Article Text

Abstract
Background Radiotherapy is the first-line treatment for patients nasopharyngeal carcinoma (NPC), but its therapeutic efficacy is poor in some patients due to radioresistance. Adoptive T cell-based immunotherapy has also shown promise to control NPC; however, its antitumor efficacy may be attenuated by an immunosuppressive tumor microenvironment. Exosomes derived from γδ-T cells (γδ-T-Exos) have potent antitumor potentials. However, it remains unknown whether γδ-T-Exos have synergistic effect with radiotherapy and preserve their antitumor activities against NPC in an immunosuppressive tumor microenvironment.
Methods γδ-T-Exos were stained with fluorescent membrane dye, and their interactions with NPC were determined both in vitro and in vivo. NPC cell deaths were detected after treatment with γδ-T-Exos and/or irradiation. Moreover, effects of γδ-T-Exos on radioresistant cancer stem-like cells (CSCs) were determined. The therapeutic efficacy of combination therapy using γδ-T-Exos and irradiation on NPC tumor progression was also monitored in vivo. Finally, the tumor-killing and T cell-promoting activities of γδ-T-Exos were determined under the culture in immunosuppressive NPC supernatant.
Results γδ-T-Exos effectively interacted with NPC tumor cells in vitro and in vivo. γδ-T-Exos not only killed NPC cells in vitro, which was mainly mediated by Fas/Fas ligand (FasL) and death receptor 5 (DR5)/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathways, but also controlled NPC tumor growth and prolonged tumor-bearing mice survival in vivo. Furthermore, γδ-T-Exos selectively targeted the radioresistant CD44+/high CSCs and induced profound cell apoptosis. The combination of γδ-T-Exos with radiotherapy overcame the radioresistance of CD44+/high NPC cells and significantly improved its therapeutic efficacy against NPC in vitro and in vivo. In addition, γδ-T-Exos promoted T-cell migration into NPC tumors by upregulating CCR5 on T cells that were chemoattracted by CCR5 ligands in the NPC tumor microenvironment. Although NPC tumor cells secreted abundant tumor growth factor beta to suppress T-cell responses, γδ-T-Exos preserved their direct antitumor activities and overcame the immunosuppressive NPC microenvironment to amplify T-cell antitumor immunity.
Conclusions γδ-T-Exos synergized with radiotherapy to control NPC by overcoming the radioresistance of NPC CSCs. Moreover, γδ-T-Exos preserved their tumor-killing and T cell-promoting activities in the immunosuppressive NPC microenvironment. This study provides a proof of concept for a novel and potent strategy by combining γδ-T-Exos with radiotherapy in the control of NPC.
- head and neck neoplasms
- immunotherapy
- radiotherapy
- tumor microenvironment
- t-lymphocytes
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Nasopharyngeal carcinoma (NPC) is one of the most prevalent and aggressive tumors in Southeast Asia. Epstein-Barr virus (EBV) exposure, dietary habits, and genetic factors are involved in NPC etiology.1 Radiotherapy is the most critical approach for NPC treatment with promising therapeutic efficacy in early-stage patients who have an overall 5-year survival rate of around 85%. Unfortunately, patients with NPC are usually diagnosed at advanced stage because of the difficulty in early diagnosis. Due to the radioresistance in patients with advanced NPC, the overall 5-year survival rate is less than 50%, and local reoccurrence or distant metastasis is common.1 NPC cancer stem-like cells (CSCs), such as CD44+ NPC cells, have high self-renewal and stem-like cell properties.2 3 Accumulating pieces of evidence suggest that NPC CSCs are radioresistant4 and associated with tumor recurrence, relapse, and metastasis.5 Immunotherapy, that is, adoptive cytotoxic T cell-based therapy, has also shown promise to control NPC.6 However, NPC tumor cells can modulate tumor microenvironment to promote tumorigenesis7 and immunosuppression,8 which can facilitate therapeutic resistance against radiotherapy and cell-based immunotherapy.9 10 Therefore, it is urgent to develop new therapeutic strategies that synergize with radiotherapy and overcome an immunosuppressive NPC microenvironment.
Exosomes are endosome-originated small extracellular vesicles (20–200 nm) that shuttle lipids, proteins, and nucleic acids in intercellular communications.11 As cell-derived nanoparticles, exosomes have high bioavailability, biostability, biocompatibility, and cargo-loading capacity, and can achieve targeting specificity to deliver antitumor agents.12 Exosome-based cancer therapies have also shown supplementary advantages in combination with other approaches.13 14 Previously, studies have shown that exosomes derived from immune cells have antitumor potentials. For example, exosomes derived from dendritic cells (DC-Exos) can enhance antitumor immunity through their carried immunostimulatory molecules.15 In addition, natural killer cell-derived exosomes (NK-Exos) have cytotoxic effects against different cancers through the contained killer molecules (eg, perforin and Fas ligand).16 However, despite the antitumor potentials of DC-Exos and NK-Exos, their applications in clinical practice are still limited, partially due to the difficulty of expanding DC and NK cells in large-scale ex vivo, leading to the uncertainty in exosome production.17
γδ-T cells (γδ-T-Exos) are innate-like T cells with lytic activities that are independent of major histocompatibility complex.18 Previously, we and others have demonstrated that γδ-T-Exos can directly eradicate stressed cells and have potent antiviral19–23 and antitumor activities.18 24–28 Peripheral γδ-T-Exos usually express Vδ2 and can be activated by phosphoantigens, leading to rapid expansion.29–32 We have also demonstrated that either adoptive transfer of ex vivo expanded γδ-T-Exos or targeted activation of γδ-T cells in vivo can control EBV-associated B-cell lymphoma,25 indicating the potential of γδ-T-Exos-based immunotherapy against EBV-associated tumors. Compared with NK cells and DC cells, γδ-T cells are much easier to be expanded in large-scale in vitro. Our previous studies demonstrated that phosphoantigen pamidronate selectively activated and expanded more than 200-fold of γδ-T-Exos in human peripheral blood.25 33 More importantly, we have recently found that exosomes derived from γδ-T-Exos carried robust antitumor molecules and controlled EBV-associated B-cell lymphoma and gastric carcinoma.24 However, it is not clear whether γδ-T-Exos also have antitumor effects against NPC because the pathogenesis of NPC is much different from EBV-associated B-cell lymphoma and gastric carcinoma.34 In addition, it remains unknown whether γδ-T-Exos can synergize with radiotherapy and preserve antitumor activities in the immunosuppressive NPC microenvironment.
In this study, we demonstrated that γδ-T-Exos not only eradicated NPC cells effectively but also preserved their tumor-killing and T cell-promoting activities in the immunosuppressive NPC microenvironment. Additionally, radiotherapy enhanced the targeted delivery of γδ-T-Exos to NPC tumors, while γδ-T-Exos improved radiosensitivity by eradicating the NPC CSCs. In a reciprocal manner, γδ-T-Exos can synergize with radiotherapy to control NPC. Our study provides a strong preclinical proof of principle for a novel and potent strategy using the combination therapy of radiotherapy and γδ-T-Exos in the control of NPC.
Methods
Cell culture
All NPC cell lines were kind gifts from our long-term collaborator, Professor S W Tsao (The University of Hong Kong). NP69 is an immortalized normal nasopharyngeal epithelial cell line. HK-1 cells are EBV-negative NPC cells. NPC43 is an NPC cell line newly established from an EBV-positive undifferentiated patient with NPC.35 C666-1 is a classical EBV-positive undifferentiated NPC cell line. NP69 cells were cultured with keratinocyte serum-free medium supplemented with bovine pituitary extract and human recombinant epidermal growth factor (ThermoFisher Scientific). HK-1 and C666-1 cells were cultured with 10% fetal bovine serum (FBS)– Roswell Park Memorial Institute (RPMI) medium. NPC43 cells were maintained in 10% FBS–RPMI supplemented with 4 µM Y27632 (Enzo Life Sciences, USA). Ionizing irradiation was carried out by a Gammacell 3000 irradiator (Nordion International).
