Article Text

Abstract
Tumor-targeted CD40 agonism represents an attractive strategy for cancer immunotherapy (CIT) as it promotes dendritic cell (DC) activation and concomitant tumor-specific T cell priming without causing systemic side effects. We developed the bispecific CD40 agonistic antibody CEA-CD40, which triggers CD40 stimulation exclusively in the presence of carcinoembryonic antigen (CEA), a glycoprotein specifically expressed on tumor cells. In this study, we demonstrate that CEA-CD40 can enable potent in vitro DC activation and consecutive T cell cross-priming in a CEA-specific manner. Furthermore, we provide evidence that CEA-CD40 increases colocalization of CEA+ tumor material and DCs. Using CEA+ tumor-derived extracellular vesicles (EVs), which are known to be an excellent tumor antigen source, we show that CEA-CD40 mediates delivery of CEA+ EVs to DCs. Importantly, our data indicates that this fosters acquisition of tumor EV major histocompatibility complex I/peptide complexes by DCs, consequently improving CD8+ T cell priming against EV-associated antigen in vitro. Thus, we provide mechanistic evidence for a dual mode of action of CEA-CD40 for CIT: we suggest that CEA-CD40 has the potential to activate DCs and in addition can promote their loading with tumor antigen derived from EVs to trigger tumor-specific T cell cross-priming.
- adaptive immunity
- dendritic cells
- drug evaluation, preclinical
- immunotherapy
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The CD40 receptor plays a crucial role for activating dendritic cells (DCs) and enhancing their T cell (cross-) priming capabilities.1 2 Consequently, agonistic anti-CD40 antibodies have been developed to harness CD40’s potential to induce adaptive immune responses for cancer immunotherapy (CIT). However, the broad CD40 receptor expression on a wide range of hematopoietic and non-hematopoietic cells causes both side and sink effects that restrict the efficacy of CD40 agonists.3–7 Employing the bispecific FAP-CD40 antibody, we recently demonstrated that tumor-targeted CD40 agonism can overcome these limitations.8 FAP-CD40 induces CD40 agonism exclusively in the presence of fibroblast activation protein α (FAP), which is selectively expressed in the tumor stroma on cancer-associated fibroblasts. We could show that by limiting CD40 agonism to the FAP+ tumor area, side effects were abrogated, allowing the systemic administration of high therapeutic doses to induce potent antitumor immunity.8 Alternatively to targeting CD40 agonistic antibodies to the tumor stroma, direct targeting to antigens specifically expressed on tumor cells represents a promising strategy with several tumor cell targets being currently explored.9 10 We developed a CD40 agonist targeted to carcinoembryonic antigen (CEA), which is highly and specifically expressed by tumor cells in various epithelial tumor indications.11 Apart from inducing CEA-dependent DC activation, we speculated that CEA-CD40 might foster tumor-specific T cell priming via a second mode of action: simultaneous binding of CEA-CD40 to CD40 on DCs and CEA+ tumor material could potentially facilitate tumor antigen delivery to DCs.
In this context, delivery of tumor-derived extracellular vesicles (EVs) seemed in particular relevant. The occurrence of CEA+ tumor-derived EVs has been reported in patients with different CEA+ tumor indications.12–14 Furthermore, tumor EVs represent an excellent source of tumor antigen for tumor-specific T cell priming. Injection of EVs isolated from in vitro autologous tumor cell cultures prior to tumor challenge has been shown to protect mice from tumor establishment. The prophylactic antitumor effects of EVs were superior to those of irradiated tumor cells or tumor lysates. Moreover, in various investigated murine tumor models, administration of EV-loaded DCs induced better antitumor immunity in comparison to administration of EVs alone.15–19 These data provide a solid rationale for enhancing the delivery of tumor-derived EVs to DCs. Of note, Squadrito and colleagues have previously demonstrated that cancer cell-derived EVs can be effectively targeted to DCs, when DCs are engineered to express a chimeric receptor for specific tumor EV recognition. With this receptor engineering approach, the ability of DCs to prime CD8+ T cells against EV-derived antigens could be strongly improved.20 Employing the bispecific CEA-CD40 antibody for a potential in vivo targeting of EVs to DCs might represent a clinically feasible alternative to previously explored strategies that rely on in vitro generation and adoptive transfer of EV-loaded DCs or receptor-engineered EV-recognizing DCs. In this study, we provide in vitro evidence that CEA-CD40 indeed increases the delivery of tumor-derived CEA+ EVs to DCs, consequently enhancing cross-priming of T cells against antigen contained in the EVs. Thus, we show for the first time mechanistic data indicating that a tumor-targeted CD40 agonist specific for a tumor surface antigen can potentially promote tumor-specific T cell priming via a dual mode of action by (1) enhancing DC activation and (2) increasing tumor antigen delivery to DCs.
Material and methods
Molecules and reagents
Recombinant biotinylated human CEA (A2-B2 domain), the human FAP-CD40 and human CEA-CD40 antibodies, as well as the murine surrogate molecule CEA-moCD40 were produced at Roche Innovation Center Zurich (RICZ) and Munich (RICM) according to the protocols previously described in.8
Human CEA-CD40 and the murine surrogate CEA-moCD40 possess the same molecular design and properties as the previously described human FAP-CD40 and murine FAP-moCD40 antibodies8 but contain an anti-CEA binding domain instead of a FAP binding site.
In brief, CEA-CD40 and CEA-moCD40 consist of a huIgG1 with Fc-silencing PGLALA mutations21 and two CD40 binding Fv regions recognizing human CD40 via the 0817 domain (CEA-CD40) or murine CD40 via the FGK4.5 domain (CEA-moCD40). For monovalent CEA binding, one anti-CEA Fab fragment (recognizing the A2-B2 CEA region) was fused to the C-terminal part of the huIgG1 (see online supplemental figure 1A for a schematic overview of the molecules).
Supplemental material
Cell lines
Cells were maintained under sterile conditions at 37°C in a humidified incubator (5% CO2) and passaged regularly on reaching 80% confluency. MC38 cells were obtained from City of Hope. MC38-CEA cells were generated by City of Hope.22 MC38-CEA-ovalbumin (OVA) cells were generated by RICZ as described previously.8 HEK-huCD40 cells were obtained from InvivoGen (hkb-cd40). CD8+ DC tumor-derived MuTu DC1s (MuTu DC1 1940,23) were obtained from Prof. Acha-Orbea (University of Lausanne). MART-1 reporter cells were generated by Promega: Jurkat cells were engineered to stably express CD8 and the MART-1-specific TCR DMF5, while endogenous CD4 and the endogenous TCR were knocked out. In addition, a Luciferase gene under control of the IL-2 promoter was introduced. MV3 cells were received from DJ Ruiter (Amsterdam, Netherlands24) and engineered at RICZ to stably express CEA. MS-5 cells were obtained from DSMZ (ACC 441). All cell lines used were routinely checked for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, LT07-118). Cells were used for EV production or experiments 1–3 weeks after thawing.
