT-PCR. Data are presented as means±SD. (D) t-SNE plots (left) of cells from our testing cohort and two GEO datasets (GSE161277 and GSE178318). Each dot and color, respectively, represent a single cell or a cell types. Cells were clustered into several major subclusters based on biological annotation. The CSF1R expression level in each subclutters were shown by the Violin plot (right). (E) Spearman’s rank correlation analysis was used to assess statistical relationships between CSF1R mRNA expression and CD11b, CD163, CD206 from TCGA datasets. (F) Kaplan-Meier curves plot the high and low (most significant split) expression of CSF1R (high: n=130; low: n=467; p=0.049, log-rank test) against overall survival of patients with colorectal cancer in the TCGA cohort. (G) CSF1R methylation and mRNA expression in DLD1, HCT-8, and SW620 cells treated with or without 5-AZA were measured using quantitative methylation-specific PCR (qMSP) and RT-PCR. Data are means±SD. *p<0.05, **p<0.01, ***p<0.001 were determined by a two-sided unpaired Student’s t-test. (H) CSF1R methylation status at FMS-intronic regulatory element (FIRE) loci in monocytes cells (M2, THP-1, U937) and CRC cells (DLD1, HCT-8, HCT116, SW620, CACO2) was tested using qMSP.
DNA methylation on an intronic FIRE enhancer is responsible for CSF1R expression
Next, we sought to investigate the mechanism of the imbalanced CSF1R expression between cancer cells and macrophages. Despite the CSF1R gene lacking CpG islands, it is well identified that DNA methylation, particularly on an FIRE enhancer located in intron 2, is crucial for the CSF1R expression in macrophages.30 Here, we treated low-CSF1R expressed DLD1, HCT-8, and SW620 cells with 5-AZA, a DNA methyltransferase inhibitor, to assess the impact of DNA methylation on CSF1R expression. Methylated specific PCR analysis confirmed a lower methylation status on the FIRE region in 5-AZA-treated CRC cells. A pronounced CSF1R re-expression occurs when DNA methylation is depleted in these cancer cells (figure 3G). Despite lacking a validated study, we still observed a restoration of CSF1R expression in a concentration-dependent manner in murine CT26 and MC38 cells exposed to different concentrations of 5-AZA (online supplemental figure 3D). We proposed that the methylation located in