Article Text

Abstract
Background The efficacy of atezolizumab (A) and/or bevacizumab (B) with carboplatin/paclitaxel (CP) chemotherapy was explored in the phase III, randomized IMpower150 study in patients with non-squamous non-small cell lung cancer (NSCLC) according to KRAS mutations (mKRAS) and co-occurring STK11, KEAP1, or TP53 mutations.
Methods Mutation status was determined by circulating tumor DNA next-generation sequencing. Overall survival (OS) and progression-free survival (PFS) were analyzed in a mutation-evaluable intention-to-treat population (MEP; n=920) and SP263 (programmed cell death ligand 1 (PD-L1)) biomarker-evaluable population (n=774).
Results Within the mKRAS population (24.5% of MEP), ABCP showed numerical improvements vs BCP in median OS (19.8 vs 9.9 months; HR 0.50; 95% CI 0.34 to 0.72) and PFS (8.1 vs 5.8 months; HR 0.42; 95% CI 0.29 to 0.61)—greater than with ACP (OS: 11.7 vs 9.9 months; HR 0.63; 95% CI 0.43 to 0.91; PFS: 4.8 vs 5.8 months; HR 0.80; 95% CI 0.56 to 1.13) vs BCP. Across PD-L1 subgroups in mKRAS patients, OS and PFS were longer with ABCP vs BCP, but OS with ACP was similar to BCP in PD-L1-low and PD-L1-negative subgroups. Conversely, in KRAS-WT patients, OS was longer with ACP than with ABCP or BCP across PD-L1 subgroups. KRAS was frequently comutated with STK11, KEAP1, and TP53; these subgroups conferred different prognostic outcomes. Within the mKRAS population, STK11 and/or KEAP1 mutations were associated with inferior OS and PFS across treatments compared with STK11-WT and/or KEAP1-WT. In mKRAS patients with co-occurring mSTK11 and/or mKEAP1 (44.9%) or mTP53 (49.3%), survival was longer with ABCP than with ACP or BCP.
Conclusions These analyses support previous findings of mutation of STK11 and/or KEAP1 as poor prognostic indicators. While clinical efficacy favored ABCP and ACP vs BCP in these mutational subgroups, survival benefits were greater in the mKRAS and KEAP1-WT and STK11-WT population vs mKRAS and mKEAP1 and mSTK11 population, suggesting both prognostic and predictive effects. Overall, these results suggest that atezolizumab combined with bevacizumab and chemotherapy is an efficacious first-line treatment in metastatic NSCLC subgroups with mKRAS and co-occurring STK11 and/or KEAP1 or TP53 mutations and/or high PD-L1 expression.
- lung neoplasms
- biomarkers
- tumor
- immunotherapy
- programmed cell death 1 receptor
- tumor biomarkers
Data availability statement
Data are available on reasonable request. Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Mutations in the Kirsten rat sarcoma viral oncogene homolog (mKRAS) oncogene are a major driver of nonsquamous non-small cell lung cancer (NSCLC) and occur in ≈25%–40% of patients (≈5%–10% in the Asian population), with the glycine 12 to cysteine (G12C) activating mutation demonstrating the highest prevalence.1–4 KRAS is frequently comutated with the serine/threonine kinase 11 (STK11), kelch-like ECH associated protein 1 (KEAP1), and tumor protein 53 (TP53) tumor suppressor genes, but it is generally mutually exclusive with mutations in the epidermal growth factor receptor (EGFR) gene.2–4 In patients with NSCLC, tumors bearing mutations in STK11 (mSTK11) and KEAP1 (mKEAP1) were recently shown to be associated with poor prognosis and variable response to treatment, including immune checkpoint inhibitors (anti-programmed cell death ligand 1 (PD-L1)/programmed cell death 1 protein (PD-1)).1–3 5 However, exploratory analysis of KEYNOTE-042 found that pembrolizumab monotherapy was associated with improved overall survival (OS) when compared with chemotherapy, regardless of STK11 and KEAP1 mutational status; however, patient populations were small.6 Combining treatments such as immune checkpoint inhibitors with chemotherapy and/or targeted therapy may overcome the challenges associated with treating NSCLC in difficult-to-treat patient groups, including those with KRAS-bearing tumors and comutations in STK11 and/or KEAP1.7
Atezolizumab is a humanized engineered immunoglobulin G1 monoclonal antibody that blocks the immune checkpoint protein PD-L1 from binding to the PD-1 and B7.1 receptors, thereby restoring tumor-specific immunity.8 9 In addition to its known antiangiogenic effects, bevacizumab’s inhibition of vascular endothelial growth factor (VEGF) has immune modulatory effects, including normalization of tumor vasculature, reprogramming of the tumor microenvironment from immune-suppressive to immune-permissive, and promotion of dendritic cell maturation.7 10–12 In combination with bevacizumab and chemotherapy, atezolizumab’s T-cell-mediated cancer cell killing may be further enhanced through both reversal of VEGF-mediated immunosuppression and chemotherapy-induced cell death.12 13 In clinical trials that combined anti-PD-L1 and anti-VEGF therapies, synergy has been observed that resulted in positive outcomes and benefits to patients over each therapy alone.7 10 14
The randomized, phase III IMpower150 study evaluated atezolizumab plus carboplatin/paclitaxel chemotherapy (ACP) or atezolizumab plus bevacizumab plus carboplatin/paclitaxel chemotherapy (ABCP) vs bevacizumab plus carboplatin/paclitaxel (BCP).15 16 Among randomized patients with no EGFR or anaplastic lymphoma kinase (ALK) alterations (intention-to-treat wild-type (ITT-WT) population), ABCP was associated with significant improvements in progression-free survival (PFS) and OS compared with BCP.15 ABCP continued to show benefit vs BCP in an updated OS analysis with an additional ≈20 months of follow-up.17 ABCP also prolonged OS and PFS vs BCP in an exploratory subgroup analysis of patients with EGFR-sensitizing mutations.16 Although studies of immune checkpoint inhibitors alone or with chemotherapy have demonstrated survival benefit in patients with mKRAS tumors,6 18–20 it remains unclear how co-occurring mutations—including mSTK11, mKEAP1, and mTP53—affect prognosis and predictive outcomes following immune checkpoint blockade. It is, therefore, imperative to determine whether differential responses to treatment and consequent effects on survival outcomes exist among patients with KRAS-mutant tumors harboring different combinations of comutations.