To generate KOs of CEA and B2m in MC38-CEA-OVA cells, single guide RNAs (sgRNAs) with the following target DNA sequences were used:
CEA: 5’-AACATCATCCAGAATGACAC-3’ (Invitrogen, CRISPR620861_SGM)
B2m: 5’-CCGAGCCCAAGACCGTCTAC-3’ (Thermo)
Scrambled control: 5’-GTACGTCGGTATAACTCCTC-3’ (Thermo)
0.3 nmol of sgRNA were mixed with 10 µg Cas9 protein (Thermo, A36498) and incubated for 10 min at room temperature to allow stable complex formation. 2×105 MC38-CEA-OVA cells were subsequently transfected with the sgRNA/Cas9 complexes using a 4D-NucleofectorTM system with the SF Cell Line 4D-Nucleofector X Kit (Lonza). KOs were evaluated by flow cytometry 3 days post-transfection and cells were sorted using a BD FACSAria III cell sorter to obtain cell populations with a complete KO. Stability of the KO was confirmed by flow cytometry analysis 3 weeks after the sort.
Flow cytometry
Flow cytometry staining of cells and analysis of flow cytometry data was performed as described previously.8 For staining of EVs, EVs were incubated for 3 hours at room temperature with anti-CD81-coated Dynabeads (Beads: Thermo, 10 608D; anti-CD81: Biolegend, 104903) in 0.1 µm filtered phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (Miltenyi, 130-091-376). Dynabeads were washed using a magnet for separation of the beads. Subsequently, a blocking step was performed by adding 0.1 µm filtered PBS containing 5% EV-depleted fetal bovine serum (FBS) (gibco, A2720803) for 30 min at room temperature to the beads. Next, staining antibodies and Calcein AM violet (Thermo, C34858) for labeling of intact EVs as described by Gray et al25 were added in PBS/5% EV-depleted FBS for 20 min at room temperature. After a washing step, EV-coated beads were acquired by flow cytometry. For EV surface marker analysis, we gated on Calcein violet+ beads. A list of antibodies used for flow cytometry can be found in online supplemental table 1.
Mice
Female mice were used for isolation of spleens. Mice were maintained under specific-pathogen-free condition with daily cycles of 12 hours light and 12 hours darkness according to committed guidelines (GV-Solas; Felasa; TierschG). Use of mice for organ isolation was reviewed and approved by the local government (ZH 002/18). OT-1 (C57BL/6-Tg(TcraTcrb)1100Mjb/Crl) and C57BL/6 J mice were acquired from Charles River France. HuCD40tg mice (described previously in8), expressing both the human and murine CD40 receptor, were generated by Taconic by oocyte pronuclear injection of a bacterial artificial chromosome (BAC) construct encoding the human CD40 receptor under control of the human CD40 promoter. A founder strain with only one copy of the BAC transgene was selected. HuCD40tg mice were provided by the Baylor research institute and bred by Charles River.
Isolation of DCs and CD8+ T cells from murine spleens
DCs and CD8+ OT-1 T cells were isolated from murine spleens using mouse CD11c UltraPure microbeads (Miltenyi, 130-108-338) and the mouse CD8a+T Cell Isolation Kit (Miltenyi, 130-104-075) as described previously.8
Expansion of CD34+ cord blood cells
CD34+ cord blood stem cells (STEMCELL, 70008) were expanded for 7 days in StemSpan SFEM medium (STEMCELL, 09650) supplemented with 20 ng/mL huIL-3 (Peprotech, 200-03), 100 ng/mL huSCF (Peprotech, 300-07), 100 ng/mL huFlt3L (Peprotech, 300-19) and 50 ng/mL huTPO (Peprotech, 300-18).
DC differentiation from CD34+ cord blood cells
MS-5 stromal cells were treated for 3 hours with 10 µg/mL proliferation-inhibiting Mitomycin (Sigma, M4287). After three washing steps, 2.5×105 MS-5 cells were added per well of a 96-well flat bottom plate in 100 µl MS-5 medium (MEM-alpha (gibco, 15 070-063) supplemented with 10% FBS (gibco, 16140), 1% Penicillin-Streptomycin (gibco, 11548876) and 2 mM Sodium Pyruvate (gibco, 11 360-039). After 1 day, 1.3×105 expanded cord blood stem cells were added to the Mitomycin-treated MS-5 cells in 100 µL MS-5 medium supplemented with 5 ng/mL huGM-CSF (Peprotech, 300-03), 5 ng/mL huIL-4 (Peprotech, 200-04), 40 ng/mL huSCF and 200 ng/mL huFlt3L. 6 days after initiation of the coculture, cells were re-fed with 50 µL of MS-5 medium containing 12.5 ng/mL huGM-CSF, 12.5 ng/mL huIL-4, 100 ng/mL huSCF and 500 ng/mL huFlt3L. 4 to 7 days later, differentiated cord blood-derived DCs were used for sorting of DC1s.
Binding assay
To analyze binding of CEA-CD40 to CEA-expressing cells and huCD40tg DCs, binding assays were performed as described previously.8
DC activation, cross-presentation (DEC205-OVA) and MC38-OVA cell killing assays
DC activation assays with CEA-coated beads or MC38-CEA cells, cross-priming assays using DEC205-targeted OVA and OT-1 T cells, as well as subsequent MC38-OVA cell killing assays shown in figure 1 were performed according to the protocols described previously.8

The bispecific CEA-CD40 molecule induces CEA-dependent DC activation and concomitant T cell priming. (A) Schematic representation of the CEA-targeted CD40 agonist: CEA-CD40 is a human IgG1 with Fc-silencing PGLALA mutations, two N-terminal anti-human CD40 domains and one C-terminally fused human CEA binding moiety. (B) Binding of CEA- and FAP-targeted CD40 agonists to MC38 cells recombinantly expressing CEA or to MC38 wild type cells and to splenic DCs isolated from huCD40tg or wild type C57BL/6 J mice. Baseline correction was performed by subtracting mean fluorescence intensity (MFI) values of secondary antibody only conditions. For the curve fit nonlinear least squares regression using a variable slope model was applied. Shown are mean and SD values of technical duplicates. (C) Splenic DCs isolated from huCD40tg mice were incubated with different concentrations of CEA-CD40 antibody and CEA-coated or non-coated beads. DC activation was measured after 24 hours. Mean and SD values for the MFI of the representative DC activation markers CD86, CD70 and CD40 (murine CD40, as CEA-CD40 binding interferes with transgenic human CD40 detection) of technical duplicates are shown. (D) OVA-pulsed huCD40tg DCs were incubated with CEA-CD40, in presence of CEA-coated or non-coated beads. OT-1 T cells were added to the DCs and OT-1 T cell counts were determined after 72 hours of DC/T cell coculture. Depicted are mean and SD values of technical duplicates. (E) OT-1 T cells from the 5 nM CEA-CD40 plus CEA-coated beads conditions were coincubated with red fluorescent OVA+ MC38 cells. Equivalent numbers of non-primed, freshly isolated OT-1 T cells were used as a control. Killing of MC38-OVA cells was determined by incucyte analysis of the target cell area. Mean and SD values of technical triplicates are depicted. Significance at 150 hours was calculated using unpaired two-tailed t-test. Results were confirmed in three (B, C) or two (D, E) independent experiments. ***p≤0.001. CEA, carcinoembryonic antigen; DC, dendritic cell; FAP, fibroblast activation protein; OVA, ovalbumin.