the FIRE enhancer is responsible for the discrepancy in CSF1R expression between CRC cells and monocytes. We hereby deployed a comparison of CSF1R methylation in a cell line panel comprizing monocytes and CRC cell lines, showing that the FIRE region in the CSF1R gene was abnormally hypermethylated in cancer cells but being hypomethylated in monocytes, including M2-like macrophage, THP-1, and U937 (figure 3H). These results suggest that DNA hypermethylation in FIRE enhancer is responsible for CSF1R silence in CRC cells and that the hypomethylation in the region sweeps away the obstacle of CSF1R expression in macrophages.
CSF1R overexpression in CRC cells blunts the activation of CSF1R in macrophages
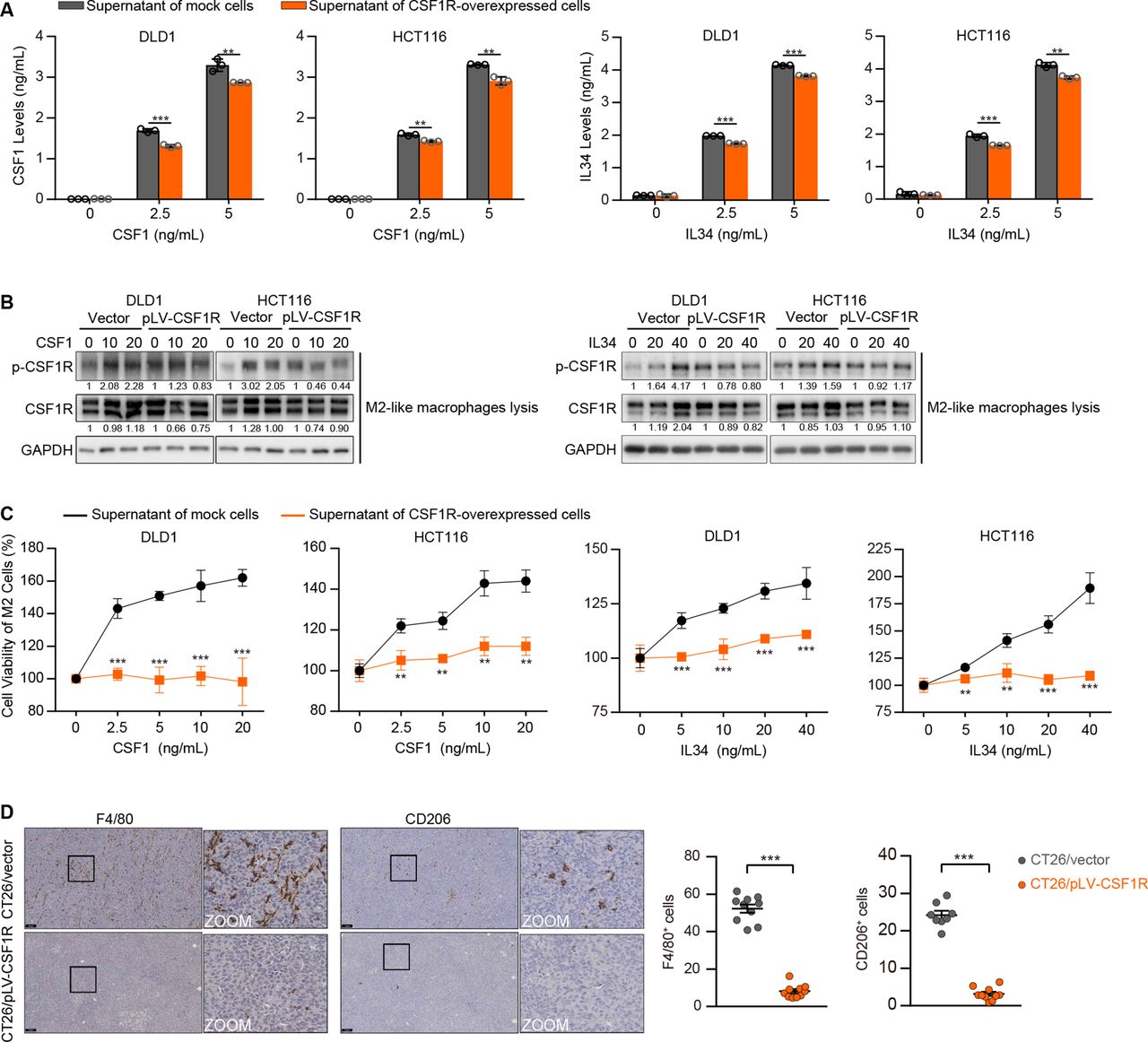
Given that CRC cells silence CSF1R, which differs sharply from TAMs, we reasoned that the event might assist TAM activation by preventing competition for the limited ligands in the tumor microenvironment rather than just helping the cells evade the tumor-suppressive effect. The ELISA assay results showed that the supernatant CSF1 and IL34 levels were dramatically decreased in the CSF1 or IL34-supplemented medium of CSF1R-overexpressing tumor cells (figure 4A). To mimic a straightforward tumor microecosystem, we constructed a co-culture system with M2-like macrophages and CRC cells utilizing transwell with cell impermeable micropores. Consequently, when M2-like macrophages were co-cultured with CSF1R-overexpressing CRC cells, the effect of CSF1 and IL34 on CSF1R activation was diminished in these macrophages (figure 4B). In addition, the CSF1 or IL34-supplemented supernatant of the parental empty vector-expressing cells pronouncedly stimulated the growth of M2-like macrophages, whereas the supernatant of the CSF1R-overexpressed CRC cells could not equivalently promote the proliferation (figure 4C). In CSF1R-overexpressed CT26 xenografts transplanted in mice bearing an intact immune system, the F4/80 (a macrophage marker) and CD206 (an M2 macrophage marker) immunostaining showed a notable reduction of the tumor-infiltrating F4/80+ and CD206+ cells (figure 4D), demonstrating that CSF1R overexpression in CRC cells dampens M2 TAM infiltration in vivo.
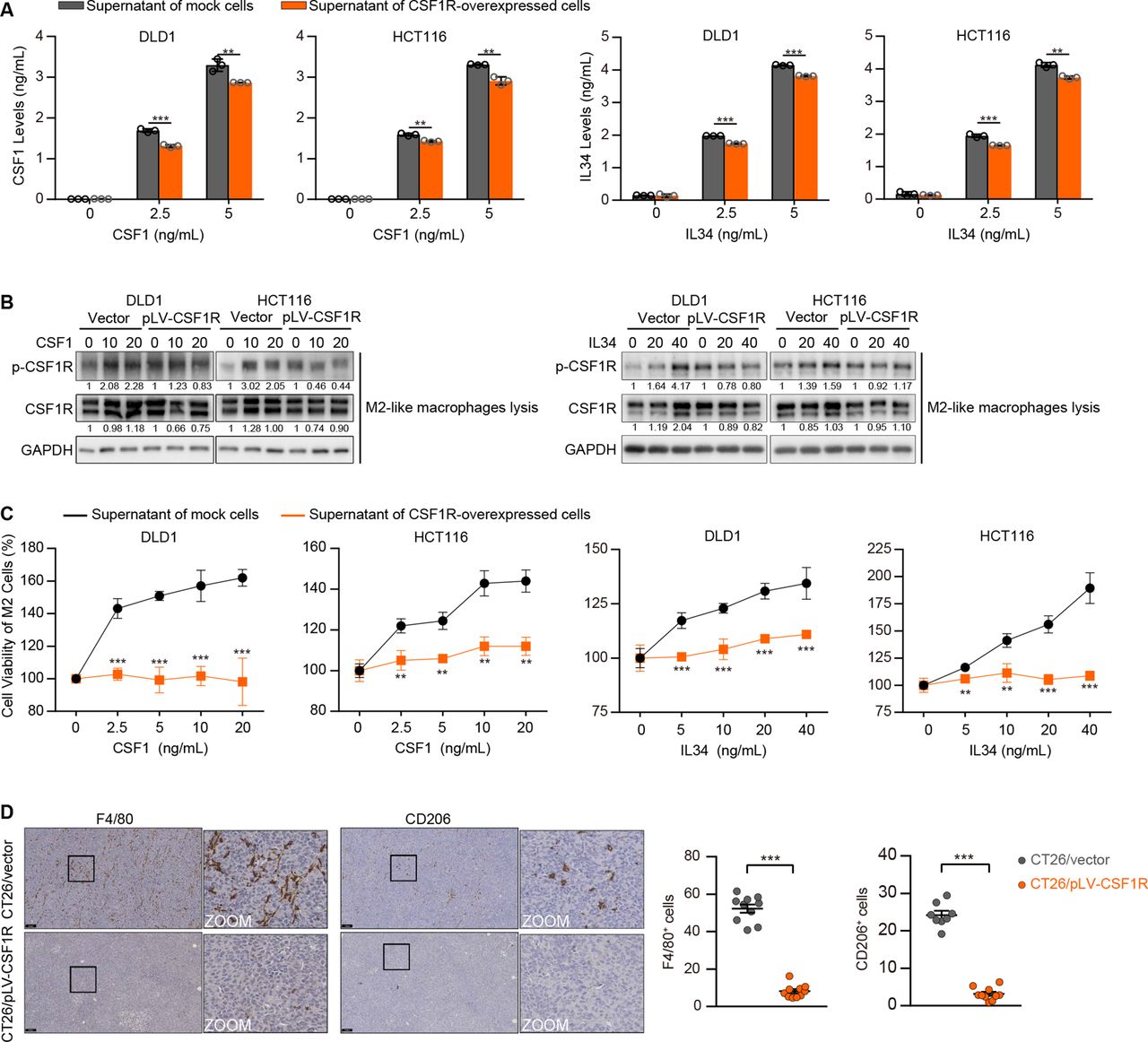