This retrospective analysis of the IMpower150 trial explored efficacy endpoints within the mKRAS population by PD-L1 status and by co-occurring mSTK11, mKEAP1, and mTP53 subgroups in patients with nonsquamous NSCLC in the first-line setting.
Methods
Study design and patients
IMpower150 was an international, open-label, randomized, phase III trial of ACP or ABCP vs BCP in 1202 patients with NSCLC enrolled from 240 study centers across 26 countries (NCT02366143; figure 1A). Chemotherapy-naive patients with stage IV metastatic nonsquamous NSCLC and measurable disease at baseline per Response Evaluation Criteria in Solid Tumors V.1.1 were eligible for inclusion in the study if they also had a baseline Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1 and available tumor tissue for biomarker testing. All patients provided written informed consent. Further detailed information on patient eligibility criteria and study design methodology were published elsewhere.15 16
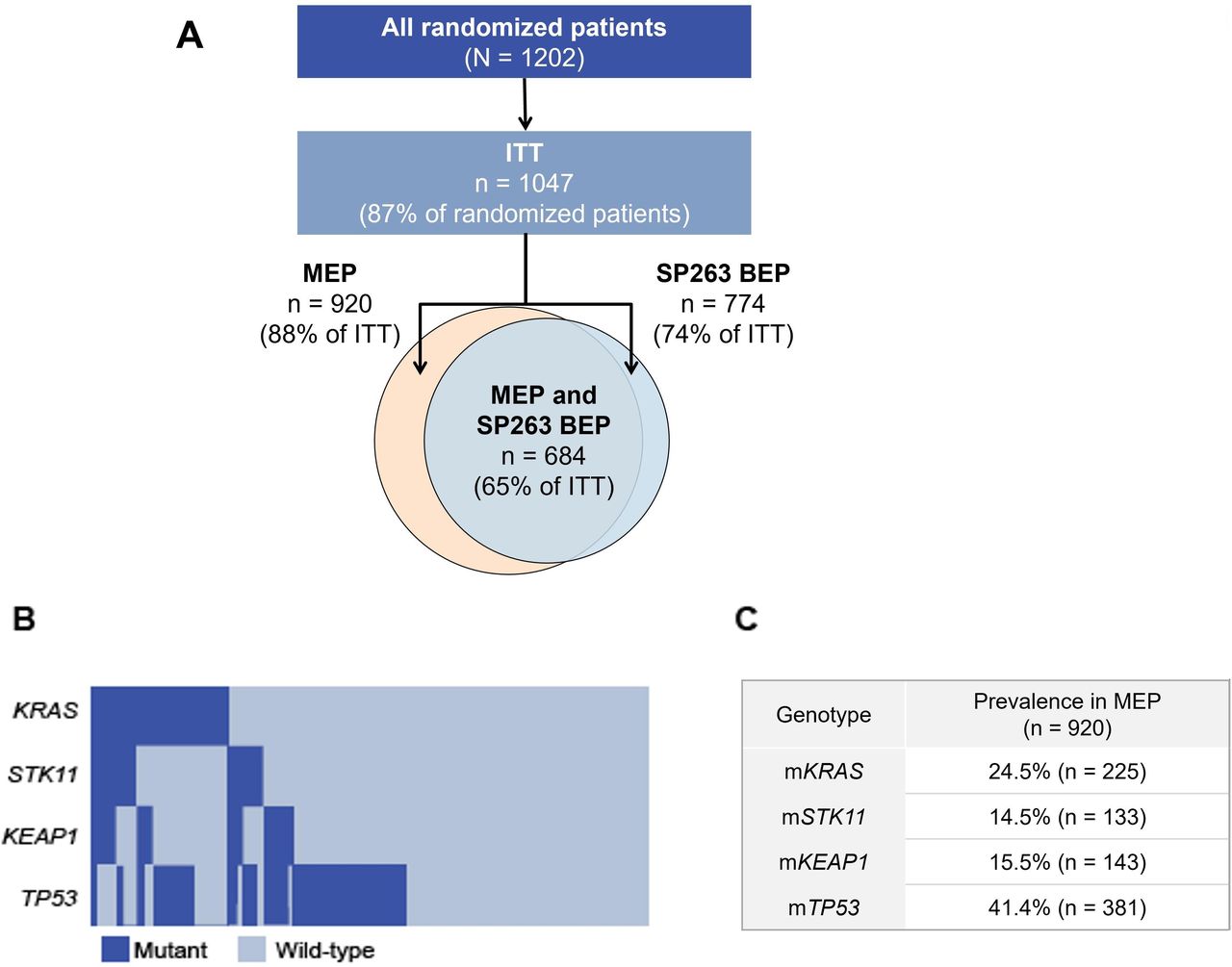
Overall study flow (A) and distribution (B) and prevalence (C) of mutation subpopulations in the MEP. Disposition of randomized, ITT, MEP, and SP263 BEP patient populations included in this analysis (A). Oncoplot (B) and prevalence (C) of KRAS, STK11, KEAP1, and TP53 mutations in the MEP population. ITT, intention-to-treat; MEP, mutation-evaluable population; KEAP1, kelch-like ECH associated protein 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; SP263 BEP, SP263 biomarker-evaluable population; STK11, serine/threonine kinase 11; TP53, tumor protein 53; WT, wild-type.