CEA-coated beads and DC colocalization
The 2.0×105 MuTu DCs were seeded on a glass slide and allowed to adhere for 24 hours. Subsequently fluorescent streptavidin beads (Spherotech, SVFP-106-5) were coated with biotinylated CEA according to the manufacturer’s instructions. The bead number was determined by flow cytometry and CEA-coated or non-coated beads were added to the MuTu DCs in a 80:1 bead: cell ratio. Subsequently, 3 nM of CEA-moCD40 were added and MuTu DCs were incubated together with beads and antibody for 2 hours at 37°C. MuTu DCs were washed with PBS and fixed using Cytofix Fixation buffer (BD, 554655) according to the manufacturer’s protocol. The glass slide with the fixed cells was mounted on a coverslip with Fluoromount-G (Thermo, 00-4958-02) and cells were imaged the next day by confocal microscopy using a Zeiss inverted LSM 800 microscope (Plan-Apochromat 63 x/1.4 Oil DIC M27 objective) and the Zen software (Zeiss). Images were analyzed with Imaris (Bitplane; Oxford Instruments).
EV isolation
For production of EVs MC38-CEA-OVA, MC38-CEAKO-OVA, MC38-CEA-OVA-B2MKO and MV3-CEA cells were cultured for 4 days in 0.1 µm filtered Dulbecco’s Modified Eagle Medium (gibco, 41965062) +10% EV-depleted FBS +1% Penicillin-Streptomycin. HLA-A*02:01+ MV3-CEA cells were pulsed with 1 µM HLA-A*02:01-restricted short MART-1 peptide (ELAGIGILTV, Proimmune) 2 hours before EV isolation. Supernatants were harvested and EVs were isolated according to the protocol published by Squadrito et al.20 For ultracentrifugation, a Thermo Sorvall Ultracentrifuge with a SureSpin 630 rotor was used.
Size distribution and concentration of the EV isolates were determined with nanoparticle tracking analysis using a NanoSight NS300 instrument (Malvern Panalytical). For the NanoSight analysis, five subsequent measurements over 60 s were performed per sample and samples were diluted between 1:100 and 1:1000 in order to obtain concentrations of 40–80 particles per frame.
Negative staining transmission electron microscopy
Carbon/formvar-coated grids were used for imaging. The grids were glow-discharged and subsequently the EV sample was loaded by floating the grid on a drop of sample for 5 min. The grids were washed twice by floating on a drop of deionized water. Excess water was wicked away and the grids were placed on a drop of 2% uranyl acetate for 30 s. Excess stain was wicked away using a filter paper. Imaging was done using JEOL 1230 transmission electron microscope at an acceleration voltage of 80 kV.
Measurement of EV delivery to DCs
EVs were incubated with 20 ng/mL of Alexa Fluor 555 NHS Ester dye in a 0.1 M sodium bicarbonate solution for 1 hour at room temperature with continuous rotation to covalently stain EV lipid membrane proteins. Unbound dye was removed from the EVs by size exclusion chromatography using qEV columns (iZON, SP2). 1×109 labeled EVs were added to 0.25×105 splenic huCD40tg DCs together with 10 nM FAP-CD40 or CEA-CD40 antibodies. After a 3 hour incubation, DCs were washed and resuspended in flow cytometry staining buffer (eBioscience, 00-4222-57) containing 0.2 µg/mL 4′,6-diamidino-2-phenylindole (Roche, 10236276001). The signal of the labeled EVs was immediately quantified in the population of viable DCs employing flow cytometry.
OT-1 T cell priming with EVs derived from OVA-expressing tumor cells
The 0.25×105 splenic huCD40tg DCs were incubated with 1×109 EVs in combination with 10 nM FAP-CD40 or CEA-CD40 for 3 hours. Alternatively, HEK-huCD40 cells were used instead of DCs. To prevent proliferation, HEK-huCD40 cells were irradiated at 30 Gray using a RS 2000 irradiator (Rad Source Technologies) prior to incubation with EVs and antibodies. Subsequently, DCs or HEK-huCD40 cells were washed and 0.5×105 carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen, C34554)-labeled splenic CD8+ OT-1 T cells were added to the DC cultures. T cell numbers, activation and proliferation were measured 72 hours later by flow cytometry. Alternatively, for measurement of DC activation after incubation with EVs and bispecific antibodies, DC activation was analyzed 24 hours after EV addition. As a positive activation control DCs were incubated with artificially crosslinked CEA-CD40 (CEA-CD40 XL) using an anti-human Fc F(ab’)2 fragment for crosslinking as described previously.8
MART-1 reporter cell assay
Differentiated cord blood-derived DC1s (CD45+CD14-Clec9A+CD141+ cells) were sorted with a FACSAriaTM III (BD Bioscience with DIVA software) cell sorter. Sorted DC1s were rested overnight. 0.4×105 DC1s were incubated with 1×108 – 1×109 MV3-CEA-MART-1 EVs in combination with 10 nM FAP-CD40 or CEA-CD40 for 3 hours. Subsequently, DC1s were washed and 0.8×105 MART-1 Luciferase reporter T cells were added. After 6 hours, TCR signaling was measured by Bioluminescence assay using the Bio-Glo-NL Luciferase Assay System (Promega, J3082) according to the manufacturer’s instructions. Alternatively, for measurement of human DC activation after incubation with EVs and bispecific antibodies, DC activation was analyzed 9 hours after EV addition. As a positive activation control human DC1s were incubated with 10 µg/mL poly(I:C) (InvivoGen, vac-pic).