Expansion of γδ-T-Exos and generation of γδ-T-Exos
γδ-T-Exos were expanded for exosome isolation as described previously.24 25 36 Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy donors from Hong Kong Red Cross. Briefly, human PBMCs from healthy donors were cultured in 10% FBS-supplemented RPMI-1640 medium and stimulated with 9 µg/mL pamidronate at days 0 and 3. Human recombinant interleukin (IL)-2 (Invitrogen) was added every 3 days from day 3 in a final concentration of 200 IU/mL. After 14–20 days, γδ-T-Exos (purity >95%) were washed with phosphate-buffered saline (PBS) and further cultured in exosome-free 10% FBS–RPMI medium, in which FBS-derived exosomes have been removed by ultracentrifugation for 18 hours at 100,000×g (SW32Ti rotor, Beckman). Forty-eight hours later, the conditioned medium was harvested for exosome isolation.
Exosome isolation and characterization
Exosomes were isolated by differential ultracentrifugation at 4°C as described previously.7 24 First, the conditioned medium was centrifuged at 300×g for 10 min, 2000×g for 10 min and 10,000×g for 30 min. Then, the supernatant was filtered using 0.22 µm syringe filter and subjected to ultracentrifugation at 100,000×g for 70 min (SW32Ti rotor, Beckman). The ultracentrifuged pellets were resuspended in PBS and centrifuged at 100,000×g for 70 min again. Finally, the exosome-containing pellets were dissolved in PBS. The protein concentration of exosomes was determined by a BCA Protein Assay Kit (Pierce, Bonn). In some experiments, γδ-T-Exos were preincubated for 30 min with the following antibodies or corresponding matched isotype controls: anti-FasL, anti-TRAIL (BioLegend) and washed by ultracentrifugation to remove the non-bound antibody.
Interaction of γδ-T-Exos with recipient cells
γδ-T-Exos were labeled with carboxyfluorescein succinimidyl ester (CFSE, Sigma-Aldrich) or 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR, Invitrogen) to monitor their interactions with recipient cells. After staining with the fluorescent dyes, exosomes were washed twice with PBS by recentrifugation at 100,000×g for 70 min to remove excess dyes. Finally, the fluorescence-labeled exosomes were resuspended in PBS for further use. In some experiments, ultracentrifuged pellets were isolated from non-conditioned FBS–exosome-free medium and labeled as described previously to serve as control. To evaluate the uptake efficacy of γδ-T-Exos, NPC cells were treated with CFSE-labeled γδ-T-Exos, and the intensity of CFSE was detected by flow cytometry after the indicated time.
Accumulation of γδ-T-Exos in NPC tumors
To assess the accumulation of γδ-T-Exos into the NPC tumor site, exosomes were labeled with DiR as described earlier. The DiR-labeled γδ-T-Exos were intraperitoneally injected into C666-1 or NPC43 tumor-bearing Rag2−/−γc−/− mice. Twenty-four hours later, the fluorescent signal of DiR in NPC tumor tissues was detected ex vivo using an In Vivo Imaging System (Caliper Life Sciences). In some experiments, the mice were irradiated at 0 or 4 Gy 3 days before the injection of DiR-labeled γδ-T-Exos. This time frame was chosen since a previous study identified maximum in vivo uptake of nanoparticles by tumors at 72 hours postirradiation.37
Cell apoptosis assay
NP69, HK-1, NPC43 or C666-1 cells were treated with PBS, γδ-T-Exos, γδ-T-Exos-depleted conditioned medium, or the ultracentrifuged pellets from non-conditioned FBS–exosome-free medium without γδ-T-cell components. Eighteen or 24 hours later, the cell apoptosis was detected using an Annexin V Apoptosis Detection Kit with propidium iodide (PI; BioLegend) as we did before.38 In some experiments, γδ-T-Exos were stained with CFSE before incubation with NPC cells, and the cell death was determined at the indicated time. In some experiments, γδ-T-Exos were preincubated with blocking anti-FasL, anti-TRAIL antibodies or isotype control (BioLegend). In some experiments, activated caspase-3 was detected in permeabilized NPC cells after 4-hour exposure to γδ-T-Exos using an antiactive-caspase-3 monoclonal antibody (BD Pharmingen).39 In some experiments, NPC cells were irradiated at 0, 1, or 3 Gy before treatment with γδ-T-Exos; then the cell apoptosis was determined as described earlier. In some experiments, NPC cells were treated with γδ-T-Exos or PBS in the presence or absence of 50% NPC supernatant.
Tumor growth factor beta (TGF-β) and C-C chemokine ligand 5 (CCL5) secretion assay
To determine NPC-secreting cytokines, NPC cells were cultured in 5% FBS–RPMI complete medium. After 48 hours, the culture supernatants of NPC cells were harvested. Then, the concentrations of TGF-β and CCL5 in culture supernatant were detected using human TGF-β assay and human proinflammatory chemokine assay (BioLegend). In some experiments, NPC cells were irradiated at 0 or 3 Gy before culturing.
T-cell migration into the NPC microenvironment in vitro and in vivo
A 5.0 μm pore size of Transwell System (Corning Costar) was used to determine the chemotactic activity of CD3 T cells as described previously.24 25 Purified CD3 T cells were treated with γδ-T-Exos or an equal volume of PBS. Forty-eight hours later, the T cells were harvested and added in the upper chamber of the Transwell System. C666-1 cell-derived supernatants were added into the lower chamber. Four hours later, the migrated CD3 cells to the lower chamber were detected by flow cytometry using counting beads (Molecular Probes, USA). In some experiments, the γδ-T-Exos-pretreated T cells were preincubated using blocking anti-CCR5 antibody or isotype control (clone 2D7, BD) for 30 min before being added into the upper chamber.
To determine the in vivo migration of CD3 T cells in the NPC tumor site, the γδ-T-Exos-pretreated T cells were stained with DiR and then incubated with neutralizing anti-CCR5 antibody, isotype control, or no antibody (−) for 30 min. The CD3 T cells were then administered intravenously into C666-1 tumor-bearing mice. Twenty-four hours later, NPC tumors were excised, and the intensity of DiR in tumor tissues was ex vivo detected by In Vivo Imaging System (Caliper Life Sciences).
T-cell proliferation and interferon gamma (IFN-γ) expression assays
CD3 T cells of healthy donors were purified from PBMCs using a Pan T Cell Isolation kit (Miltenyi Biotec). For proliferation assay, purified CD3 T cells were stained with CFSE (Sigma-Aldrich) and treated with CD3/CD28 agonist (Miltenyi Biotec) in the presence or absence of 50% NPC supernatant. Seven days later, T-cell proliferation and IFN-γ expression were determined by flow cytometry as described previously.40 In some experiments, CFSE-labeled CD3 T cells were cultured with allogeneic γδ-T-Exos in the presence or absence of 50% NPC supernatant for 7 days. To test intracellular IFN-γ expression, CD3 T cells were treated with phorbol myri-state acetate (100 ng/mL, Sigma-Aldrich), ionomycin (1 µg/mL, Sigma- Aldrich), and brefeldin A (BFA, 10 µg/mL; Sigma-Aldrich) for 6 hours. Then the CD3 T cells were harvested and subjected to intracellular staining of IFN-γ. In some experiments, LMP1-specific T cells were selected from EBV-seropositive huPBMCs using a CD137 microbeads kit (Miltenyi Biotec, USA) after 24 hours of stimulation by LMP1 peptide pool (Miltenyi Biotec). The selected cells were treated with allogeneic Vδ2-T-Exos or PBS and incubated with 100 IU/mL IL-2 for 2 weeks. The cells were then challenged with LMP1 peptide pool for 6 hours, with addition of BFA 2 hours later. LMP1-specific T cells were detected on flow cytometry by staining of CD3 and IFN-γ. The cell numbers were calculated using counting beads (Molecular Probes).
Establishment and treatment of NPC tumors in Rag2−/−γc−/− mice
C57BL/10SgAiRag2−/−γc−/− (Rag2−/−γc−/−) mice were cultivated in the Laboratory Animal Unit of The University of Hong Kong. NPC tumor models were established as previously described.35 41 Briefly, C666-1 (1.0×107/mouse) or NPC43 cells (1.5×107/mouse) were mixed with equal volume of Matrigel (Corning) and injected subcutaneously into Rag2−/−γc−/− mice 6–8 weeks old. Mice with palpable tumors were randomly grouped for further treatment. Mice were age-matched and gender-matched, and there were no evident differences in tumor volume among the groups. Equal volume of PBS (control) or γδ-T-Exos were intraperitoneally administrated and followed up as indicated. To compare the therapeutic efficacy of γδ-T-Exos administered through different delivery routes, intraperitoneal, intratumoral or intravenous injection was performed. In some experiments, the NPC tumor-bearing mice were irradiated at 0 or 4 Gy before the administration of γδ-T-Exos. The tumor volume and mouse survival were monitored and calculated at indicated time. Tumor volume was calculated as length×(width)2×0.52. Mice with tumor diameter reaching 17 mm were sacrificed and counted as dead according to the regulation in The University of Hong Kong. At endpoints, tumor tissues were harvested and photographed, then subjected to immunohistochemical analysis.