The coprimary endpoints were PFS and OS in the ITT-WT population, which excluded patients with EGFR or ALK genomic alterations.15 In this post hoc study, exploratory survival analyses were undertaken in the population of patients without EGFR or ALK genomic alterations (herein referred to as the ITT population) and mutation-evaluable population (MEP) from the third/final OS clinical cut-off date. PD-L1 expression was analyzed in the SP263 biomarker-evaluable population (SP263 BEP).
Treatment and assessments
Patients were randomized (1:1:1) to ACP, ABCP, or BCP. Induction chemotherapy was administered for four or six cycles, as determined by the investigator before randomization, every 21 days. The number of chemotherapy cycles patients actually received may have differed based on factors such as toxicities and disease progression. On day 1 of each 21-day cycle, treatments were administered intravenously as follows: 1200 mg atezolizumab; 15 mg/kg bevacizumab; area under the concentration–time curve of 6 mg/mL per minute carboplatin; and 200 mg/m² paclitaxel (patients of Asian ethnicity were given 175 mg/m²). After the induction phase, patients continued bevacizumab until unmanageable toxicity or disease progression (ABCP or BCP) or atezolizumab until loss of clinical benefit (ABCP or ACP).
Key exploratory efficacy endpoints of this IMpower150 subgroup analysis were investigator-assessed PFS per Response Evaluation Criteria in Solid Tumors V.1.1 and OS. Safety was assessed in all patients who received at least 1 dose of study treatment. Adverse events were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events, V.4.0.
Investigations
The mutation status of KRAS, STK11, KEAP1, and TP53 was determined by blood-based circulating tumor DNA next-generation sequencing (Foundation Medicine, Cambridge, Massachusetts, USA) from baseline plasma samples. Mutations included known, likely, and unknown functional impact status; synonymous mutations were excluded.
For this analysis of IMpower150, PD-L1 expression in tumor cells (TC) was analyzed in archival or fresh tumor tissue by the VENTANA SP263 immunohistochemistry assay (Ventana Medical Systems, Tucson, AZ, USA). PD-L1-positive expression was defined as staining on TC ≥1%, whereas PD-L1 high was defined as TC ≥50%.
Statistical analysis
Kaplan-Meier curves and associated medians were estimated for survival outcomes in the MEP, SP263 BEP, and mutation-defined subpopulations. For each survival comparison, HRs and corresponding 95% CIs were calculated from unstratified Cox proportional models.
Results
Disposition and baseline characteristics of the ITT and MEP populations
Of the 1202 patients enrolled in IMpower150, 1047 patients were included in the ITT population (data cut-off date: September 13, 2019; figure 1A). Among the ITT population, 920 and 774 patients were included in the MEP and SP263 BEP, respectively. Of the 920 MEP patients, 684 (65% of ITT) were also deemed SP263 BEP. The median follow-up duration in the ITT population was 39.4 months.
Among MEP patients, 24.5% (n=225), 14.5% (n=133), 15.5% (n=143), and 41.4% (n=381) had mKRAS, mSTK11, mKEAP1, and mTP53 tumors, respectively (figure 1B,C). All mutational subgroups in the MEP are shown in online supplemental figure S1. In the MEP, G12C (9.8% of MEP), glycine 12 to aspartate (3.8%), and glycine 12 to valine (3.7%) were the most frequently occurring KRAS mutations. Within the mKRAS population, 44.9% (101/225) of mKRAS patients also had co-occurring mutations in STK11 and/or KEAP1, and 49.3% (111/225) of mKRAS patients had co-occurring mutations in TP53 (online supplemental figure S2).
Supplemental material
Baseline characteristics were generally well balanced between treatment arms across mutation-defined patient subgroups and consistent between the MEP and ITT population (table 1). Higher ECOG PS, median baseline sum of longest diameter of target lesion, and baseline liver metastases were observed in the mKRAS, mSTK11, mKEAP1, and mTP53 populations compared with the overall MEP or ITT population. Smoking history was associated with mKEAP1, mSTK11, and mKRAS. Elevated C-reactive protein levels, a poor prognostic factor, appeared highest in mKEAP1 and mSTK11 populations compared with other mutational subgroups and overall MEP. Safety was similar between the MEP and ITT population (online supplemental table S1).