Statistical analysis
Statistical significance was evaluated using the GraphPad Prism V.8.4.2 (GraphPad Software, San Diego, California USA) with the statistical test indicated. P values of or below 0.05 were considered statistically significant. Significance levels are indicated as: ns=not significant=p>0.05; *=p≤0.05; **=p≤0.01; ***=p≤0.001; ****=p≤0.0001.
Illustrations
Illustrations were created with BioRender.com.
Results
CEA-CD40 induces CD40 agonism in a CEA-specific manner
The molecular properties of the bispecific CEA-CD40 antibody closely resemble those of the previously described FAP-CD40 molecule.8 In brief, CEA-CD40 targets human CD40 bivalently with two N-terminal CD40 binding sites and contains one C-terminally fused CEA binding domain (CEA refers to human CEA, as there is no murine homologue). To induce CD40 signaling, CD40 agonistic antibodies usually require oligomerization via Fcγ receptor (FcγR) crosslinking.26 However, FcγR binding of CEA-CD40 is abrogated by PGLALA mutations within the antibody’s Fc region21 (see figure 1A and online supplemental figure 1A for an illustration of the targeted CD40 agonistic antibodies). The purpose of this molecular design is to allow CEA-CD40 crosslinking solely by CEA+ cells. In contrast, crosslinking by systemically expressed FcγRs is abolished. Therefore, we envisioned that on binding of CEA-CD40 antibodies to CEA-expressing tumor cells, antibody clustering and consequently CD40 receptor multimerization and CD40 signaling are induced, while peripheral CD40 activation is eliminated (see online supplemental figure 1B for a schematic illustration of this mode of action).
For an initial characterization of CEA-CD40, we assessed its binding specificities. Binding of CEA-CD40 to CEA was confirmed using a MC38 murine colon adenocarcinoma cell line with recombinant CEA expression (MC38-CEA). Only CEA-CD40 but not the FAP-targeted anti-CD40 antibody bound to MC38-CEA. In contrast, CEA-CD40 did not bind to MC38 wild type cells lacking CEA expression (figure 1B). As they contain identical anti-human CD40 binding domains, CEA-CD40 and FAP-CD40 showed equivalent binding to splenic DCs isolated from mice transgenic for the human CD40 receptor (huCD40tg mice,8). No binding of CEA-CD40 or FAP-CD40 to splenic DCs of wild type C57BL/6 J mice was detected, since the anti-human CD40 moiety of the targeted CD40 agonists is not cross-reactive to the murine CD40 receptor (figure 1B).
To test CEA-CD40’s potential to induce CEA-dependent CD40 agonism, we coincubated splenic DCs of huCD40tg mice with CEA-CD40 and either CEA-coated vs non-coated polystyrene beads or MC38-CEA vs MC38 wild type cells. Indeed, CEA-CD40 induced a pronounced dose-dependent upregulation of various DC activation markers, such as CD86, CD70 and CD40, in the presence of CEA-coated beads or CEA-expressing cells. Conversely, DC activation was absent when non-coated beads or MC38 wild type cells were used (figure 1C and online supplemental figure 1C). We further demonstrated that CEA-dependent DC activation can induce effective T cell cross-priming. For this, we loaded DCs with OVA via the antigen uptake receptor DEC-205. Targeting of antigen to DEC-205 enables efficient processing of antigen for cross-presentation on major histocompatibility complex I (MHCI).27 Subsequently, we added CEA-CD40 in combination with CEA-beads or non-coated beads to the OVA-pulsed DCs. CD8+ OT-1 T cells, which recognize the OVA-derived SIINFEKL peptide when presented on MHCI of the H-2kb haplotype, were coincubated with the DCs. In accordance with our DC activation data, CEA-CD40 strongly enhanced T cell expansion in conditions with CEA-coated but not with non-coated beads (figure 1D). Of note, in contrast to freshly isolated OT-1 T cells, OT-1 T cells expanded in response to CEA-CD40-mediated DC activation were able to efficiently induce killing of OVA+ MC38 tumor cells (figure 1E). Taken together, our data show that CEA-CD40 can mediate a potent CEA-dependent DC activation, resulting in increased CD8+ T cell priming.
CEA-CD40 enhances delivery of CEA+ tumor-derived EVs to DCs
We next evaluated whether CEA-CD40 could enhance antigen cross-presentation and subsequent T cell priming not only by increasing DC activation but also by promoting delivery of tumor antigen to DCs. To investigate CEA-CD40’s potential to deliver CEA+ material to DCs, we coincubated MuTu DC1s, a murine green fluorescent protein-expressing DC cell line derived from splenic CD8+ conventional DCs,23 with fluorescent CEA-coated beads and the murine CEA-moCD40 surrogate molecule (recognizing human CEA and murine CD40, see online supplemental figure 1A for a scheme of the murine surrogate antibody). After 2 hours of incubation, a strong colocalization of DCs with CEA-coated beads but not with non-coated beads was observed by confocal imaging (figure 2A). Based on these findings, we subsequently explored whether CEA-CD40 can enhance not only the delivery of CEA+ beads but also of CEA+ tumor-derived EVs to DCs, since EVs are known to be an extraordinary good source of tumor antigen for induction of tumor-specific T cell priming.15–19