CSF1R-overexpressing colorectal cancer (CRC) cells suppress the activation of the CSF1R expressed in tumor-associated macrophages (TAMs) and affect TAM proliferation in co-culture systems. (A) CSF1 and IL34 levels were detected by ELISA in the supernatant medium of DLD1 and HCT116 cells with or without CSF1R overexpression. Data are means±SD. (B) The stimulation of CSF1R in M2-like macrophages co-cultured with transfected DLD1 or HCT116 cells was detected via assessing CSF1R phosphorylation (Tyr723). (C) The proliferation of M2-like macrophages was tested by CCK-8 assays after treating with the CSF1 or IL34-supplemented supernatant of transfected DLD1 or HCT116 cells. Data are means±SD. (D) F4/80 and CD206 IHC representative images and quantifications of transfected CT26 tumors. Scale bars represent 100 µm. Data are means±SD. Significant results were obtained by a two-tailed unpaired Student’s t-test (A, C–D). *p<0.05, **p<0.01, ***p<0.001, ‘NS’ indicates no significance. IHC, immunohistochemistry.
CSF1R blockade by a neutralizing antibody is effective in depleting M2 TAMs and stimulate cancer immunity in vivo
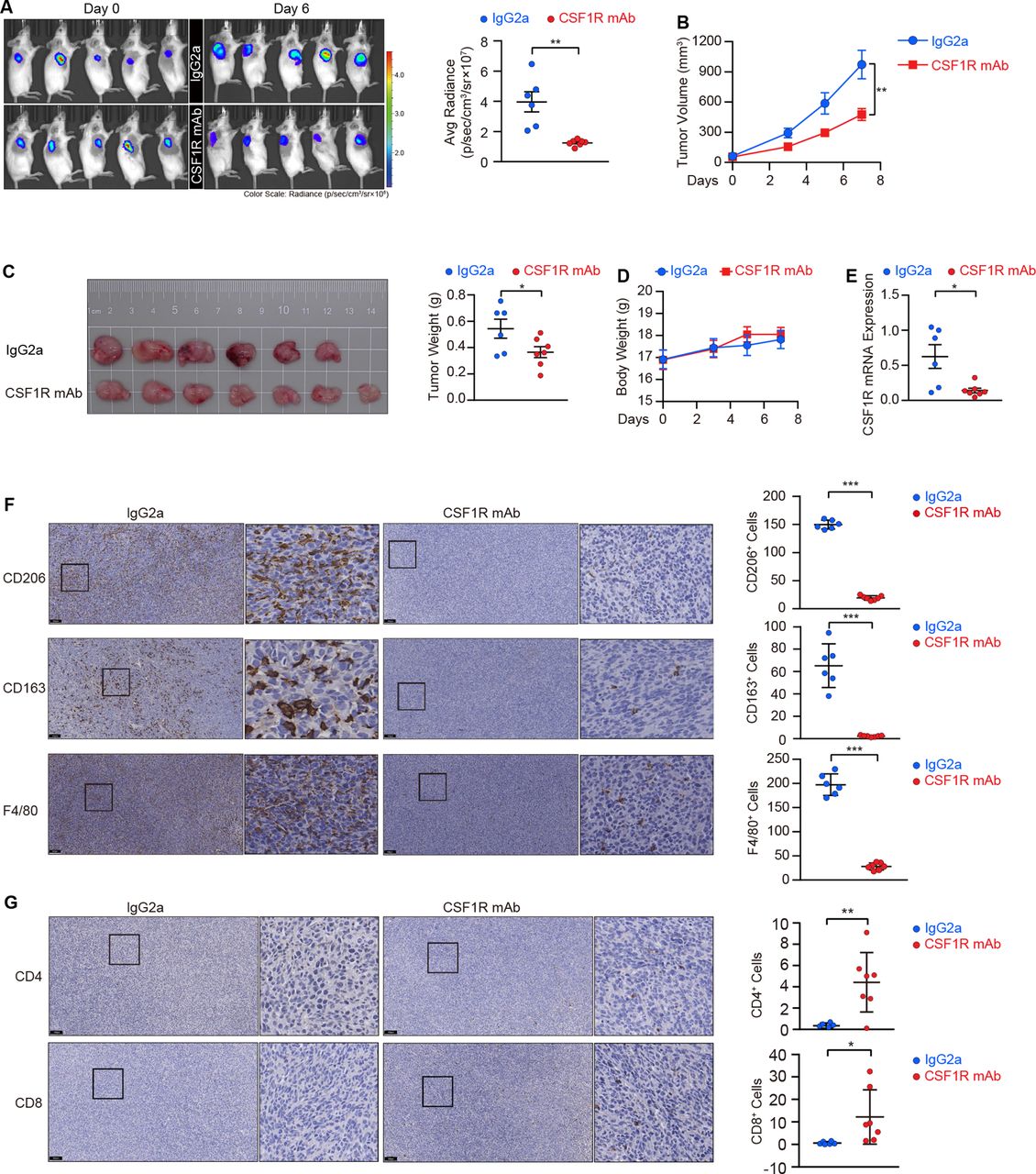
Given that the homeostatic survival and infiltration of M2 TAMs require CSF1R-mediated stepwise signaling activation, we employed CT26 cells to establish a preclinical model for functionally evaluating the effect of CSF1R blockage using a neutralizing antibody. As shown in figure 5A–C, the mice were pre-administrated with the CSF1R neutralizing antibody before the tumors cells were transplanted, and we maintained the antibody delivery once the tumors cells were subcutaneously seeded into mice, resulting in a deceleration of the tumor growth in these mice. Interestingly, the body weight monitoring reflected a good tolerance of the antibody in these mice (figure 5D). The neutralized antibody-treated tumors displayed a decline in CSF1R gene expression (figure 5E), confirming the efficacy of the neutralization in eliminating CSF1R-dependent M2 TAMs in vivo. By detecting M2 TAMs using the common macrophage marker F4/80 as well as two M2 macrophage markers, CD206 and CD163, we confirmed that M2 TAMs were depleted in tumors following treatment with the neutralizing antibody (figure 5F). A recent study demonstrates that TAMs can restrain tumor-specific CD4+ and CD8+ T cell infiltration, obstructing the effectiveness of immunotherapy.30 Herein, two effector lymphocyte populations were enriched in tumors from mice receiving neutralizing antibody treatment, with a 12.7-fold enrichment in CD4+ T cells and a 20.3-fold enrichment in CD8+ T cells in these mice compared with the control group (figure 5G).