Histological and immunohistochemical analysis
Mice organs or tumor tissues were fixed with 10% formalin and embedded in paraffin for sectioning. The sections were subjected to immunohistochemistry analysis. Ki67 was detected using anti-human Ki67 antibody (Abcam) and visualized by a diaminobenzidine detection kit (Maixin Biotech) as we did before.24 25
Flow cytometric analysis
Surface staining of cells was performed using the following antibodies: anti-Fas (DX2), anti-DR5 (DJR2-4), anti-CD44 (IM7), anti-CD3 (HIT3a), anti-CD4 (RPA-T4), and anti-CD8 (SK1). For the intracellular staining, cells were fixed, permeabilized, and followed by staining with antiactive caspase 3 antibody (BD, USA) or anti-IFN-γ (BioLegend) as described previously.25 42 All samples were detected using a FACSLSR II Flow Cytometer (BD Biosciences) and analyzed by FlowJo software (Tree Star).
Statistics
Quantitative data were expressed as mean±SEM. Two groups comparison was analyzed by Mann-Whitney U test. One-way analysis of variance (ANOVA) with Bonferroni correction was used for multiple groups’ comparison. For multiple variables, the effects of each variable and their interactions were analyzed using two-way ANOVA. The mice survival was compared using Kaplan-Meier log-rank test. Two-tailed test was used for all analyses. A p value of <0.05 was regarded as significant. Specific sample size, p value, and details for each test were described in the figures or figure legends.
Results
γδ-T-Exos effectively interact with NPC cells and induce tumor cell apoptosis
Similar to our previous study, exosomes were isolated from human γδ-T-Exos through differential ultracentrifugation.24 To investigate the interaction of γδ-T-Exos with NPC cells, γδ-T-Exos were labeled with CFSE and incubated with NPC cells (C666-1 or NPC43). Ultracentrifuged pellets isolated from non-conditioned FBS–exosome-free medium were used as controls. Flow cytometry analysis revealed that γδ-T-Exos effectively interacted with NPC cells within 6 hours, peaked at 24 hours, and retained in tumor cells after 72 hours (figure 1A,B). To determine the accumulation of γδ-T-Exos in tumor site, C666-1 or NPC43 cells were injected subcutaneously into Rag2−/−γc−/− immunodeficient mice to build NPC tumor models as described previously.35 43 γδ-T-Exos or controls labeled with lipophilic dye DiR were then injected intraperitoneally into C666-1 or NPC43 tumor-bearing mice. Twenty-four hours later, the analysis using the In Vivo Imaging System identified significantly higher fluorescent signals in the NPC43 tumors from mice treated with DiR-labeled γδ-T-Exos compared with those from the mice treated with DiR-labeled controls (online supplemental figure S1A,B). Similarly, higher fluorescent signals were also detected in C666-1 tumors from mice treated with DiR-labeled γδ-T-Exos than those from the mice treated with DiR-labeled controls (online supplemental figure S1C,D). Taken together, our findings indicate that γδ-T-Exos can effectively accumulate in NPC tumors.
Supplemental material

Exos effectively interact with NPC cells and induce tumor cell apoptosis. NPC43 cells were treated with CFSE-labeled Exos at different doses and cultured for different time as indicated. The ultracentrifuged pellets isolated from non-conditioned FBS–exosome-free medium were labeled with CFSE parallelly and used as Ctr. (A) Representative figures of CFSE intensity in NPC43 cells. The CFSE intensity (B), apoptosis (Annexin V+) (C) and necrosis (AnnexinV− PI+) (D) of NPC43 cells were detected by flow cytometry. C666-1 cells were treated with Exos at different doses for 24 hours, then the cell apoptosis and necrosis were detected by flow cytometry. (E) Representative figures of AnnexinV and PI expression in C666-1 cells, apoptosis (F) and necrosis (G) in C666-1 cells are shown. (H) Apoptosis of NP69, NPC43 or C666-1 after treatment with Exos (Exos) and calculated as relative to those treated with PBS (Ctr). Quantitative data are mean±SEM of four biological replicates. (B–D) Statistical analysis was determined by two-way ANOVA. (F–H) Statistical analysis was performed using one-way ANOVA with Bonferroni correction. **P<0.01, ***P<0.001. ANOVA, analysis of variance; CFSE, carboxyfluorescein succinimidyl ester; Ctr, control; Exos, exosomes derived from γδ-T cells; MFI, median fluorescence intensity; NS, not significant.
The apoptosis (Annexin V+) and necrosis (AnnexinV− PI+) of NPC cells were also detected after γδ-T-Exos treatment. Data showed that γδ-T-Exos induced significant apoptosis of NPC43 cells in time-dependent and dose-dependent manners (figure 1C), while their effects on NPC43 cell necrosis were not evident (figure 1D). γδ-T-Exos also caused significant C666-1 cell apoptosis in a dose-dependent manner (figure 1E,F) but did not affect C666-1 necrosis (figure 1G). The necrotic fractions of NPC cells were generally less than 1%, regardless of γδ-T-Exos treatment (figure 1D,G), suggesting that the apoptotic cells could well represent the cell death in these assays. Compared with NPC tumor cells, the responsiveness of NP69 to γδ-T-Exos-induced apoptosis was much lower (figure 1H), indicating the selective killing effects of γδ-T-Exos against tumor cells than immortalized normal epithelial cells.
We also tested the killing effects of γδ-T-Exos against EBV-negative NPC cells because there is also a substantial fraction of EBV-negative patients with NPC.44 The treatment of EBV-negative NPC cell line (HK-1) with γδ-T-Exos also resulted in tumor cell apoptosis (online supplemental figure S2). Hence, our data indicate that γδ-T-Exos can effectively interact with and kill both EBV-positive and negative NPC cells.
γδ-T-Exos induce NPC tumor cell apoptosis through death receptor ligation
The differential ultracentrifugation we used to isolate exosomes is the most widely applied method and is better to remove the miscellaneous proteins than precipitation kits/polymer methods as suggested in the guideline proposed by the International Society for Extracellular Vesicles.45 In our previous study, we confirmed the successful isolation of γδ-T-Exos through the analysis of size distribution, morphology, and exosomal markers.24 To further determine whether the tumor cell-killing effects of our preparations were entirely due to γδ-T-Exos, NPC cells were treated with PBS, ultracentrifuged pellets from same amount of FBS–exosome-free conditioned or non-conditioned medium. The apoptosis and necrosis of NPC cells were detected 24 hours later. Data showed that the pellets isolated from the non-conditioned medium did not affect the NPC cell death (figure 2A–D). However, compared with either PBS or non-conditioned pellets, the conditioned pellets (which referred to γδ-T-Exos in our study) induced significant apoptosis of NPC43 and C666-1 cells (figure 2A,C). The NPC cell necrosis was not affected by neither conditioned nor non-conditioned pellets (figure 2B,D). These data suggested that the NPC cell-killing effects of ultracentrifuged pellets were caused by γδ-T-Exos components.

Exos induce apoptosis of NPC tumor cells through death receptor ligation. NPC43 or C666-1 cells were treated with PBS, ultracentrifuged pellets isolated from FBS–exosome-free non-conditioned medium or conditioned medium for 24 hours. Then, cell apoptosis (Annexin V+) and necrosis (AnnexinV− PI+) were detected by flow cytometry. Apoptosis (A) and necrosis (B) of NPC43 cells. Apoptosis (C) and necrosis (D) of C666-1 cells. NPC43 cells were treated PBS, Exos-depleted CdM containing soluble factors from Exos. Apoptosis (E) and necrosis (F) of NPC43 cells were analyzed. (G) Representative figures of DR5 and Fas on NPC tumor cells determined by flow cytometry. The blue histograms represent isotype controls. (H) Apoptosis of C666-1 cells after treatment with Exos with or without pretreatment of neutralizing aFasL, aTRAIL antibodies or isotype control. Quantitative data are shown as mean±SEM of four biological replicates. Statistical analysis was performed using one-way analysis of variance with Bonferroni correction. **P<0.01, ***P<0.001. aFasL, anti-FasL; aTRAIL, anti-TRAIL; CdM, conditioned medium; DR5, death receptor 5; Exos, exosomes derived from γδ-T cells; NS, not significant.