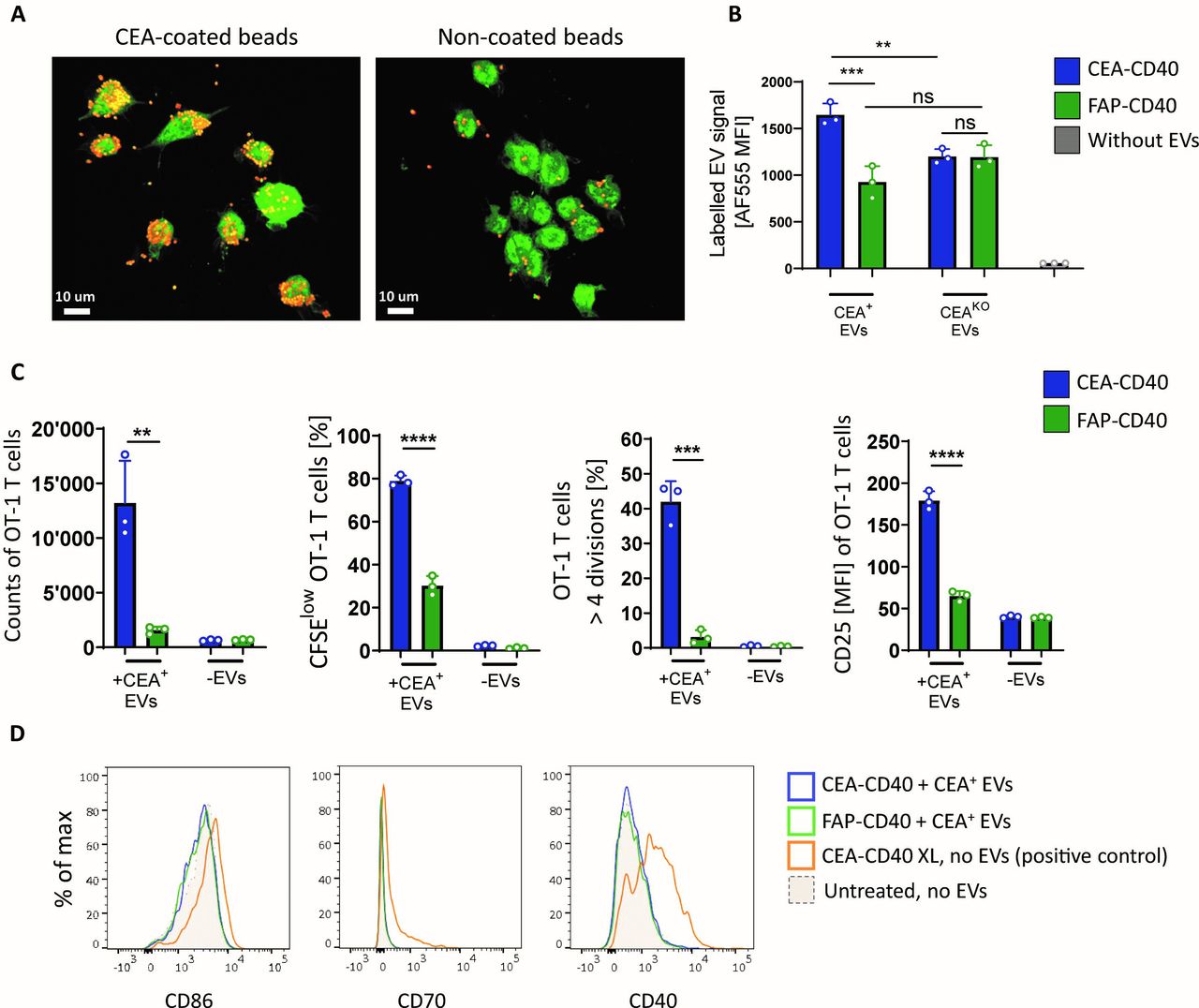
CEA-CD40 promotes delivery of CEA+ tumor-derived EVs to DCs and increases T cell cross-priming against EV-associated antigen. (A) GFP+ MuTu DC1s were coincubated with the murine surrogate molecule CEA-moCD40 and CEA-coated or non-coated red fluorescent beads. Representative confocal microscopy pictures showing colocalization of DCs and beads after 2 hours. (B) AF555-labeled EVs isolated from MC38-CEA-OVA (CEA+ EVs) or MC38-CEAKO-OVA cells (CEAKO EVs) were added to splenic huCD40tg DCs in combination with CEA-CD40 or FAP-CD40. Shown are AF555 MFIs of DCs after 3 hours. Mean and SD of technical triplicates are depicted. Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test. (C) Splenic huCD40tg DCs were incubated with MC38-CEA-OVA-derived EVs (+ CEA+ EVs) or without EVs (- EVs) and CEA-CD40 or FAP-CD40. After 3 hours, DCs were washed and CFSE-labeled OT-1 T cells were added to the DCs. T cell counts, percentage of proliferated (CFSElow) T cells, percentage of highly proliferated T cells that underwent more than four divisions and MFI of the T cell activation marker CD25 were quantified after 72 hours. Mean and SD of technical triplicates are depicted. Significance was calculated with unpaired two-tailed t-test. (D) HuCD40tg DCs were incubated with CEA+ EVs and targeted CD40 agonists as described in (C). As positive control, DCs were incubated with artificially crosslinked CEA-CD40 (CEA-CD40 XL, using an anti-human Fc F(ab’)2 fragment for crosslinking). Representative examples of the expression profile of DC activation markers CD86, CD70 and murine CD40 measured after 24 hours. Results were confirmed in at least three independent experiments. **p≤0.01; ***p≤0.001; ****p≤0.0001. ANOVA, analysis of variance; CEA, carcinoembryonic antigen; CFSE, carboxyfluorescein succinimidyl ester; DC, dendritic cell; EV, extracellular vesicle; FAP, fibroblast activation protein; MFI, mean fluorescence intensity; ns, not significant.
To answer this question, we isolated EVs from in vitro cultured MC38-CEA tumor cells expressing OVA as model antigen (MC38-CEA-OVA). Negatively stained transmission electron microscopy revealed the round/cup-shaped morphology of the EV isolates typically associated with membranous vesicles (online supplemental figure 2A). Moreover, MC38-CEA-OVA-derived EVs, which expressed the EV marker CD81, maintained CEA surface expression and were between 70 to 300 nm in size (online supplemental figure 2B,C), falling within the typical EV size range.28
We added Alexa Fluor 555 (AF555) fluorescently labeled CEA+ EVs to splenic DCs of huCD40tg mice in combination with CEA-CD40 or FAP-CD40. Addition of CEA-CD40 significantly increased the labeled EV signal in DCs after 3 hours in comparison to FAP-CD40 conditions. In contrast, when we used EVs produced by OVA-expressing tumors cells with a clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9 (CRISPR-Cas9) -based knockout (KO) of CEA (MC38-CEAKO-OVA), which are devoid of CEA expression (online supplemental figure 2B,D), no enhanced CEA-CD40-mediated EV delivery could be detected (figure 2B). Hence, our results indicate that CEA-CD40 indeed facilitates tumor-derived EV delivery to DCs in a CEA-specific manner.
CEA-CD40 promotes acquisition of CEA+ EV-derived MHCI/peptide complexes by DCs, leading to enhanced T cell cross-priming against EV-associated antigen
To investigate whether the enhanced CEA-CD40-mediated EV delivery to DCs translated into better T cell cross-priming, we incubated huCD40tg DCs with EVs derived from MC38-CEA-OVA tumor cells and CEA-CD40 or FAP-CD40 for 3 hours. Subsequently, DCs were washed and CFSE—labeled CD8+ OT-1 T cells were added to the DC cultures. Compared with FAP-CD40, CEA-CD40 greatly enhanced both proliferation and activation of OT-1 T cells. T cell numbers increased eight-fold on average when CEA-CD40 was added, consistent with a significantly higher fraction of proliferated, CFSElow T cells. Moreover, the expression of CD25 on OT-1 T cells was substantially elevated in CEA-CD40 vs FAP-CD40 conditions (figure 2C). Addition of FAP-CD40 did not affect T cell proliferation in comparison to conditions in which no antibody but only EVs were added to the DCs (online supplemental figure 2E). Importantly, the enhanced T cell priming with CEA-CD40 did not result from increased DC activation as no difference in the expression of DC maturation markers could be detected in cultures of DCs treated with CEA+ EVs and CEA-CD40 compared with untreated DCs or to DCs incubated with FAP-CD40 and EVs (figure 2D and online supplemental figure 2F). We hypothesize that, in contrast to CEA-expressing cells, CEA+ EVs cannot induce CEA-CD40 crosslinking and concomitant DC activation as their low CEA surface density and their small size are insufficient to trigger CEA-CD40 clustering.