Baseline demographics and characteristics
Efficacy by mKRAS status and by PD-L1 subgroup
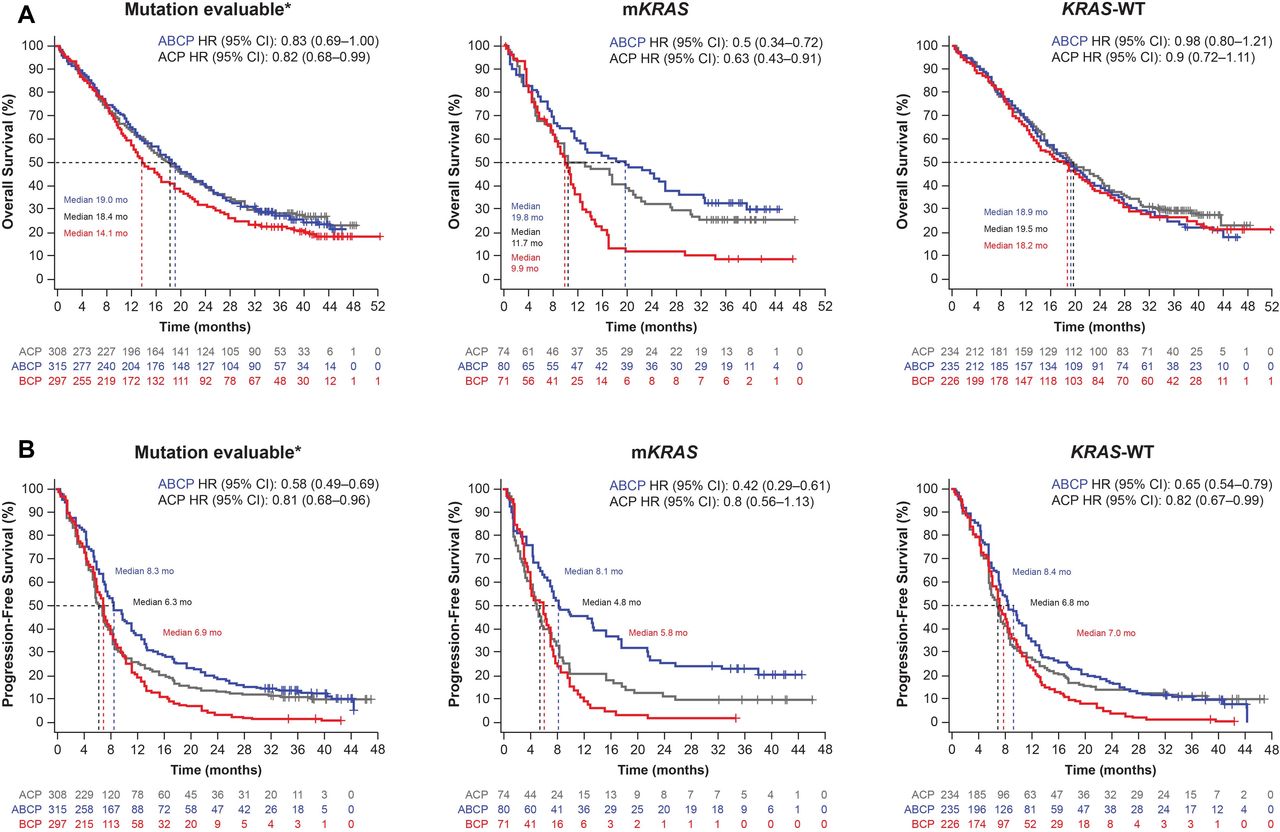
As shown in figure 2A,B, efficacy in the ABCP and ACP arms vs the BCP arm was observed in the mKRAS population. Across treatment arms, median OS of 19.8 (ABCP), 11.7 (ACP), and 9.9 (BCP) months and median PFS of 8.1 (ABCP), 4.8 (ACP), and 5.8 (BCP) months were observed. Both the ABCP and ACP arms demonstrated greater survival improvements compared with the BCP arm in this population. However, compared with BCP, the ABCP arm showed numerically greater survival than the ACP arm in mKRAS patients: OS (HR 0.50; 95% CI 0.34 to 0.72 vs HR 0.63; 95% CI 0.43 to 0.91) and PFS (HR 0.42; 95% CI 0.29 to 0.61 vs HR 0.80; 95% CI 0.56 to 1.13).

Survival in patients with and without KRAS mutations. Kaplan-Meier estimates of OS (A) and PFS (B) among the MEP and KRAS populations by treatment arm. All HRs are vs BCP. *Within the ITT population. ABCP, atezolizumab plus bevacizumab plus carboplatin/paclitaxel chemotherapy; ACP, atezolizumab carboplatin/paclitaxel; BCP, bevacizumab plus carboplatin/paclitaxel; KRAS, Kirsten rat sarcoma viral oncogene homolog; mKRAS, mutations in KRAS; MEP, mutation-evaluable population. WT, wild-type.
In KRAS-WT patients, median OS was 18.9 months in the ABCP arm, 19.5 months in the ACP arm, and 18.2 months in the BCP arm. In contrast to the mKRAS subgroups, KRAS-WT patients demonstrated no apparent OS improvement with ABCP (HR 0.98; 95% CI 0.80 to 1.21) or ACP (HR 0.90; 95% CI 0.72 to 1.11) vs BCP. Across treatment arms in the KRAS-WT population, median PFS values were 8.4 (ABCP), 6.8 (ACP), and 7.0 (BCP) months; PFS was greater in the ABCP arm (HR 0.65; 95% CI 0.54 to 0.79) than in the ACP arm (HR 0.82; 95% CI 0.67 to 0.99) relative to the BCP arm.