To assess the possibility that NPC cell death may be affected by other soluble factors in conditioned medium of γδ-T-Exos, which could be hidden in the ultracentrifuged pellets, NPC43 cells were treated with PBS, γδ-T-Exos-depleted conditioned medium containing γδ-T-Exos-secreted soluble factors or γδ-T-Exos. The data indicated that γδ-T-Exos-depleted conditioned medium did not affect the NPC cell death when compared with PBS treatment (figure 2E,F). However, γδ-T-Exos consistently induced NPC cell apoptosis but not necrosis (figure 2E,F). Therefore, the killing effects of the ultracentrifuged pellets against NPC cells were due to γδ-T-Exos rather than the other hidden soluble factors isolated from conditioned medium.
Next, we explored the underlying mechanisms of γδ-T-Exos-induced NPC cell apoptosis. NPC cells were treated with γδ-T-Exos and subjected to the detection of active caspase-3 detection after 4 hours. The results indicated that active caspase-3 was significantly increased in either C666-1 (online supplemental figure S3A,B) or NPC43 cells (online supplemental figure S3C,D) after γδ-T-Exos treatment. Considering that the caspase-3 activation is a hallmark of cell apoptosis, the NPC apoptosis induced by γδ-T-Exos was likely caspase-dependent. Additionally, we found that C666-1 and NPC43 cells expressed high level of surface death receptor 5 (DR5) and Fas (figure 2G). Since robust death-inducing ligands (FasL, TRAIL) were identified on γδ-T-Exos,24 we investigated the involvement of Fas/FasL or DR5/TRAIL pathway in this interaction. Blockade of TRAIL/DR5 or Fas/FasL pathway using anti-FasL or anti-TRAIL monoclonal antibody inhibited the γδ-T-Exos-induced NPC cell apoptosis, and the combined pretreatment using both neutralizing antibodies led to higher inhibition (figure 2H). These findings indicate that γδ-T-Exos can induce the apoptosis of NPC tumor cells through Fas/FasL and DR5/TRAIL pathways.
γδ-T-Exos inhibit the progression of NPC tumors in vivo
To determine the therapeutic effects of γδ-T-Exos against NPC tumors in vivo, C666-1 or NPC43 tumor-bearing mice were administered intraperitoneally with equal volume of γδ-T-Exos or PBS (control) as indicated (figure 3A). The mice were euthanized 21 days post-treatment; the tumors were then excised, photographed, and weighed. The tumor size was significantly smaller in γδ-T-Exos-treated mice than that in control mice (figure 3B). Moreover, the tumor weight was reduced by around 80% (p<0.01) in mice receiving γδ-T-Exos compared with those in control mice (figure 3C,D). Consistently, γδ-T-Exos effectively reduced the tumor volume of NPC tumors (figure 3E,F). Next, the effects of delivery routes of γδ-T-Exos on antitumor efficacy were determined. NPC tumor-bearing mice were administered with γδ-T-Exos via intravenous, intraperitoneal, or intratumoral routes. The data indicated that γδ-T-Exos delivered via these routes resulted in similar therapeutic efficacies against NPC tumor by reducing tumor sizes, tumor volume, and tumor weight (figure 3G,I).
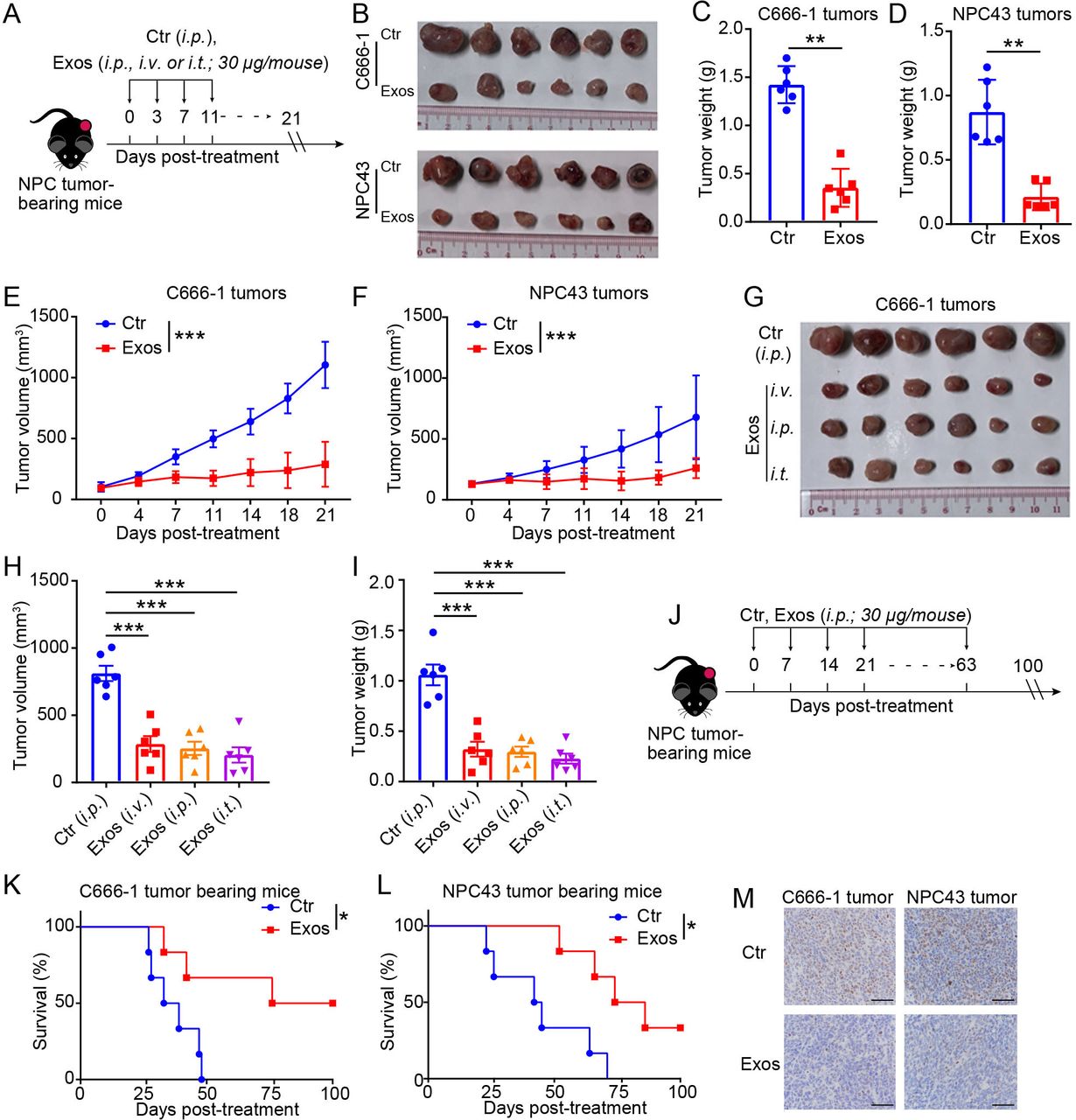
Exos inhibit the progression of NPC tumors in vivo. (A) In vivo therapeutic effects of Exos on NPC tumors were determined as indicated. The NPC tumor-bearing mice were established by subcutaneous injection of C666-1 or NPC43 cells in Rag2−/−γc−/− mice. Exos (30 μg/mouse) or equivalent volume of Ctr were injected into tumor-bearing mice as indicated (n=6). Tumor volume was measured post-treatment. At endpoints, tumors were excised and photographed. Excised tumors (B) of NPC xenografts. Tumor weight (C) and tumor volume (E) of C666-1 xenografts. Tumor weight (D) and tumor volume (F) of NPC43 xenografts. C666-1 tumor-bearing Rag2−/−γc−/− mice were also treated with Exos or Ctr through intraperitoneal, intratumoral or intravenous route (n=6). At endpoints, tumors were excised and photographed. Excised tumors (G), tumor volume (H), and tumor weight (I) of C666-1 xenografts. (J) C666-1 or NPC43 tumor-bearing Rag2−/−γc−/− mice were intraperitoneally injected with Exos or Ctr as indicated (n=6). (K,L) Survival of mice after treatment. (M) Representative immunohistochemical analysis of human Ki67 in sections of NPC xenografts at endpoints. Scale bars: 50 µm. For statistical significances, (C,D) were determined by Mann-Whitney U test; (E,F) were determined by two-way ANOVA; (H,I) were determined by one-way ANOVA with Bonferroni correction; (K,L) were determined by Kaplan-Meier log-rank test. *P<0.05, **P<0.01, ***P<0.001. ANOVA, analysis of variance; Ctr, control; Exos, exosomes derived from γδ-T cells; NPC, nasopharyngeal carcinoma.