In line with our previous data showing enhanced CEA-CD40-induced delivery of CEA+ but not CEAKO EVs to DCs, increased CEA-CD40-mediated T cell priming was dependent on the presence of CEA on the EVs: superior T cell proliferation was abrogated when DCs were incubated with CEA-CD40 and EVs derived from MC38-CEAKO-OVA cells (online supplemental figure 2G).
Next, we aimed to better understand the mechanisms underlying the priming of T cells against EV-associated antigen in both presence and absence of CEA-CD40.
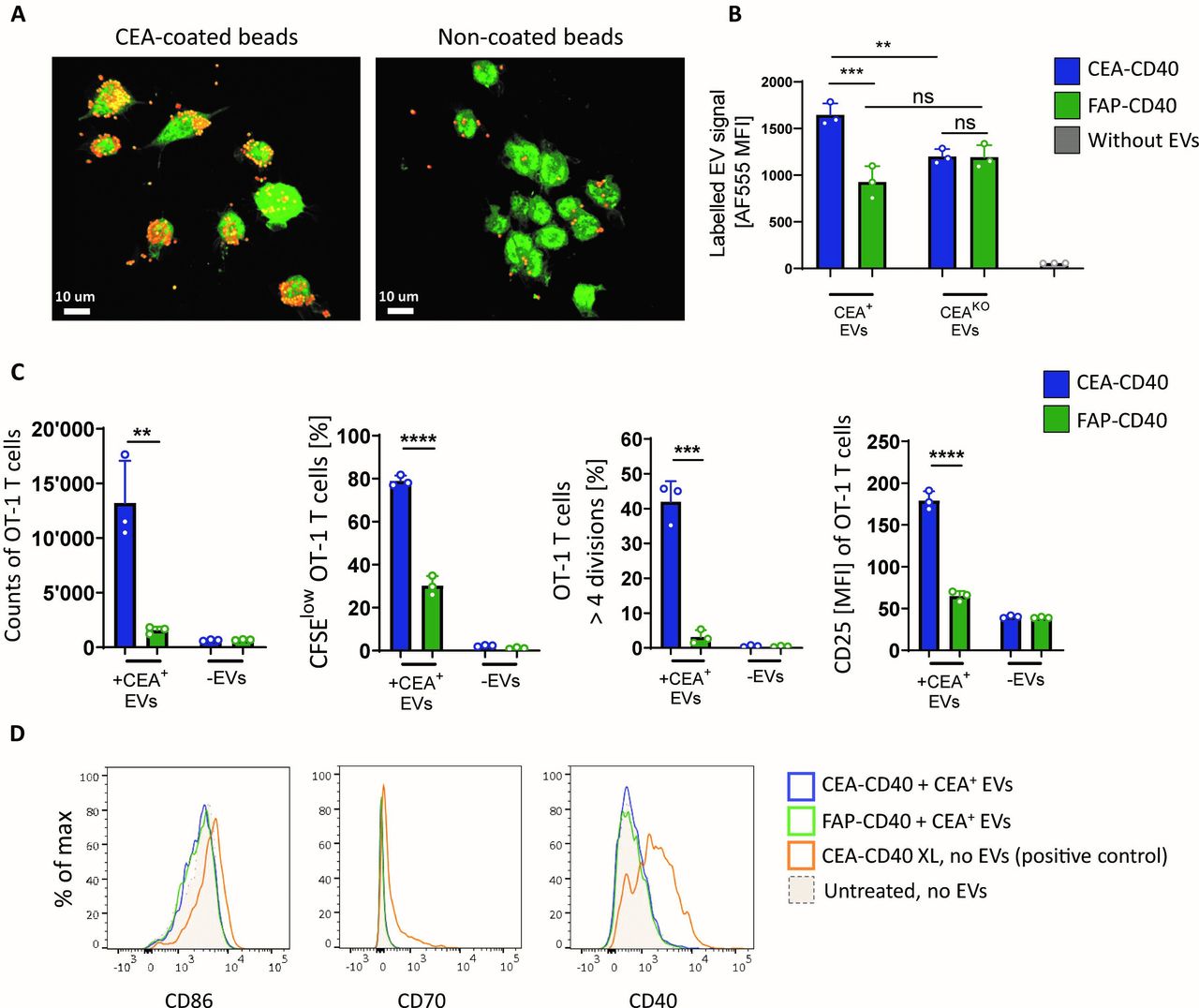
We found that prolonged incubation of MC38-CEA-OVA-derived EVs over 72 hours without DCs but with OT-1 T cells alone, did not result in a significant T cell proliferation (figure 3A and online supplemental figure 2E), implying a necessity of presentation of EV-associated antigens by DCs for effective T cell priming.

CEA-CD40 enhances cross-priming against CEA+ tumor EV antigen by promoting acquisition of EV-derived MHCI-peptide complexes by DCs. (A) Splenic huCD40tg DCs were incubated with MC38-CEA-OVA-derived EVs alone or in combination with CEA-CD40 or FAP-CD40. After 3 hours, DCs were washed and CFSE-labeled OT-1 T cells were added to the DCs. Alternatively, MC38-CEA-OVA-derived EVs were directly added to OT-1 T cells. Representative CFSE dilutions as measure of T cell proliferation after 72 hours are shown. (B) HuCD40tg DCs or HEK cells recombinantly expressing human CD40 (HEK-huCD40) were incubated with MC38-CEA-OVA-derived EVs (+ CEA+ EVs) or without EVs (- EVs), CEA-CD40 or FAP-CD40 and OT-1 T cells as described in (A). OT-1 T cell proliferation was measured after 72 hours. (C) HuCD40tg DCs were incubated with EVs from MC38-CEA-OVA cells (CEA+B2M+ EVs) or MC38-CEA-OVA-B2MKO cells (CEA+B2MKO EVs), CEA-CD40 or FAP-CD40 and OT-1 T cells as described in (A). Depicted is OT-1 proliferation after 72 hours. (B, C) graphs show mean and SD values of technical triplicates. Significances were calculated with one-way ANOVA using Tukey’s multiple comparisons test. Results were confirmed in three independent experiments. ANOVA, analysis of variance; CEA, carcinoembryonic antigen; CFSE, carboxyfluorescein succinimidyl ester; DC, dendritic cell; EV, extracellular vesicle; MHCI, major histocompatibility complex I; OVA, ovalbumin; ****=p≤0.0001.