Consistent with previously published literature,1 mKRAS tumors were enriched for high PD-L1 expression (TC ≥50%) compared with the KRAS-WT population and overall MEP/SP263 BEP (figure 3A). In mKRAS patients with high PD-L1 expression (TC ≥50%), a similar prolonged OS was observed for patients treated with both ABCP (median 23.9 months; HR 0.40; 95% CI 0.19 to 0.85) and ACP (median 19.9 months; HR 0.35; 95% CI 0.17 to 0.74) compared with BCP (median, 9.9 months) (figure 3B). In contrast, mKRAS patients with low or negative PD-L1 expression demonstrated greater OS in the ABCP arm than in the ACP arm. For patients with low PD-L1 expression (TC 1-<50%), the HR was 0.37 (95% CI 0.15 to 0.91; median OS, 17.5 months) for ABCP and 0.83 (95% CI 0.36 to 1.90; median OS, 4.8 months) for ACP vs BCP (median OS, 5.0 months) (figure 3B). For patients with negative PD-L1 expression (TC <1%), the HR was 0.43 (95% CI 0.21 to 0.90; median OS, 22.4 months) for ABCP and 0.95 (95% CI 0.49 to 1.83; median OS, 7.9 months) for ACP vs BCP (median OS, 8.7 months) (figure 3B). In contrast, KRAS-WT patients with high (TC ≥50%) and low (TC 1-<50%) PD-L1 expression demonstrated greater OS in the ACP arm than in the ABCP or BCP arm (online supplemental figure S3). In mKRAS patients, median PFS was longer in the ABCP arm than in the ACP or BCP arms in the PD-L1-high, PD-L1-low, and PD-L1-negative subgroups (figure 3C). PFS improvements in the ABCP vs BCP arm were similar among patients with PD-L1-high (HR 0.36; 95% CI 0.17 to 0.74), PD-L1-low (HR 0.22; 95% CI 0.08 to 0.60), and PD-L1-negative (HR 0.42; 95% CI 0.20 to 0.86) expression.

PD-L1 prevalence in the overall BEP and KRAS-defined populations and survival according to PD-L1 expression status in patients with KRAS mutations. PD-L1 prevalence in the MEP/SP263 BEP and KRAS subgroups (A), and Kaplan-Meier estimates of OS (B) and PFS (C) among the mKRAS population according to SP263 PD-L1 status. All HRs are vs BCP. ABCP, atezolizumab plus bevacizumab plus carboplatin/paclitaxel chemotherapy; ACP, atezolizumab carboplatin/paclitaxel; BCP, bevacizumab plus carboplatin/paclitaxel; KRAS, Kirsten rat sarcoma viral oncogene homolog; MEP, mutation-evaluable population; mKRAS, mutation in KRAS; PD-L1, programmed cell death ligand 1; SP263 BEP, SP263 biomarker-evaluable population; TC, tumor cells.
Effect of comutations on clinical efficacy in patients with or without mKRAS
Efficacy was evaluated in patients with individual mutations in STK11, KEAP1, and TP53, independent of comutation status (online supplemental figure S4). Similar to previous reports, STK11 and KEAP1 mutations were associated with overall poorer PFS and OS prognosis; patients with STK11/KEAP1 double mutation had the worst prognosis (online supplemental figure S5). Patients with mKEAP1 status showed no OS improvement with ABCP (median 11.4 months; HR 0.92; 95% CI 0.59 to 1.44) and limited improvement with ACP (median 6.9 months; HR 1.51; 95% CI 0.96 to 2.37) when compared with BCP (median 11.7 months). In mSTK11 patients, longer OS was seen in the ABCP arm (median 12.1 months; HR 0.71; 95% CI 0.44 to 1.13) and similar OS in the ACP arm (median 7.7 months; HR 1.01; 95% CI 0.64 to 1.58) vs the BCP arm (median 9.9 months). In patients with TP53-mutated tumors, an OS improvement was observed with both ABCP (median 18.9 months; HR 0.72; 95% CI 0.54 to 0.95) and ACP (median 14.3 months; HR 0.91; 95% CI 0.69 to 1.20) vs BCP (median 11.2 months), and the patients in the ABCP arm had longer OS than those in the ACP arm. A similar trend in PFS was observed across all mutational subgroups, whereby the ABCP arm demonstrated the longest PFS; limited PFS improvement was observed in the ACP arm compared with the BCP arm.

Survival and PD-L1 expression status in patients with KRAS mutations according to STK11/KEAP1 mutational status. Kaplan-Meier estimates of OS (A), PFS (B), and PD-L1 expression status (C) in mKRAS patients and co-occurring STK11/KEAP1 mutation or WT status. All HRs are vs BCP. ABCP, atezolizumab plus bevacizumab plus carboplatin/paclitaxel chemotherapy; BCP, bevacizumab plus carboplatin/paclitaxel; IHC, immunohistochemistry; KEAP1, kelch-like ECH associated protein 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; MEP, mutation-evaluable population; mKRAS, KRAS mutations; PD-L1, programmed cell death ligand 1; STK11, serine/threonine kinase 11; TC, tumor cells; WT, wild-type.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival and PD-L1 expression status in patients with KRAS mutations according to TP53 mutational status. Kaplan-Meier estimates of OS (A), PFS (B), and PD-L1 expression status (C) in mKRAS patients and co-occurring TP53 mutation or WT status. HRs are vs BCP. ABCP, atezolizumab plus bevacizumab plus carboplatin/paclitaxel chemotherapy; BCP, bevacizumab plus carboplatin/paclitaxel; BEP, biomarker-evaluable population; IHC, immunohistochemistry; KRAS, Kirsten rat sarcoma viral oncogene homolog; mKRAS, mutation in the Kirsten rat sarcoma viral oncogene homolog; MEP, mutation-evaluable population; PD-L1, programmed cell death ligand 1; TC, tumor cells; TP53, tumor protein 53; WT, wild-type.