Moreover, we investigated whether γδ-T-Exos could prolong the survival of NPC tumor-bearing mice (figure 3J–M). Either C666-1 or NPC43 tumor-bearing mice were injected intraperitoneally with γδ-T-Exos or control (figure 3J). The mice were followed up for 100 days after treatment. Compared with the control group, the injection of γδ-T-Exos significantly prolonged the survival of NPC tumor-bearing mice (figure 3K,L). The tumor cells in γδ-T-Exos-treated mice were also identified with lower proliferative capability, because immunophenotypical analysis found fewer Ki-67 positive cells in the residual tumors from γδ-T-Exos-treated mice than those from control mice (figure 3M). Taken together, our results demonstrate that γδ-T-Exos can control NPC tumor growth and prolong the survival of tumor-bearing mice.
γδ-T-Exos synergize with radiotherapy to eradicate NPC tumor cells
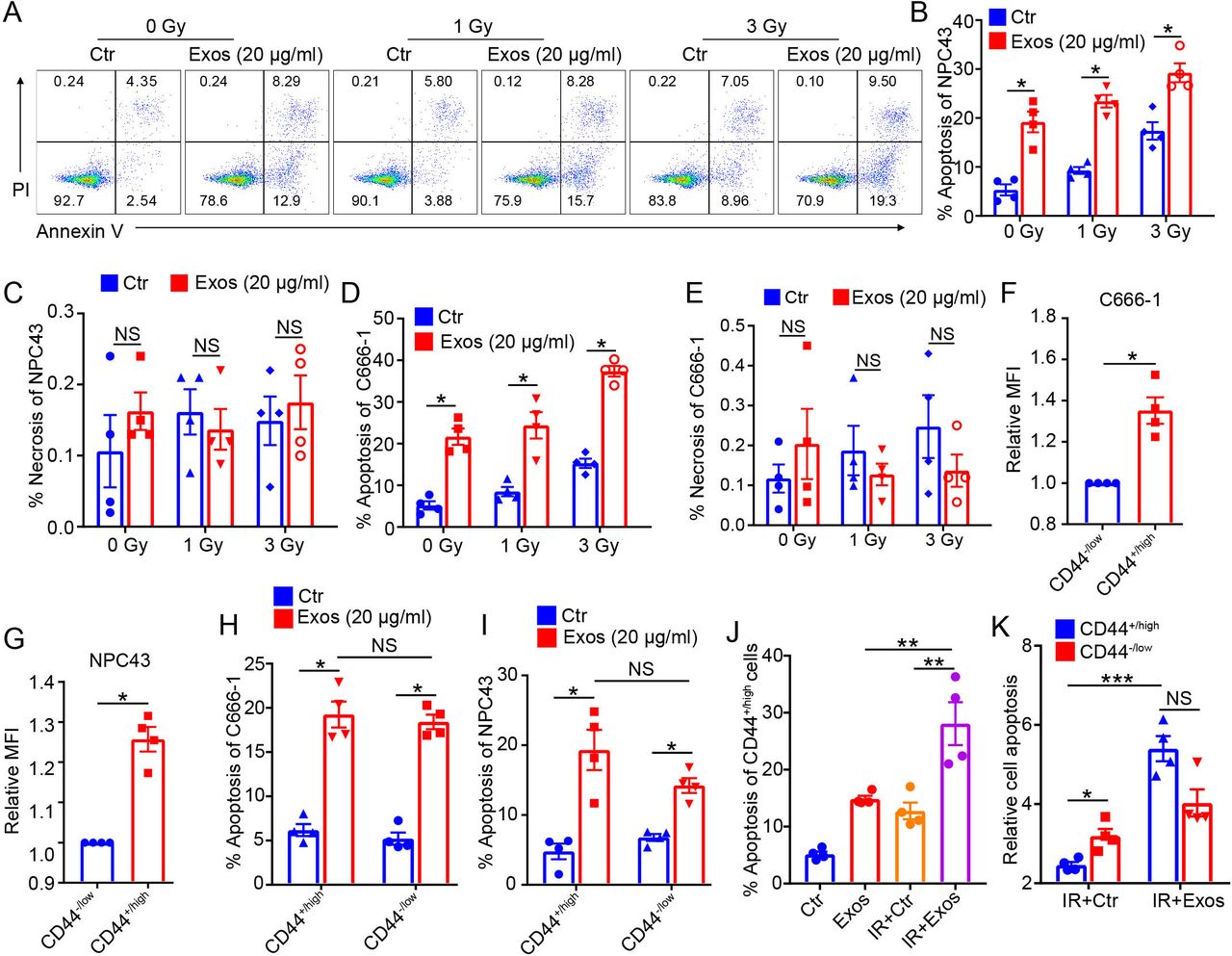
Since our in vitro and in vivo experiments showed that γδ-T-Exos have potent antitumor potential against NPC, and considering that radiotherapy is the primary treatment for NPC, we wondered if γδ-T-Exos had synergistic effects with radiotherapy. NPC tumor cells were irradiated at different doses and incubated with γδ-T-Exos, then the cell death was detected by flow cytometry. The data showed that irradiation induced the apoptosis of NPC cells in a dose-dependent manner (figure 4A,B,D). More importantly, combination of irradiation and γδ-T-Exos resulted in significantly higher NPC43 cell apoptosis than irradiation or γδ-T-Exos monotherapy (figure 4). The effects of irradiation, γδ-T-Exos, or combination therapy on NPC43 cell necrosis were not evident (figure 4C). Similarly, combination therapy using irradiation and γδ-T-Exos increased the C666-1 cell apoptosis when compared with the monotherapies (figure 4D). The necrosis of C666-1 cells was also not affected by neither the combination therapy nor monotherapies (figure 4E). Additionally, the combined effect of irradiation and γδ-T-Exos was also determined in the EBV-negative NPC cell line (HK-1). Synergistic effect of irradiation and γδ-T-Exos was also observed in the induction of HK-1 cell apoptosis (online supplemental figure S4A), while HK-1 cell necrosis was not changed after treatment (online supplemental figure S4B). Therefore, our findings demonstrate that γδ-T-Exos can synergize with radiotherapy to eradicate NPC tumor cells by inducing tumor cell apoptosis.

Exos synergize with radiotherapy to eradicate NPC tumor cells. NPC43 or C666-1 NPC tumor cells were IR at 0, 1, or 3 Gy, then cultured in the presence of PBS (Ctr) or Exos (20 µg/mL). Twenty-four hours later, the cell apoptosis (Annexin V+) and necrosis (AnnexinV−PI+) were detected by flow cytometry. (A) Representative figures of Annexin V and PI expression in NPC43 cells. Apoptosis (B) and necrosis (C) in NPC43 cells. Apoptosis (D) and necrosis (E) in C666-1 cells. Fluorescent intensity of CFSE in CD44+/high or CD44−/low C666-1 (F) or NPC43 (G) cells were determined after incubation with CFSE-labeled Exos for 24 hours. Apoptosis of CD44+/high or CD44−/low C666-1 (H) or NPC43 (I) cells after treatment with Exos or Ctr. (J) Apoptosis of CD44+/high NPC43 cells after IR at 3 Gy or not, in the presence of Exos (20 µg/mL) or Ctr. (K) NPC43 cells were IR at 3 Gy then treated with Exos (20 µg/mL) or Ctr. Apoptosis of CD44+/high or CD44−/low NPC cells relative to those treated with control alone is shown. Quantitative data are shown as mean±SEM of four biological replicates. (B–G) Statistical significances were determined by Mann-Whitney U test. (H–K) Statistical significances were determined by one-way analysis of variance with Bonferroni correction. *P<0.05, **P<0.01, ***P<0.001. CFSE, carboxyfluorescein succinimidyl ester; Ctr, control; Exos, exosomes derived from γδ-T cells; IR, irradiated; MFI, median fluorescence intensity; NPC, nasopharyngeal carcinoma; NS, not significant.