Nevertheless, we questioned whether DCs could be dispensable for the observed enhanced T cell priming mediated by CEA-CD40. We considered the possibility that CEA-CD40 might induce an accumulation of EVs on the surface of any CD40+ cell and this increased density of surface-bound MHCI/SIINFEKL+ EVs might be sufficient to trigger augmented OT-1 T cell proliferation in our experimental setup.
Thus, we repeated our T cell cross-priming studies using human embryonic kidney 293 cells expressing high levels of recombinant human CD40 (HEK-huCD40) (online supplemental figure 3A) instead of huCD40tg DCs. When HEK-huCD40 cells were incubated with the MC38-CEA-OVA-derived EVs, no T cell priming could be detected either in presence or in absence of CEA-CD40 (figure 3B). Hence, CEA-CD40-mediated accumulation of EVs on CD40-expressing cells devoid of professional antigen-presenting cell functions was insufficient to induce OT-1 T cell priming.
To further investigate the process of EV antigen acquisition by DCs following CEA-CD40-mediated EV delivery, we isolated EVs from MC38-CEA-OVA cells with a KO of the MHCI subunit beta-2 microglobulin (B2m) (MC38-CEA-OVA-B2MKO), which are impaired in the formation of stable MHCI/antigen complexes and therefore lack MHCI/SIINFEKL surface expression (online supplemental figure 3B). When adding these B2MKO EVs to DCs, OVA-specific T cell proliferation was abolished in both CEA-CD40 and FAP-CD40 control conditions (figure 3C).
Therefore, both basal T cell cross-priming with EV-associated antigen in absence of CEA-CD40 and enhanced cross-priming due to increased CEA-CD40-mediated EV delivery seemed to rely on acquisition of EV-derived MHCI/peptide complexes by DCs.
Thus, taken together, our data suggests that the enhanced delivery of CEA+ tumor-derived EVs to DCs via CEA-CD40 can increase transfer of EV-associated MHCI/peptide antigen to DCs and consequently promote T cell cross-priming.
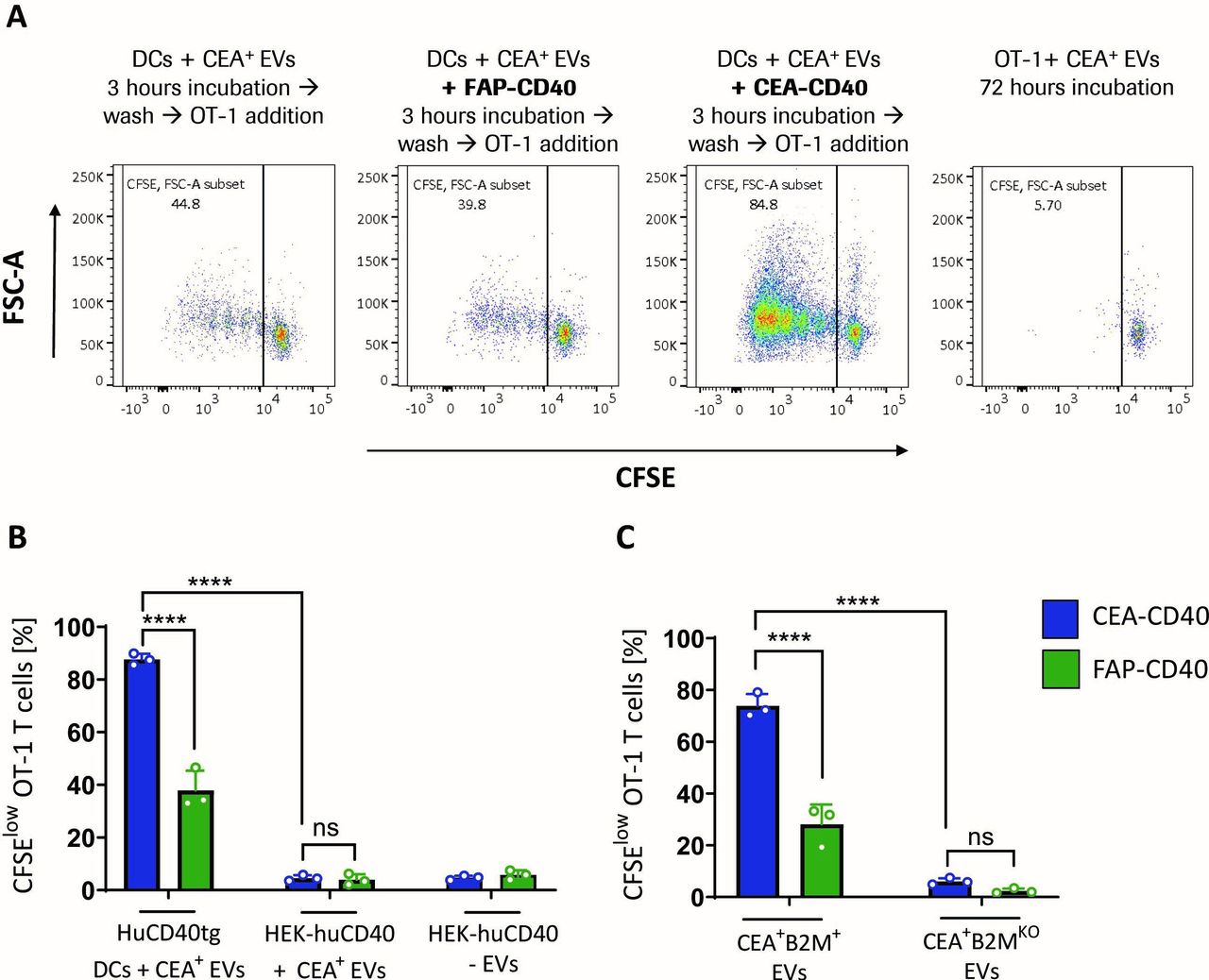
Finally, we aimed to test whether the findings we made using the murine OVA/OT-1 model system could be reproduced in an experimental setup employing human immune cells and a human tumor antigen. For this, we pulsed the CEA+ human melanoma cell line MV3-CEA with the short melanoma antigen recognized by T cells 1 (MART-1) peptide (ELAGIGILTV). Peptide pulsing was necessary as MV3 cells do not express any of the T cell-defined melanoma antigens commonly used in immuno-oncology studies.29 EVs were isolated from MV3-CEA-MART-1 tumor cells and incubated with human cord blood-derived DC1s in combination with CEA-CD40 or FAP-CD40. Human DCs were washed after 3 hours and subsequently MART-1-specific reporter T cells were added. MART-1 reporter cells are Jurkat T cells engineered to express CD8 and the MART-1-specific TCR DMF5, while the endogenous TCR and CD4 were knocked out. In addition, MART-1 reporter cells express Luciferase under control of the IL-2 promoter. Therefore, MART-1-specific TCR activation and consecutive IL-2 signaling could be measured by bioluminescence assay (see experimental scheme in figure 4A). In accordance with our previous results, FAP-CD40 did not induce higher IL-2 signaling compared with untreated conditions. In contrast, CEA-CD40, while not affecting DC activation, triggered significantly increased MART-1-specific TCR activation 6 hours after DC-T cell coculture (figure 4B,C). Thus, CEA-CD40’s effects could be recapitulated in the context of a human model system, indicating that tumor EV delivery to DCs and concomitant enhanced tumor-specific T cell cross-priming might indeed represent a therapeutically relevant mode of action of tumor cell-targeted CD40 agonists.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CEA-CD40 fosters human DC-mediated T cell cross-priming against human tumor EV-derived antigen. (A) Scheme of the experimental setup of the human cross-priming assay: human DC1s were differentiated and sorted from human cord blood stem cells. Human MV3-CEA melanoma cells were pulsed with the short MART-1 peptide. Tumor-targeted CD40 agonists and EVs derived from MV3-CEA-MART-1 tumor cells were incubated with the DC1s for 3 hours. MART-1 luciferase reporter T cells, expressing the MART-1-specific TCR DMF5 and a luciferase gene under control of the IL-2 promoter, were added to DC1s. TCR activation and concomitant IL-2 signaling were measured after 6 hours by bioluminescence assay. (B) Increase in IL-2 signaling of MART-1-specific T cells cocultured with DCs treated with MV3-CEA-MART-1-derived EVs (+ CEA+ EVs) or without EVs (- EVs) and CEA-CD40 or FAP-CD40. Results were normalized to the corresponding untreated conditions in which no antibody was added. Shown are mean and SD values of technical triplicates. Statistical significance was calculated using unpaired two-tailed t-test. Results are representative of three experiments. (C) Cord blood-derived DC1s were incubated with CEA+ EVs and targeted CD40 agonists as described in (B). As positive control, DCs were incubated with poly(I:C). Shown is the expression profile of the representative DC activation markers CD70, CD80 and CD86 after 9 hours. Results were confirmed using two different cord blood donors. CEA, carcinoembryonic antigen; DC, dendritic cell; EV, extracellular vesicle; FAP, fibroblast activation protein; ***=p≤0.001.