Patients with mKRAS tumors are often classified and treated as a single population; however, numerous mKRAS comutations—including STK11, KEAP1, and TP53—are frequently found in NSCLC.2 3 Considering the numerical differences in median OS and published prognostic associations of individual TP53 and STK11/KEAP1 mutants, clinical efficacy and PD-L1 status in the mKRAS and comutated STK11/KEAP1 or TP53 subgroups were evaluated. In patients with mKRAS and co-occurring mSTK11 and/or mKEAP1 tumors (figure 4A), a longer OS was observed in the ABCP arm (median, 11.1 months; HR 0.60; 95% CI 0.34 to 1.03) than in the ACP arm (median, 7.9 months; HR 0.87; 95% CI 0.52 to 1.45) vs the BCP arm (median 8.7 months). A similar effect was also observed with PFS: ABCP (median 6.0 months; HR 0.49; 95% CI 0.28 to 0.84) and ACP (median 3.2 months; HR 0.88; 95% CI 0.54 to 1.46) vs BCP (median 3.4 months) (figure 4B). However, in KRAS-WT patients with mSTK11 and/or mKEAP1 tumors, OS was not improved with ABCP (median, 13.2 months; HR 1.04; 95% CI 0.66 to 1.64) or ACP (median, 9.0 months; HR 1.39; 95% CI 0.83 to 2.33) vs BCP (median 12.5 months) (online supplemental figure S6).
In the BEP, which included patients with and without mKRAS, a PFS improvement was observed in patients with mKEAP1 and STK11-WT tumors with ABCP vs ACP or BCP; however, no difference in OS was observed between treatment arms (online supplemental figure S7). In patients with KEAP1-WT and mSTK11 tumors, PFS improvements were seen in the ACP arm and ABCP arm vs the BCP arm. This effect was not observed for OS.
Patients with mKRAS and STK11-WT and KEAP1-WT comutation status showed similar OS improvements between the ABCP (median 26.2 months; HR 0.43; 95% CI 0.26 to 0.72) and ACP (median 21.0 months; HR 0.43; 95% CI 0.25 to 0.74) arms vs the BCP arm (median 10.7 months) (figure 4A). In contrast, the mKRAS, STK11-WT and KEAP1-WT patient population had longer PFS in the ABCP arm (median 15.2 months; HR 0.36; 95% CI 0.22 to 0.59) than in the ACP arm (median, 7.4 months; HR 0.64; 95% CI 0.39 to 1.05) vs the BCP arm (median 6.9 months) (figure 4B). Although clinical efficacy favored ABCP and ACP vs BCP in these subgroups, median survival and overall clinical efficacy was greater in the mKRAS and KEAP1-WT and STK11-WT population than in the mKRAS and mKEAP1 and mSTK11 comutation population, suggesting both prognostic and predictive effects.
Because of the observed efficacy differences between the mKRAS subpopulations, we also examined whether differences existed between baseline PD-L1 TC expression. mKRAS tumors bearing co-occurring mSTK11 and/or mKEAP1 were associated with reduced PD-L1 expression compared with the overall MEP/SP263 BEP group, whereas mKRAS patients with STK11-WT and KEAP1-WT status correlated with high PD-L1 expression (figure 4C).
OS and PFS were also examined in mKRAS patients with or without co-occurring mutations in mTP53 (figure 5). Among patients with tumors bearing mKRAS and co-occurring mTP53, overall OS improvements favored ABCP (median 30.6 months; HR 0.37; 95% CI 0.21 to 0.65) and ACP (median 11.7 months; HR 0.67; 95% CI 0.40 to 1.14) compared with BCP, with the greatest improvement demonstrated in the ABCP arm (figure 5A). Median PFS was also greater in the ABCP arm (14.3 months; HR 0.26; 95% CI 0.15 to 0.47) than in the ACP arm (4.6 months; HR 0.68; 95% CI 0.40 to 1.14) (figure 5B).
In patients with mKRAS and TP53-WT tumors, overall OS improvements favored ABCP (median 13.4 months; HR 0.67; 95% CI 0.40 to 1.12) and ACP (median 12.1 months; 0.61; 95% CI 0.36 to 1.04) vs BCP (median 10.7 months), with similar OS between ABCP and ACP (figure 5A). In this subgroup, median PFS was 5.2 months in the ACP arm (HR 0.95; 95% CI 0.59 to 1.54) and 7.3 months in the ABCP arm (HR 0.67; 95% CI 0.40 to 1.10) compared with 7.0 months in the BCP arm (figure 5B). As observed for mKRAS tumors with co-occurring mSTK11 and/or mKEAP1, mKRAS tumors showed differential PD-L1 expression depending on TP53 status. mKRAS tumors with co-occurring mTP53 were enriched for high PD-L1 expression compared with the overall MEP/SP263 BEP population and mKRAS TP53-WT tumors. Conversely, mKRAS tumors with TP53-WT status had PD-L1 prevalence rates similar to those of the overall MEP/SP263 BEP population (figure 5C).
Discussion
We present survival findings from a retrospective exploratory analysis of the efficacy of ABCP in mKRAS, mSTK11, mKEAP, and mTP53 mutation and comutation subgroups from the IMpower150 all-comer nonsquamous NSCLC patient population. Overall, patients with mKRAS tumors demonstrated greater OS and PFS improvements with ABCP than with ACP or BCP, regardless of comutations. However, it should be noted that a higher proportion of patients treated with BCP (vs ABCP and in some cases ACP) had liver metastases across the mutation subgroups. These results are consistent with reported survival improvements with immune checkpoint inhibitors in KRAS-mutant NSCLC.6 18–20 In contrast, similar survival improvements were not observed across treatment arms in the KRAS-WT population in this analysis. ACP and ABCP demonstrated no notable OS and PFS benefit vs BCP in patients with KRAS-WT tumors but it should be noted that the BCP arm overperformed with respect to median OS compared with historical controls for chemotherapy-treated KRAS-WT patients.18 19 From previous studies, it remains unclear how underlying comutations affected outcomes after immune checkpoint blockade. In the mutation-evaluable IMpower150 population, mSTK11, mKEAP1, and mTP53 were frequently comutated with mKRAS and, similar to the overall mKRAS population, were observed to have greater survival with ABCP than with ACP or BCP.