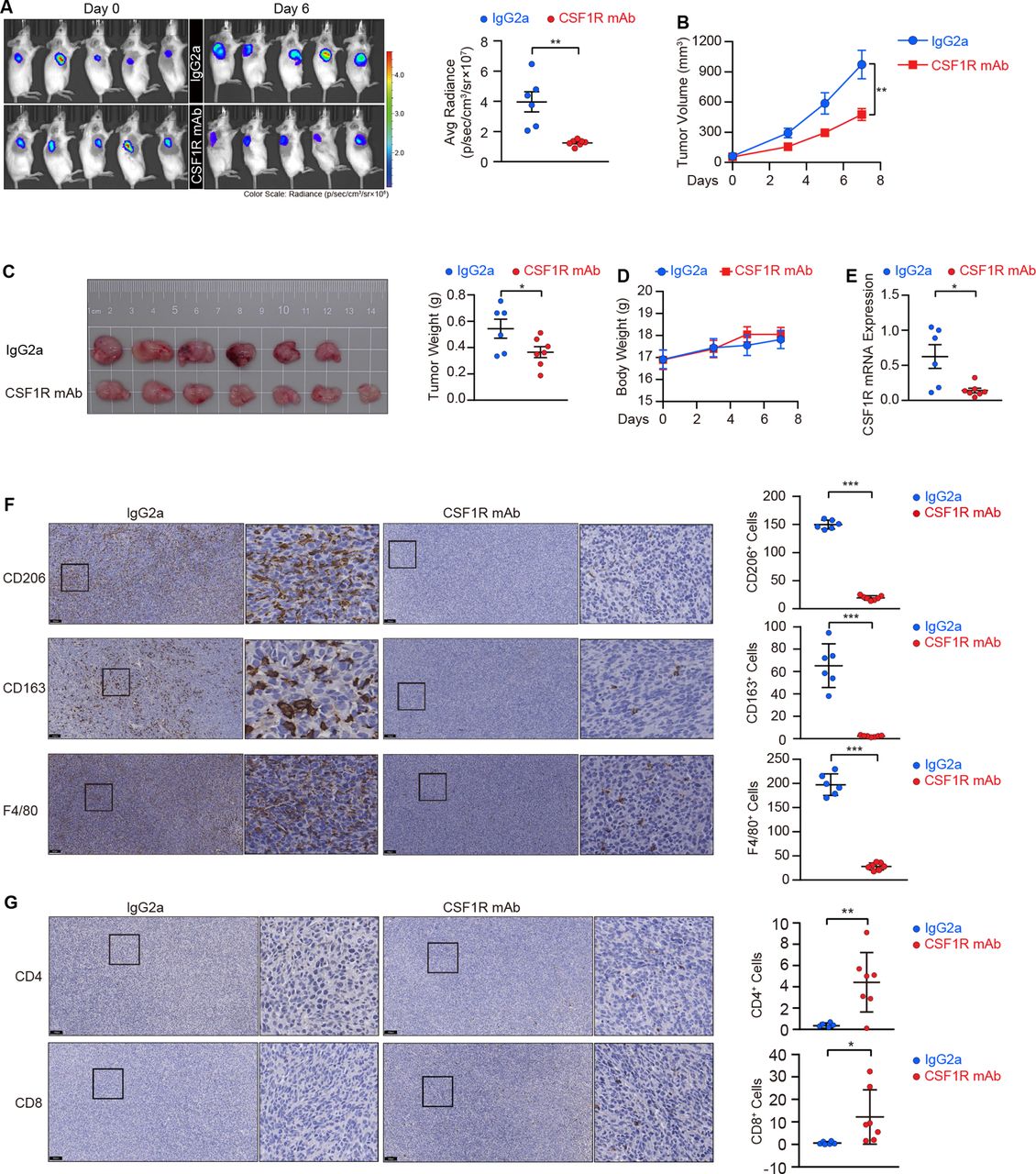
CSF1R neutralization depletes tumor-associated macrophages (TAMs) and suppresses colorectal cancer (CRC) tumor growth in vivo. (A) Scanning images and quantifications of subcutaneous xenografts in mice treated with the CSF1R neutralizing antibody (n=7) or IgG2a (n=6) in an IVIS. Data are means±SEM. (B, C) Tumor volumes (B), tumor panorama (C), and tumor weights (C) of the xenografts. Data are presented as means±SEM. (D) Body weights of mice were monitored during administration. Data are means±SEM. (E) CSF1R mRNA expression in the resected tumors was validated by RT-PCR. Data are means±SD. (F) Representative IHC images and corresponding quantitation of macrophages in the resected tumors marked by CD206, CD163, and F4/80. Scale bars, 100 µm. Data are means±SD. (G) CD4+ and CD8+ T cells were stained by IHC, and representative images and their corresponding quantitation were presented. Scale bars, 100 µm. Data are means±SD. Statistical significance (A, C, E, F, G) was determined by the Student’s t-test. The values at the end-point were statistically analyzed (B).*p<0.05, **p<0.01, ***p<0.001, ‘NS’ indicates no significance. IHC, immunohistochemistry; IVIS, in vivo imaging system.
PLX3397 targeting CSF1R blocks tumor growth in vivo
We further investigated an alternative CSF1R blockade to find a more safe and effective therapy to debate CRC using the FDA-approved small molecular inhibitor PLX3397. When THP-1 cells were differentiated into M2-like macrophages that displayed a robust increase in CSF1R expression, these macrophages had a greater sensitivity to PLX3397 (figure 3A, online supplemental figure 6A). In M2-like macrophages, the inhibitor successfully restrained CSF1 and IL34-induced CSF1R activation in vitro. (figure 6A). In a panel of murine (CT26 and MC38) and human (CACO2, HT-29, T84, SW48, SW480, COLO205) CRC cell lines, the cytotoxic activities of PLX3397 were moderate with IC50 at a range of 3.7–18.4 µM (online supplemental figure 6B,C). We next evaluated its antitumor activity on MC38 tumors in C57BL/6J mice with the intact immune system. Owing to the efficacy of PLX3397, tumors in experimental mice were substantially smaller than tumors in control mice (figure 6B,C). In line with the CSF1R neutralizing antibody, the oral administration of the drug was well tolerated over the treatment with no significant weight loss (figure 6D). We validated the effective ablation of M2 TAMs in tumor tissues from PLX3397-treated mice, which were marked using CD206 and F4/80 antibodies (figure 6E). Similar to the CSF1R neutralizing antibody, PLX3397 treatment enriched the infiltration of CD4+ and CD8+ T cells in tumor foci, with a 1.86-fold enrichment in CD4+ T cells and an 8-fold enrichment in CD8+ T cells in these mice compared with mice receiving vehicle (figure 6E).

PLX3397 inhibits the CSF1R activation in M2-like macrophages in vitro, restraining tumor growth and liver metastasis in vivo. (A) The inhibitory effect of PLX3397 on CSF1R stimulation (phospho-Tyr723) in M2-like macrophages treated with CSF1 or IL34. (B, C) Photo (B) and tumor mass (C) of MC38 xenografts in mice treated with (PLX3397, n=10) or without (Vehicle, n=8) PLX3397. Data with means±SEM are shown. (D) Body weights of mice were monitored before and after treatment with PLX3397. Data are presented as means±SEM. (E) Representative IHC images and quantitative data of CD206, F4/80, CD4 and CD8 positive cells in the subcutaneous MC38 xenografts. Scale bars, 100 µm. *p<0.05, **p<0.01, ***p<0.001, ‘NS’ indicates no significance. (F) Scanned images and quantitative radiance of liver metastasis in mice treated with vehicle (n=8), PLX3397 (n=10), and 5-FU (n=10) in an IVIS. Data are presented as means±SEM. (G, H) Liver weights (G) and the number of liver metastases (H) were quantified. Data are presented as means±SEM. (I, J) Representative photos (I) and pathological sections (J) of liver metastases. (K) Body weights of mice were monitored during administration. Significant results were obtained by two-tailed unpaired Student’s t-test (C–E), one-way ANOVA by Tukey’s multiple comparisons test (F, G, H,) and the values at the end-point (K). ANOVA, analysis of variance; IHC, immunohistochemistry; IVIS, in vivo imaging system.