NPC CSCs, which generally express CD44, are radioresistant and the major contributors to therapeutic failure.2 4 Both NPC cell lines (C666-1 and NPC43) used in this study contained CD44-expressing subsets (online supplemental figure S5). We then evaluated the therapeutic effects of γδ-T-Exos on CD44+/high or CD44−/low NPC cells. Interestingly, γδ-T-Exos had higher efficacy to target CD44+/high NPC cells than CD44−/low NPC cells (figure 4F,G). Moreover, γδ-T-Exos induced significant cell apoptosis in both CD44+/high and CD44−/low NPC cells (figure 4H,I). The combined treatment of NPC cells with γδ-T-Exos and irradiation significantly enhanced the cell apoptosis of CD44+/high NPC cells when compared with the monotherapies (figure 4J). Unlike irradiation that showed attenuated apoptosis-inducing effect on CD44+/high NPC cells (figure 4K), combination of γδ-T-Exos with irradiation overcame the therapeutic resistance of CD44+/high NPC cells and greatly enhanced their apoptosis (figure 4K). Therefore, these findings indicate that γδ-T-Exos can improve the therapeutic effects of radiotherapy against NPC cells by overcoming the therapeutic resistance of NPC CSCs.
Radiotherapy increases the accumulation of γδ-T-Exos into NPC tumors
To determine whether radiotherapy can enhance the targeted delivery of γδ-T-Exos to NPC tumors, NPC tumor cells were irradiated at 0, 1 or 3 Gy, and then incubated with CFSE-labeled γδ-T-Exos for 24 hours. The uptake of γδ-T-Exos by NPC cells increased by ~20% to 60% after irradiation, compared with non-irradiated NPC cells (figure 5A,B). To confirm this effect in vivo, DiR-labeled γδ-T-Exos were injected intraperitoneally into NPC tumor-bearing mice 3 days after irradiation. After 24 hours postinjection of γδ-T-Exos, ex vivo detection using an In Vivo Imaging System found more DiR signal in the tumors from mice receiving irradiation than those from mice without irradiation (figure 5C,D). Therefore, these findings suggest that radiotherapy can enhance the uptake of γδ-T-Exos by NPC tumors.

Radiotherapy enhances NPC tumors to uptake Exos that promote T-cell migration into tumor microenvironment. NPC43 (A) or C666-1 (B) cells were irradiated at 0, 1, or 3 Gy, then cultured with CFSE-labeled Exos (20 µg/mL) isolated from four biological replicates. Twenty-four hours later, the fluorescent intensity of CFSE in NPC cells were determined by flow cytometry. The NPC43 (C) or C666-1 (D) tumor-bearing mice (n=3 or 4) were irradiated at 0 or 4 Gy and 3 days later were injected with DiR-labeled Exos (30 µg/mouse). Twenty-four hours postinjection, the tumors were excised and imaged ex vivo by In Vivo Imaging System. Ultracentrifuged pellets isolated from non-conditioned FBS–exosome-free medium without Exos components were used as Ctr. CCL5 in the CM or culture SNs of C666-1 (E) or NPC43 (F) cells. CCL5 in the culture SNs of C666-1 (G) or NPC43 (H) cells 24 hours after irradiation at 0 or 3 Gy. (I) Human CD3 T cells pretreated with PBS (Ctr) or Exos were cultured in the upper chamber of a Transwell assay; SNs from C666-1 cells were added to the bottom chamber. Four hours later, migration of Exos-pretreated CD3 T cells was calculated. (J) Exos-pretreated CD3 T cells were incubated with neutralizing aCCR5, isotype Ctr, or no antibody (−) for 30 min before added into the upper chamber. PBS-pretreated CD3 T cells were used as Ctr. (K,L) Exos-pretreated CD3 T cells were stained with DiR and then incubated with neutralizing aCCR5, isotype Ctr, or no antibody (−) for 30 min. PBS-pretreated CD3 T cells stained with DiR were used as Ctr. The human CD3 T cells were then administered intravenously into C666-1 tumor-bearing mice (n=4). Twenty-four hours later, the NPC tumors were excised, and migration of human CD3 T cells in tumor tissues was ex vivo detected by In Vivo Imaging System. (K) Ex vivo detection of DiR signal in tumor tissues. (L) Analysis of DiR intensity in tumor tissues. Quantitative data are shown as mean±SEM (n=4). (A–D,I,J,L) Statistical analysis was determined by one-way analysis of variance with Bonferroni correction. (E–H) Statistical analysis was determined by Mann-Whitney U test. *P<0.05, **P<0.01, ***P<0.001. aCCR5, antibody against CCR5; Avg, average; CCL5, C-C chemokine ligand 5; CFSE, carboxyfluorescein succinimidyl ester; CM, complete medium; Ctr, control; DiR, dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide; Exos, exosomes derived from γδ-T cells; MFI, median fluorescence intensity; NPC, nasopharyngeal carcinoma; NS, not significant; SN, supernatant.
γδ-T-Exos promote T-cell migration into tumor microenvironment
In our previous study, we found that EBV-associated lymphoma and gastric carcinoma cells secreted abundant CCR5 chemokine ligands, and γδ-T-Exos promoted T-cell migration toward the tumor microenvironment of these cancers by enhancing CCR5 expression on T cells.24 However, whether γδ-T-Exos display similar effects on NPC cells remains unclear. In this study, we demonstrated that NPC cells secreted robust CCL5 (figure 5E,F), which is chemotactic for T cells by interacting with CCR5. In addition, irradiation did not change the secretion of CCL5 from NPC cells (figure 5G,H).
Next, we determined the effects of γδ-T-Exos on T-cell migration into the NPC tumor microenvironment. In a Transwell chemotaxis system, pretreatment of T cells with γδ-T-Exos enhanced T-cell migration toward NPC supernatant in a dose-dependent manner (figure 5I). The T-cell migratory activity toward NPC microenvironment was mediated by the CCR5 on T cells because the addition of neutralizing anti-CCR5 antibody inhibited the cell migration (figure 5J). Consistent with the in vitro observation, human T cells could be recruited into NPC tumors in vivo (figure 5K,L). However, blockage of CCR5 using the neutralizing antibody significantly prevented T-cell migration to NPC tissues (figure 5K,L), suggesting that the CCR5 pathway mediated the T-cell migration into NPC tumor in vivo. Taken together, these data indicate that γδ-T-Exos can promote T-cell migration into NPC tumor microenvironment.
γδ-T-Exos synergize with radiotherapy to control NPC tumors in vivo
Our data shown in figures 4 and 5 indicated that γδ-T-Exos and radiotherapy have reciprocal antitumor potentials against NPC cells in vitro. To confirm this effect in vivo, we combined γδ-T-Exos with radiotherapy to treat the NPC tumor-bearing mice. The C666-1 tumor-bearing mice were irradiated at 0 or 4 Gy and then administered with γδ-T-Exos intraperitoneally or their control as indicated (figure 6A). Compared with the control, irradiation or γδ-T-Exos monotherapy reduced the tumor size and tumor volume of C666-1 xenografts (figure 6B,C). Tumor weight was also decreased by the monotherapies (figure 6D). However, combination therapy using irradiation and γδ-T-Exos displayed the highest therapeutic efficacy against C666-1 tumors over both monotherapies (figure 6B–D).

Exos synergize with radiotherapy to Ctr NPC tumor growth in vivo. C666-1 or NPC43 tumor-bearing Rag2−/−γc−/− mice (n=5) were IR at 0 or 4 Gy and then injected intraperitoneally with Exos (20 µg/mouse) or Ctr according to the schemes in (A,E). Excised tumors (B), tumor volume (C), and tumor weight (D) of C666-1 xenografts after treatment. Excised tumors (F), tumor volume (G), and tumor weight (H) of NPC43 xenografts after treatment. Statistical analysis of tumor volume was determined by two-way ANOVA. Statistical analysis of tumor weight was determined by one-way ANOVA with Bonferroni correction. *P<0.05, **P<0.01, ***P<0.001. ANOVA, analysis of variance; Ctr, control; Exos, exosomes derived from γδ-T cells; IR, irradiated; NPC, nasopharyngeal carcinoma; NS, not significant.