Notably, in our analysis, it was demonstrated that relative survival improvements in the mKRAS population were associated with the underlying PD-L1 status and the presence and type of additional comutations. In particular, PD-L1 expression was enriched among the mKRAS population, which aligns with existing evidence of an association between KRAS-mutant tumors and increased PD-L1 expression.1 Both PD-L1-high and PD-L1-low mKRAS subgroups demonstrated OS improvement with ABCP, whereas ACP was less beneficial in the PD-L1-low or negative subgroups. Median OS with ACP was shorter in the mKRAS PD-L1-low subgroup than the PD-L1-negative subgroup (4.8 vs 7.9 months, respectively). This discrepancy may be attributed to the small patient numbers in each treatment arm. The differences in OS improvements between the ABCP and ACP arms are likely to be driven by the contribution of bevacizumab. However, IMpower150 was designed and statistically powered to compare ABCP and ACP to BCP; therefore, caution must be exercised when comparing differences between ABCP and ACP. In addition to its established anti-angiogenic effects, bevacizumab further enhances atezolizumab’s T-cell-mediated killing by inhibiting VEGF-related immunosuppression, promoting T-cell tumor infiltration and creating a favorable tumor microenvironment for T-cell reinvigoration.7 10–13 Specifically, in low or no PD-L1–expressing tumors, atezolizumab may enhance T-cell priming in the lymph node through blockade of the PD-L1/B7.1 interaction.20–24 Furthermore, reprogramming of the tumor microenvironment from an immune suppressive to immune stimulatory state through VEGF inhibition by the addition of bevacizumab may facilitate interferon gamma–mediated induction of PD-L1 expression on TC and render the tumor further amenable to PD-L1 inhibition.25
Consistent with prior reports of STK11 and KEAP1 as poor prognostic indicators,26 the findings from these analyses demonstrated that patients with mKRAS and comutations in STK11 and/or KEAP1 had an overall poorer prognosis than patients with STK11-WT and KEAP1-WT status, regardless of the treatment combination they received. Notably, the findings suggest a possible correlation between biomarker and comutation status with respect to survival outcomes in the atezolizumab arms versus BCP. The adverse impact of STK11 and/or KEAP1 mutations was enhanced in patients treated with either ACP or ABCP, suggesting a strong negative predictive effect of STK11 and/or KEAP1 mutations on clinical outcomes with atezolizumab containing regimens. A marked OS improvement with ABCP was observed in patients with mKRAS and co-occurring mTP53 tumors, whereas no apparent OS improvements were observed with ABCP among patients with mKRAS tumors in the presence of comutations associated with poor prognosis (mSTK11 and mKEAP1). Notably, mKRAS and mTP53 tumors had elevated PD-L1 expression, whereas mKRAS and co-occurring mSTK11 and mKEAP1 tumors had reduced PD-L1 expression. A previous retrospective analysis also demonstrated noteworthy clinical benefit with a checkpoint inhibitor among patients with high PD-L1-expressing tumors harboring mKRAS and mTP53 comutations; this effect was attributed to an underlying increased sensitivity to PD-1 inhibition conferred by this double-mutant phenotype.27 Together, these results suggest that the addition of bevacizumab to atezolizumab may be the preferred treatment strategy for KRAS and TP53 comutated NSCLC.
Smoking is strongly associated with genetic heterogeneity in mKRAS tumors and confers a greater mutational burden and higher frequency of co-occurring mutations in TP53 or STK11 than never smoking.28 In this analysis, the mKRAS population and other mutation subgroups were enriched for smokers and patients with other known poor prognostic factors (such as ECOG PS status of 1 and higher median sum of longest diameter of target lesion or C-reactive protein levels) compared with the overall MEP or ITT population. The adverse effect of these prognostic factors was evident for OS in the BCP arm, which was markedly worse in mKRAS patients (median 9.86 months) than in the KRAS-WT population (median 18.23 months). Additionally, the enrichment of higher PD-L1 expression in mKRAS tumors (vs KRAS-WT tumors) may also account for the observed differences in treatment outcomes.
The current findings from this study offer insights into the personalized treatment of patients with KRAS-mutated NSCLC. Certain subgroups of mKRAS and comutations (eg, STK11/LKB1, TP53, and CDKN2A/B inactivation) are postulated to generate biological diversity in NSCLC, which, in turn, warrants a personalized approach to treatment.29 However, consistent evidence has been lacking on the utility of mKRAS as a sole predictive or prognostic biomarker for immune checkpoint inhibitor therapy,1 30 31 likely due to heterogeneity in comutations. The findings from these analyses suggest that it is plausible that consideration of mKRAS and co-occurring mutations in STK11, KEAP1, and TP53 may dictate treatment choices in the future, similar to mEGFR being a determinant of outcomes to targeted therapies with tyrosine kinase inhibitors.3 Collectively, findings from this and previous analyses of IMpower150 have shown the consistent benefits of ABCP in specific mutant subgroups ranging from patients with EGFR-mutant tumors16 to mKRAS populations with co-existing mutations in STK11, KEAP1, or TP53.