Efficacy of PLX3397 on liver metastatic CRC
A liver metastatic model was established to evaluate the efficacy of PLX3397. Like the 5-FU-based chemotherapy, chemiluminescent living imaging showed substantially weak signals in PLX3397-administrated mice compared with mice receiving vehicle (figure 6F). In bright field images, we observed a markedly lower frequency of scattered metastases and an alleviated liver tumor burden in mice receiving PLX3397 (figure 6G–I). Pathological H&E staining of liver sections further demonstrated that PLX3397 resulted in a great reduction in metastatic number and size in comparison to mice in the control group (figure 6J). In contrast to 5-FU, PLX3397 administration did not result in significant body mass loss, providing evidence for preliminary evidence of the inhibitor’s safety (figure 6K).
PLX3397 has a potential synergic effect on chemotherapy and immunotherapy in CRC treatment
In order to investigate the potential of PLX3397 as an adjuvant agent in chemotherapy, we monitored the tumor response in CT26 tumor-bearing BALB/c mice with the intact innate immune system after administering a combination of PLX3397 and 5-FU. The pharmacodynamics study showed that the monotherapy of PLX3397 was efficacious against tumor growth, which was correlated well with its activity on the MC38 tumor-bearing mice (figure 7A–C). Importantly, the combination of PLX3397 and 5-FU received an uplifting synergic effect when compared with the mice that either received PLX3397 or 5-FU (figure 7A–C). As usual, PLX3397 did not cause weight loss in this model, which was starkly contrasted with 5-FU which led to a drop in body weight (figure 7D). Immunohistochemistry (IHC) staining showed that PLX3397 gradually reduced the number of CD206+, CD163+, and F4/80+ cells in tumor tissues (figure 7E). CD8+ T cells in the experimental group showed substantial enrichment compared with mice in the control group, but we did not observe this similar enrichment in the CD4+ T cell population in this model (figure 7F).
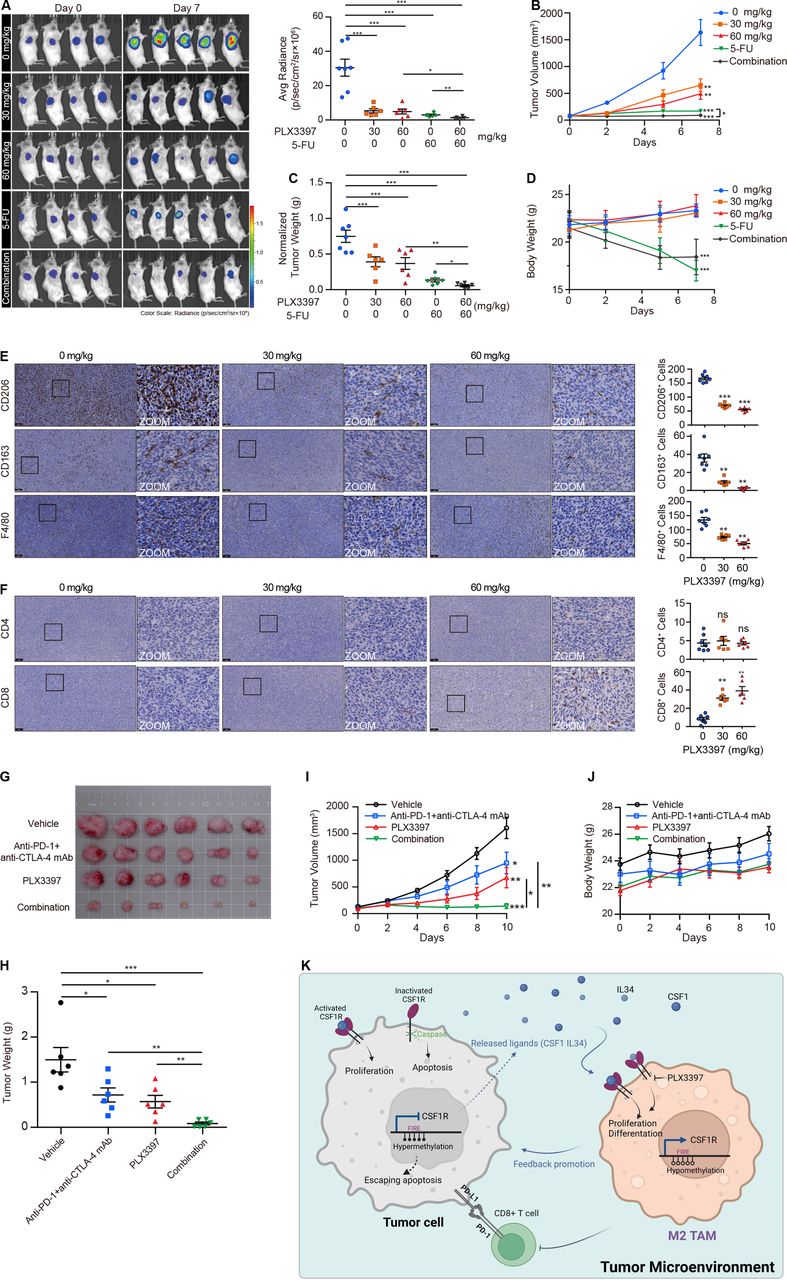
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PLX3397 combined with 5-FU or anti-PD-1 and anti-CTLA-4 mAb effectively attenuates tumor growth in vivo. (A) Bioluminescence imaging of CT26 xenografts from mice treated with PLX3397, 5-FU, and combination treatment, and then the radiance was quantified. Data are means±SEM. (B) Tumor volumes of subcutaneous xenografts. Data are means±SEM. (C) Tumor weights normalized to body weight were presented. Data are presented as means±SEM. (D) Monitoring data of mice body weights during administration. Data are means±SEM. (E, F) Representative IHC staining and corresponding quantified data of M2 TAMs (CD206, CD163, and F4/80; E) and T cells (CD4 and CD8; F) in tumor tissues. Scale bars represent 100 µm. (G–I) The photo (G), tumor weights (H), and tumor volumes (I) of MC38 xenografts from mice treated with PLX3397, anti-PD-1 plus anti-CTLA-4 mAb, and combination treatment. Data are presented as means±SEM. (J) Body weights of mice were monitored during administration. Data are means±SEM. (K) A schematic diagram summarizing the role and mechanism of CSF1R in the tumor microenvironment. *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA followed by Tukey’s multiple comparisons test. ANOVA, analysis of variance; IHC, immunohistochemistry.