Discussion
With our study, we demonstrate for the first time that CEA-CD40, a bispecific tumor-targeted CD40 agonistic antibody, which binds to CD40 and a tumor surface antigen simultaneously, can exert its T cell priming effects via a dual mode of action. CEA-CD40 triggers DC activation by CD40 stimulation in a CEA-dependent manner. Thus, CEA-CD40 promotes T cell priming via DC maturation, a mechanism that is well described for CD40 agonistic antibodies in the context of CIT.30 31 In addition, we have shown that CEA-CD40 can foster the delivery of CEA+ tumor-derived EVs to DCs and consequently enhances cross-priming of T cells against EV antigen.
While it is conceivable that CEA-CD40 increases the delivery of other types of CEA+ tumor material (e.g, tumor cell fragments), our investigation focused on tumor-derived EVs, as they are known to have an extraordinary immunogenic potential when it comes to the induction of tumor-specific T cell responses.15–19
Using B2MKO EVs, we provided evidence that in our experimental setup acquisition of EV-derived MHCI/peptide complexes by DCs is indispensable for efficient cross-priming after CEA-CD40-mediated EV delivery. These findings are in line with other studies, which suggest that EV immunogenicity relies on the presence of antigenic MHCI/peptide complexes.16 17 20 32 Of note, as EV MHCI/peptide complexes as source of antigen eliminate the need of protein antigen processing by DCs, the peptide epitopes acquired from tumor-derived EVs might differ from those generated by the DC immunoproteasome and therefore potentially enable priming of T cells specific for epitopes not covered by classical cross-presentation processes.
In summary, our in vitro data suggest a unique novel mode of action of CEA-CD40 that could extend the effects of tumor antigen-targeted CD40 antibodies beyond CD40 agonism. Thus, our findings provide a mechanistic basis for further investigations that will be required to dissect how CEA-CD40’s effects can potentially contribute to antitumor immunity in vivo.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
We thank Anton Jochner and Klaus Neff (RICM) for their contributions to supply and production of the bispecific molecules used in this study. We would like to thank Maria Panagopoulou (University of Edinburgh) for kindly sharing her comprehensive knowledge on EVs and EV-related methodologies. We thank Ines Grazina de Matos, Duvini De Silva, Philipp Fröbel, Mi He and Thuy Trinh Nguyen for their support with the cord blood DC differentiation and sorting. Thanks to Stanford Chen (RICZ) for his help during confocal imaging. We gratefully acknowledge the RICZ Cell Technologies group for the generation of recombinant cell lines. Thanks to Tobias Schmidt, Antonio Sorrentino, Janine Toggweiler and Caroline Waltzinger (RICZ) for their advice and support during the generation of CRISPR-Cas9-based KO cell lines. We are also grateful to the RICZ Biochemistry and Downstream Processing as well as in vivo pharmacology teams for the supply of reagents and organs used in this study. We thank Prof. Acha-Orbea (University of Lausanne) for providing the MuTu DC1 cell line.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @JITCancer
Contributors ES: Conceptualization, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. MR: Conceptualization, supervision, methodology, project administration, writing–review and editing. HD: Conceptualization, resources. AM: Validation, investigation, methodology, writing–review and editing, resources. PJR: Conceptualization, supervision. CT: Conceptualization, supervision, methodology, project administration, writing–review and editing. PU: Conceptualization, supervision.
Funding PJR work was financially supported by F. Hoffmann La Roche AG. Roche authors declare ownership of Roche stock (options). ES, MR, HD, CT, PJR and PU are inventors on patent applications (WO 2020/070041 and WO 2018/185045, European patent application No. EP20207768; filed on November 16, 2020 (patent application pending), European patent application No. EP 21174716.7; filed on May 19, 2021 (patent application pending)) by F. Hoffmann La Roche AG that cover tumor-targeted CD40 agonists. PJR is supported by grants from Swiss Cancer League (KFS-4404-02-2018) and the Swiss National Science Foundation (310030_182735).
Competing interests All authors were Roche employees at the time of study conduction, with exception of AM and PJR.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.