A major limitation of this retrospective exploratory analysis was that some mutation-defined subgroup sizes were small. The prevalence of mKRAS was found to be slightly lower in this study than previously published.1–4 This may be attributed to the use of blood-based mutation analysis vs using a tissue-based approach, which may underestimate the prevalence and limit sensitivity. Due to limitations in obtaining tissue at baseline, tissue mutation calls were not explored in the present study. Therefore, due to the small subgroup sizes, comparisons were not adequately powered to detect treatment differences, although exploratory endpoints were prespecified. Additionally, this analysis included patients with any alterations in KRAS, STK11, KEAP1 or TP53 regardless of functional relevance, which may be a confounding factor. It has also been reported that STK11/LKB1 functional loss can occur by nonmutational mechanisms32; however, this was not evaluated in patients in this study. Accordingly, caution should be applied in extending these findings to a clinical setting. Overall, prospective studies are essential to verify the promising findings observed in this subgroup analysis.
This exploratory analysis supports previous findings that mutation of STK11 and/or KEAP1 is associated with poorer prognosis. This analysis also suggests that atezolizumab combined with bevacizumab and chemotherapy is an efficacious first-line treatment option for patients with metastatic NSCLC, including difficult-to-treat NSCLC patient groups with mKRAS and co-occurring mutations in STK11 and/or KEAP1 and TP53.
Data availability statement
Data are available on reasonable request. Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Ethics statements
Patient consent for publication
Ethics approval
The study was performed according to the Good Clinical Practice guidelines and the Declaration of Helsinki, and the study protocol was approved by independent ethics committees at participating centers.
Acknowledgments
We thank the patients and their families. Medical writing assistance for this manuscript was provided by Anusha Bolonna, PhD, of Health Interactions and funded by F. Hoffmann-La Roche.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors participated in the data analyses, contributed to data interpretation and the writing of the manuscript, approved the final version of the submitted manuscript, and agreed to be accountable for all aspects of the report. HJW is responsible for the overall content as the guarantor.
Funding This work was supported by F. Hoffmann-La Roche/Genentech, a member of the Roche Group. Editorial support, funded by the sponsor, was provided by an independent medical writer under the guidance of the authors.
Competing interests FC reports nonfinancial support from Roche/Genentech during the conduct of the study and personal fees from Roche/Genentech, AstraZeneca, Takeda, Pfizer, Bristol Myers Squibb, Merck Sharp & Dohme, Lilly, and Bayer, outside the submitted work. RMJ reports nonfinancial support from Roche/Genentech during the conduct of the study and personal fees from Bristol Myers Squibb and Roche/Genentech, outside the submitted work. EK reports stock ownership from Genentech Inc outside the submitted work and employment by Genentech. MM reports other from Roche/Genentech outside the submitted work and employment by Roche/Genentech. TSKM reports personal fees from Abbvie, InMed Medical Communication, MD Health (Brazil), Medscape/WebMD, MoreHealth, PeerVoice, Physicians' Education Resource, P. Permanyer SL, PrIME Oncology, Research to Practice, Touch Medical Media, Curio Science, Inivata, and Berry Oncology. He reports personal fees and other from ACEA Pharma, Alpha Biopharma, Amgen, Amoy Diagnostics, BeiGene, Boehringer Ingelheim, Blueprint Medicines, CStone Pharmaceuticals, Daiichi Sankyo, Eisai, Fishawack Facilitate, Gritstone Oncology, Guardant Health, Hengrui Therapeutics, Ignyta, IQVIA, Incyte, Janssen, Lilly, Loxo-Oncology, Lunit, Mirati Therapeutics, OrigiMed, Puma Technology, Roche, Sanofi-Aventis R&D, Takeda, and Yuhan. He reports grants from Clovis Oncology, SFJ Pharmaceuticals, and XCovery; grants and personal fees from G1 Therapeutics; and grants, personal fees, and other from AstraZeneca, Bristol Myers Squibb, Merck Serono, Merck Sharp & Dohme, Novartis, and Pfizer. He reports other from Aurora, Virtus Medical Group, AstraZeneca, Hutchison Chi-Med, Sanomics, and geneDecode, outside the submitted work. SM reports employment by and stock ownership in F. Hoffmann-La Roche. MN reports grants and personal fees from Ono Pharmaceutical, Bristol Myers Squibb, Pfizer, Chugai Pharmaceutical, Eli Lilly, Taiho Pharmaceutical, AstraZeneca, Merck Sharp & Dohme, Novartis, Daiichi Sankyo, and Takeda Pharmaceutical Company; and personal fees from Boehringer-Ingelheim, Merck Biopharma, Teijin Pharma, and AbbVie, outside the submitted work. MR reports personal fees from Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer-Ingelheim, Lilly, Merck, Mirati, Merck Sharp & Dohme, Novartis, Pfizer, Roche, and Bioepis, outside the submitted work. DS reports employment by Genentech and stock ownership in F. Hoffmann-La Roche. MDT reports employment by Genentech and stock ownership in F. Hoffmann-La Roche. GS reports previous employment by Genentech. MAS reports nonfinancial support from Roche/Genentech during the conduct of the study; grants and personal fees from Roche/Genentech and AstraZeneca; personal fees from Merck, Guardant, Bristol Myers Squibb, and Bayer; and grants from Novartis, outside the submitted work. HJW reports personal fees from Genentech/Roche, AstraZeneca, Merck, Takeda, and Bristol Myers Squibb, outside the submitted work. WZ reports employment by Genentech.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.