Given the observation that the decrease of M2 TAMs and increase of CD8+ T cell infiltration with PLX3397 in CRC tissues, we asked whether PLX3397 could enhance immune efficacy in combination with immune checkpoint inhibitors. To this end, we combined PLX3397 with anti-PD-1 mAb and anti-CTLA-4 mAb in the immunocompetent C57BL/6J mice bearing MC38 tumors. Compared with the monotherapy, PLX3397 showed a potent synergic effect when it was combined with anti-PD-1 mAb and anti-CTLA-4 mAb (figure 7G–J), suggesting that PLX3397 has the potential to improve immune response in CRC immunotherapy.
Discussion
Here we first show evidence that CSF1R functions as a tumor suppressor in CRC. Indeed, CSF1R has the potential to act as either an oncogene or a tumor suppressor gene, depending on the availability of its ligands and its activated state. CSF1R predominantly functions as a tumor suppressor rather than an oncogene during CRC progression, which is conferred by its limited ligands in the tumor microenvironment. Therefore, CSF1R is one target that need to be inactivated for releasing cancer cells from CSF1R-mediated carcinostatic effects. Except for its tumor-suppressive effect, the silence of expression in cancer cells benefits M2 TAMs by making more ligands available for activating CSF1R expressed in M2 TAMs in the tumor microenvironment where ligands are scarce.
CSF1R is a member of class III RTK, which also consists of PDGFRα, PDGFRβ, c-Kit, and FLT3. These receptors have been illustrated to be implicated in driving a wide range of cancers.31 Previous evidence has demonstrated that the activation of CSF1R by its ligands induces CRC cells to manifest an epithelial-mesenchymal transition (EMT) phenotype and triggers lung metastasis.18 Our findings seem to be antagonistic; however, these paradoxes appear to be a consequence of CSF1R being a dependence receptor. In the class III RTK family, c-Kit has been found as a dependence receptor in lung cancer, neuroblastoma, and melanoma.7 It functions as a proto-oncogene via its kinase activity, whereas the receptor with kinase-inactivating mutation and the ligand-deprived wild-type receptor both exert as tumor suppressors.7 Therefore, the ligand availability in the CRC microenvironment is critical for CSF1R functions. We confirmed a low level of CSF1 and IL34 by ELISA assay in the complete cell culture medium that is a prerequisite for CSF1R to exert tumor-suppressive effect in vitro. In tumor tissues, CSF1 was found to be substantially suppressed in colorectal adenocarcinomas. Although we were unable to observe a downregulation of IL34 expression in cancerous tissues, it was nevertheless expressed at a very low level in CRC. Moreover, CSF1 has been identified as a dominant trophic factor for TAMs.32 Along this line, CSF1R is anticipated to exert its tumor-suppressive function rather than its carcinogenesis in the ligand-limited milieu. A previous report has demonstrated that genetic deficiency of CSF1 in CRC cells reduces immunosuppressive CSF1R+ TAM infiltration within tumor tissues.33 There is a possibility that the loss of CSF1 may precede the silence of CSF1R, which would induce apoptosis in CRC cells with CSF1R inactivation, and thus creates a clonal survival advantage for those CSF1R-silenced cancer cells.
CSF1R expression in gliomas has been found to be enriched in TAMs, but not within malignant glioma cells.34 We also found here that the CSF1R expression was restricted to TAMs in CRC microenvironment. Therefore, TAMs are the main source of CSF1R expression in CRC tissues. This notion clearly explains the anomalous result that the poor survival shown in patients with CRC carried highly CSF1R expression. In addition, TAMs benefit from the decrease of CSF1R expression in CRC cells by utilizing the limited and constant CSF1 or IL34 to activate the CSF1R expressed in M2 TAMs, which then acts as a cycle for carcinogenesis. This finding would deepen our understanding about these dependence receptors that tumor cells depleting them to relieve their tumor suppressive effect as well as abalienate cytokines for stromal cells, such as TAMs, to generate an adaptive environment.