To confirm this observation, NPC43 tumor-bearing mice were also established and used to evaluate the efficacy of the combination therapy (figure 6E). Similar to that in C666-1 tumor-bearing mice, combination therapy of irradiation and γδ-T-Exos also showed advantages over either irradiation or γδ-T-Exos monotherapy in inhibiting NPC43 tumor growth and reducing tumor weight (figure 6F–H). Taken together, these results demonstrate that γδ-T-Exos have synergistic effects with radiotherapy to control NPC tumor in vivo.
γδ-T-Exos preserve antitumor activities in immunosuppressive NPC microenvironment
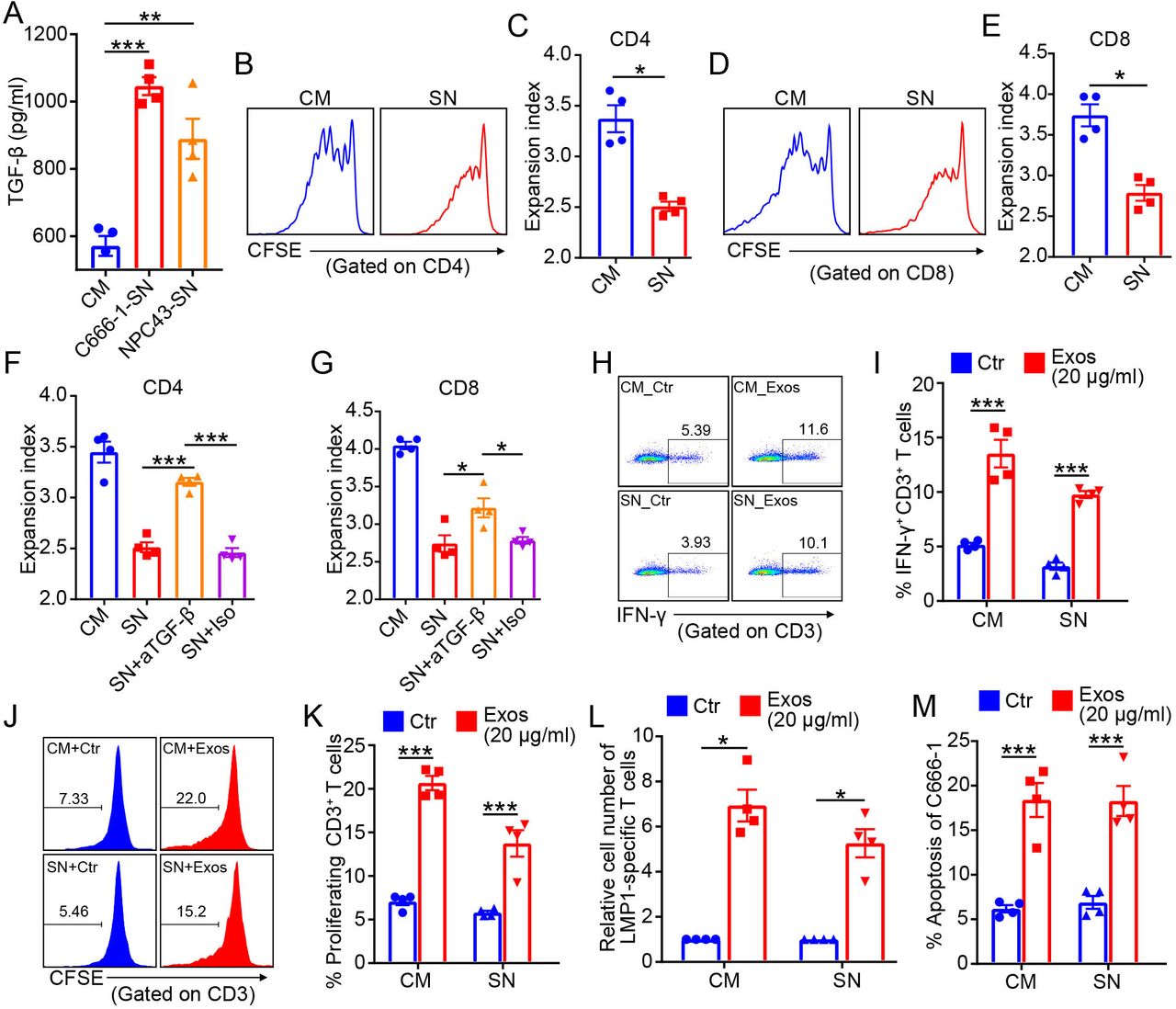
The immunosuppressive tumor microenvironment is a predominant contributor to therapeutic resistance,9 including radioresistance.10 TGF-β is one of the most important immunosuppressive cytokines in tumor microenvironment. In addition to the suppressive stromal cells like regulatory T cells and myeloid-derived suppressor cells, tumor cells are also a major source of TGF-β in tumor microenvironment.46 Notably, NPC cells secreted abundant TGF-β into the culture supernatant (figure 7A). Irradiation did not change the secretion of TGF-β from NPC cells (online supplemental figure S6A,B). The culture supernatant of NPC cells was immunosuppressive, because it significantly inhibited the T cell expansion (figure 7B–E). The CFSE dilution profiles suggested that the proliferations of CD4 and CD8 T cells were significantly suppressed by NPC supernatant (figure 7B,D). Reduced expansion indexes of CD4 and CD8 T cells were also identified after the treatment of NPC supernatant (figure 7C,E). As expected, the suppressive effects of NPC supernatant on T-cell response were mainly mediated by TGF-β, because the addition of neutralizing anti-TGF-β antibody effectively restored the CD4 and CD8 T-cell expansion (figure 7F,G).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Exos preserve antitumor activities in immunosuppressive NPC tumor microenvironment. (A) TGF-β in the CM or culture SN of C666-1 and NPC43 cells. (B–E) CFSE-stained T cells were stimulated with anti-CD3/CD28 antibodies in the presence of CM or culture SN of C666-1 cells for 7 days, then CFSE dilution was detected by flow cytometry. (B) Representative figures of CFSE dilution in CD4 T cells. (C) Expansion index of CD4 T cells. (D) Representative figures of CFSE dilution in CD8 T cells. (E) Expansion index of CD8 T cells. Expansion index of CD4 (F) and CD8 (G) T cells cultured in CM or culture SN of C666-1, with or without neutralizing anti-TGF-β antibody or Iso Ctr. Representative figures (H) and analysis (I) of intracellular IFN-γ in CD3+ T cells after 7 days cultured in CM or culture SN of C666-1 cells, in the presence of PBS (Ctr) or allogenic Exos (20 µg/mL) from four biological replicates. Representative figures (J) and analysis (K) of CD3+ T-cell proliferation after 7 days cultured in CM or culture SN of C666-1 cells, in the presence of allogenic γδ-T-Exos or Ctr. (L) Expansion of LMP1-specific T cells cultured in CM or culture SN of C666-1 cells, in the presence of Exos or Ctr) for 2 weeks. (M) Apoptosis of C666-1 cells after 18 hours cultured in CM or culture SN of C666-1 cells, in the presence of Exos or Ctr (Exos). Quantitative data are shown as mean±SEM (n=4). (A,F,G) Statistical analysis was determined by one-way analysis of variance with Bonferroni correction. (C,E,I,K–M) Statistical analysis was determined by Mann-Whitney U test. *P<0.05, **P<0.01, ***P<0.001. CFSE, carboxyfluorescein succinimidyl ester; CM, complete medium; Ctr, control; Exos, exosomes derived from γδ-T cells; IFN-γ, interferon gamma; Iso, isotype; NPC, nasopharyngeal carcinoma; SN, supernatant; TGF-β, tumor growth factor beta.
Previously, we demonstrated that γδ-T-Exos could promote T cells responses and expand the pre-existing tumor-specific T cells.24 Therefore, we wondered whether γδ-T-Exos could preserve these properties in immunosuppressive NPC microenvironment. Our data revealed that γδ-T-Exos still increased the expression of IFN-γ in T cells in immunosuppressive NPC supernatant (figure 7H,I). Furthermore, the proliferation of T cells could also be promoted by γδ-T-Exos under NPC supernatant (figure 7J,K). More importantly, γδ-T-Exos overcame the immunosuppressive NPC supernatant to promote the expansion of pre-existing LMP1-specific T cells from EBV-seropositive individuals (figure 7L), which are critical tumor-specific T cells with promising therapeutic potentials against NPC.47 In addition, the direct killing activities of γδ-T-Exos against NPC cells were not attenuated in the presence of immunosuppressive NPC-supernatant (figure 7M). Taken together, these findings indicate that γδ-T-Exos can preserve their tumor-killing and T cell-promoting activities in immunosuppressive NPC microenvironment.