During the gradual differentiation of macrophage progenitors into mature macrophages, the CSF1R expression progressively rises from its initial low level on hematopoietic stem cells.35 The sophisticated control of CSF1R expression follows a two-step mechanism during the differentiation, which involves transcription factor assembly (PU.1, RUNX1, and C/EBP binding) and chromatin remodeling within the CSF1R promoter and FIRE enhancer.35 FIRE controls the maximal expression of CSF1R in differentiated macrophages, depleting this region preferentially inhibits CSF1R expression and prevents the formation of macrophages in mice.36 Histone changes of the chromatin at the FIRE regulatory regions are regulated by DNA methylation during myeloid cell maturation.37 CSF1R expression is upregulated in myeloid cells as a result of the hypomethylation of the FIRE loci caused by AML1 or PKA.37 38 Due to the pivotal role of the FIRE enhancer in the CSF1R expression, we focused on the epigenetic alterations in this region. We found here that CSF1R expression is inversely related to the methylation located in the FIRE region, implying that DNA methylation in the FIRE region contributes to CSF1R silence in CRC. In macrophages, the DNA hypomethylation in the FIRE enhancer paves the way for expressing CSF1R. Moreover, DNA methylation in the FIRE region mediates the imbalance of CSF1R expression between tumor cells and macrophages.
M2 TAMs has been confirmed to be involved in CRC progression.39 40 Its infiltration is correlated with the effectiveness of standard-of-care therapeutics.41–43 In addition, recent studies have shown that TAMs can directly induce the T cells apoptosis, causing systemic tumorous CD4+ and CD8+ T cell depletion and restraining immunotherapy.30 44 As poor survival in a variety of tumor types is correlated with the intertumoral presence of CSF1R+ macrophages, applicating CSF1R inhibitors to target M2 TAMs is therapeutically appealing and has been proven effective in preclinical models.20 45–47 However, CSF1R neutralizing antibody has a limited curative effect.48 A resistant mechanism has been identified that the CSF1R neutralization only affects a certain percentage of C1QC+ macrophages, but the percentage of SPP1+ macrophages is unaffected, which are crucial to the progression of CRC.48 We alternatively used a small molecular CSF1R inhibitor PLX3397 to block CSF1R, it dramatically diminishes M2 TAM and enhances CD8+ T cell infiltration, reducing tumor growth and liver metastasis in vivo. Our findings consist with a previous study that the decrease of CSF1R+ TAMs within CRC tumors results in a burst of CD8+ T cell infiltration.32 In line with the observation that immunosuppressive CSF1R+ TAMs limit adaptive immunity in CRC tumors, we further illustrated a synergic effect of PLX3397 on CRC treatment in vivo when combined with chemotherapy (5-FU) as well as immunotherapy (PD1 and CTLA-4 antibodies). It is worth noting that the oral administration of PLX3397 is acceptable for mice. These attractive traits endow PLX3397 as a promising candidate for CRC treatment.
In summary, we have revealed CSF1R as a novel dependence receptor. Ligand-free CSF1R undergoes an inhibitory function, whereas it promotes tumor proliferation when the receptor binds to its ligands. We presented proof that CRC cells with CSF1R silence benefit to activate M2 TAMs by avoiding competing with ligands in the tumor microenvironment, and these TAMs in turn drive tumor progression (figure 7K). Additionally, we identified CSF1R as a promising therapeutic target for reducing M2 TAMs and enhancing tumor immunity, and PLX3397 is an efficient agent to block CSF1R and debate CRC.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Institutional Review Board of Sun Yat-sen University approved this study with reference number 2021ZSLYEC-366. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors wish to acknowledge the TCGA Research Network, the CCLE platform, and the GEO datasets for kindly sharing important data that has been accessed and shown here.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MZ, LB and XL contributed equally.
Contributors YLi and YLuo designed research, taken responsibility for the integrity andaccuracy of the data and had the full access for this study; MZ, LB, XL, SP, HB, JL, PY, and RM performed research. XW, LW, and MH contributed new reagents/analytic tools. MZ, HY and YX analyzed data; YLi and MZ wrote the paper.
Funding This work was supported by grants from the National Natural Science Foundation of China (No. 81972245 to YLuo; No. 81902877 to HY; No. 31900505 to YLi), the Natural Science Foundation of Guangdong Province (No. 2020A1515010036 to XL; No. 2021A1515010639 to XL; No. 2021A1515010134 to MH; No. 2022A1515012656 to HY), the Sun Yat-Sen University Clinical Research 5010 Program (No. 2018026 to YLuo), the Young Teacher Foundation of Sun Yat-sen University (CN) (No. 19ykpy03 to XL), the 'Five Five' Talent Team Construction Project of the Sixth Affiliated Hospital Of Sun Yat-Sen University (No.P20150227202010251 to YLuo), the Excellent Talent Training Project of the Sixth Affiliated Hospital Of Sun Yat-Sen University (No. R2021217202512965 to YLuo), the Sixth Affiliated Hospital of Sun Yat-sen University Clinical Research-'1010' Program (MH), the Program of Introducing Talents of Discipline to Universities, and National Key Clinical Discipline (2012).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.