Discussion
In this study, we demonstrated that γδ-T-Exos selectively eradicated NPC tumor cells and effectively prevented NPC tumor progression in vivo. More importantly, γδ-T-Exos overcame the therapeutic resistance of NPC CSCs by inducing notable cell apoptosis. γδ-T-Exos also preserved their direct killing activities against tumor cells and expanded pre-existing tumor-specific T cells in the immunosuppressive NPC microenvironment. Combination treatment of γδ-T-Exos with radiotherapy significantly enhanced therapeutic efficacies against NPC tumor when compared with the monotherapies.
As a type of extracellular nanoparticles, exosomes have been applied in combination with ionizing irradiation in NPC therapy. miR-34c-overexpressing exosomes derived from mesenchymal stem cells (MSCs) were found to drastically increase radiation-induced apoptosis of NPC cells and improve radiotherapy to prevent tumor progression.48 However, MSCs are functionally plastic and heterogeneous.49 MSC-derived exosomes were also found to promote tumorigenesis50 and suppress antitumor immune responses,51 which may limit their clinical application in cancer therapy. In contrast, homogeneous γδ-T-Exos can be prepared ex vivo at clinical scale using phosphoantigens,25 33 52–55 allowing sufficient production of γδ-T-Exos for clinical use. More importantly, the cytolytic activities against tumor cells and immunostimulatory properties of γδ-T-Exos are maintained even after long-term expansion.56
Tumor cells preferentially uptake nanoparticles due to the acidic tumor microenvironment.57 In a previous study, we demonstrated that γδ-T-Exos could target malignant EBV-associated tumor cells by interacting with the stressed molecules on their surface.24 γδ-T-Exos specifically induced cell death of EBV-associated B-cell lymphoma cells rather than normal B cells.24 Similarly, here we also found that γδ-T-Exos selectively attacked NPC tumor cells than immortalized normal epithelial cells. The killing activity of γδ-T-Exos against NPC was mainly mediated by Fas/FasL and DR5/TRAIL pathways, in which γδ-T-Exos carried robust FasL and TRAIL to trigger the apoptotic signaling pathways through death receptor (Fas and DR5) ligation on NPC cells. As the death receptors are rarely expressed on normal tissues,58 this mechanism of γδ-T-Exos-induced NPC apoptosis can minimize the potential toxicity against normal cells.
Here we found that radiotherapy reciprocally enhanced the γδ-T-Exos-mediated tumor control against NPC. After irradiation, increased accumulation of γδ-T-Exos in NPC tumors was observed. In support of our findings, previous studies also proved that tumor cells could uptake more nanoparticles after irradiation due to increased tumor cell endocytosis or vascular bursts.37 59 Moreover, irradiation can reduce the interstitial fluid pressure of the tumor site, leading to increased transport of nanoparticles into tumor microenvironment.60 Hence, there is a strong theoretical basis to combine nanomedicine with radiotherapy in the fight against NPC.59
CSCs have self-renewing properties and can differentiate into defined progenies. They not only initiate tumorigenesis, but also maintain tumor growth.61 CSCs are considered as the main cause of many therapeutic resistances, including radiotherapy.62 63 CD44 has been identified as a surface marker for CSCs of NPC and many other cancers.2 3 64 65 Interestingly, we found that γδ-T-Exos interacted with CD44+/high NPC CSCs in higher efficacy than CD44- NPC CSCs. As CD44 is considered as a phagocytosis receptor and also involved in cell endocytosis processes,66 67 the enhanced interaction efficacy of γδ-T-Exos with CD44+/high NPC CSCs might be due to the increased phagocytic and endocytic ability of CD44+/high NPC CSCs. Particularly, this effect can be amplified when coupled with irradiation, which can increase the accumulation of γδ-T-Exos into tumor microenvironment as shown in our current study. Similar to their parent cells that can kill CSCs efficiently,68 69 γδ-T-Exos also displayed potent cytotoxic effects on CD44+/high NPC CSCs. Combination of γδ-T-Exos with irradiation overcame the resistance of CD44+/high NPC CSCs to radiotherapy and resulted in significant tumor cell eradication. Therefore, γδ-T-Exos can supplement radiotherapy and enhance its therapeutic efficacy against NPC. Overall, our data revealed that γδ-T-Exos had synergistic effects with radiotherapy to suppress NPC in vitro and in vivo.
The antitumor T-cell immunity plays a critical role in cancer therapy. However, there are abundant soluble immunosuppressive factors, such as TGF-β, in solid tumor microenvironment8 70 71 to inhibit antitumor immune responses. We found that NPC cells secreted significant TGF-β in culture supernatants. The NPC supernatants are immunosuppressive and can dramatically inhibit T-cell responses in a TGF-β-dependent manner, suggesting that the NPC tumor microenvironment can hamper antitumor immune responses through immunosuppression. The tumor microenvironmental immunosuppression is a critical challenge for the cell-based cancer immunotherapies because it can impede the antitumor efficacy of administered immune cells12 72 or even educate them into protumoral phenotypes.73 What’s worse, radiotherapy can exacerbate the immunosuppressive tumor microenvironment by recruiting more immunosuppressive immune cells.74–76 Therefore, it is important for the combination therapy to not only display direct killing activities against NPC cells but also possess immunostimulatory effects to boost antitumor immunity in the immunosuppressive NPC microenvironment.
Theoretically, exosomes, as cell-free nanoparticles, may have advantages over cell-based immunotherapy for not being attenuated by the immunosuppressive tumor microenvironment.15 However, to our best knowledge, the antitumor efficacy of exosomes in immunosuppressive tumor microenvironment has rarely been studied. In this study, we found that NPC cells secreted abundant CCR5 ligand, and the treatment of γδ-T-Exos effectively increased T-cell infiltration into NPC tumor by upregulating CCR5 expression on T cells. In addition, γδ-T-Exos overcame the immunosuppressive effect of NPC-supernatant and effectively promoted T-cell responses. More importantly, γδ-T-Exos could preserve their immunostimulatory property in the immunosuppressive NPC microenvironment and amplify pre-existing tumor-specific T cells, which have promising antitumor potentials and can improve overall therapeutic efficacies against NPC. On the other hand, γδ-T-Exos also preserved tumor-killing activities against NPC cells under the immunosuppressive NPC microenvironment. Therefore, combination therapy using γδ-T-Exos can overcome the potential immunosuppressive effects after radiotherapy and improve overall therapeutic efficacy of radiotherapy by supplementing tumor cell killing and promoting T cellantitumor activities.
In summary, our study demonstrates a strong preclinical proof of concept using a novel therapeutic strategy by combining γδ-T-Exos with radiotherapy to treat NPC. γδ-T-Exos can not only eradicate radioresistant NPC CSCs but also preserve their tumor-killing and T cell-promoting activities in the immunosuppressive NPC microenvironment. Therefore, combination of radiotherapy with γδ-T-Exos has great potential in the treatment of NPC.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All the research protocols and animal work in this study and the experiments involving human subjects were approved by the institutional review board of The University of Hong Kong/Hospital Authority Hong Kong West Cluster (reference number UW21-133) and the Committee on the Use of Live Animals in Teaching and Research, The University of Hong Kong. Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
XW and YZ contributed equally.
Contributors WT, YL, XW, YZ conceived and designed the study; XW, YZ, XM and YC performed the experiments; XW, YZ, YL and WT interpreted the results and wrote the manuscript; CTR, SWT, GCFC, WHL and YLL provided critical suggestions for the project design and manuscript revision. WT, YL supervised the project. XW and WT are responsible for the overall content as guarantor.
Funding This work was supported in part by Health and Medical Research Fund, Food and Health Bureau, Hong Kong SAR Government (18192021); Seed Funding for Strategic Interdisciplinary Research Scheme, University of Hong Kong; General Research Fund, Research Grants Council of Hong Kong (17122519, 17126317, 17114818, 17122420 and 17104617), Hong Kong SAR, China.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.