Article Text

Abstract
Background On the basis of efficacy in mouse tumor models, multiple CD137 (4-1BB) agonist agents are being preclinically and clinically developed. The costimulatory molecule CD137 is inducibly expressed as a transmembrane or as a soluble protein (sCD137). Moreover, the CD137 cytoplasmic signaling domain is a key part in approved chimeric antigen receptors (CARs). Reliable pharmacodynamic biomarkers for CD137 ligation and costimulation of T cells will facilitate clinical development of CD137 agonists in the clinic.
Methods We used human and mouse CD8 T cells undergoing activation to measure CD137 transcription and protein expression levels determining both the membrane-bound and soluble forms. In tumor-bearing mice plasma sCD137 concentrations were monitored on treatment with agonist anti-CD137 monoclonal antibodies (mAbs). Human CD137 knock-in mice were treated with clinical-grade agonist anti-human CD137 mAb (Urelumab). Sequential plasma samples were collected from the first patients intratumorally treated with Urelumab in the INTRUST clinical trial. Anti-mesothelin CD137-encompassing CAR-transduced T cells were stimulated with mesothelin coated microbeads. sCD137 was measured by sandwich ELISA and Luminex. Flow cytometry was used to monitor CD137 surface expression.
Results CD137 costimulation upregulates transcription and protein expression of CD137 itself including sCD137 in human and mouse CD8 T cells. Immunotherapy with anti-CD137 agonist mAb resulted in increased plasma sCD137 in mice bearing syngeneic tumors. sCD137 induction is also observed in human CD137 knock-in mice treated with Urelumab and in mice transiently humanized with T cells undergoing CD137 costimulation inside subcutaneously implanted Matrigel plugs. The CD137 signaling domain-containing CAR T cells readily released sCD137 and acquired CD137 surface expression on antigen recognition. Patients treated intratumorally with low dose Urelumab showed increased plasma concentrations of sCD137.
Conclusion sCD137 in plasma and CD137 surface expression can be used as quantitative parameters dynamically reflecting therapeutic costimulatory activity elicited by agonist CD137-targeted agents.
- costimulatory and inhibitory T-cell receptors
- biomarkers
- tumor
- receptors
- immunologic
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
CD137 (4-1BB or TNFRSF9)1 was discovered as a surface glycoprotein expressed by activated but not resting T lymphocytes.2 3 It was shown to exert costimulatory functions in mouse and human T cells.3 4 CD137 is also expressed by other activated lymphocyte subsets including NK cells,5–7 but the function and immunobiology of the molecule has been mainly studied in CD8 T lymphocytes.1 8 A single ligand that belongs to the TNF family has been identified (CD137L or 4-1BBL).9 On CD8 T lymphocytes undergoing antigen activation, CD137 ligation with cognate ligand10 or agonist antibodies4 leads to protection from apoptosis and promotes proliferation, cytokine secretion, effector functions11 and metabolic adaptation.12 13 Reversal of exhaustion14 and nuclear reprogramming have been reported as well.15 Partial inefficacy in fighting viral infections has been observed in tnfrsf9−/− mice16 and in patients harboring loss of function mutations.17 18
On the basis of their costimulatory activity, CD137 agonists became a target to increase antitumor immunity. In mouse models of cancer, agonist anti-CD137 monoclonal antibodies (mAbs) induced tumor regressions contingent on CD8 T cell mediated responses.19 Such effects could also be elicited to some extent by CD137L.10 CD137 agonist antibodies show synergistic immunotherapy effects in mice on combinations with many other immunotherapeutic agents including PD-(L)1 checkpoint inhibitors,20 21 as well as with conventional chemotherapy22 23 and radiotherapy.24
In the clinic, a monoclonal human IgG4 antibody with intense agonist intrinsic activity on the CD137 receptor (Urelumab) showed single agent activity against melanoma and non-Hodgkin’s lymphoma but caused serious liver inflammation in a fraction of patients25 26 that precluded development at a full dose, even though it has been used safely at dose levels <0.2 mg/kg. Another antibody with low intrinsic agonist activity termed Utomilumab also entered clinical development26 with no liver toxicity but with weak antitumor activity.27
To elicit antitumor immunity, these agents need to reach the tumor microenvironment or tumor-draining lymph nodes and become capable of crosslinking CD137 from a solid-phase surface such as FcR-expressing neighboring leukocytes.28 However, in the case of the Urelumab agonist antibody activity on the receptor was at least partially independent from FcR involvement.29
Currently, about a dozen agents acting agonistically on CD137 are under early clinical development or late preclinical investigation.30 The theme common to all of these agents is the limitation of CD137 activity and crosslinking to the tumor microenvironment and associated lymphoid tissue. This is achieved with bispecific constructs targeting moieties expressed in the tumor microenvironment31–34 or making conditionally active versions of CD137 agonists.35 36
In all these cases, reliable biomarkers of CD137 ligation and activation are needed in sequential tumor biopsies and more conveniently in peripheral blood. In this study, we report that CD137 transcription and translation are intensely fostered by CD137 costimulation itself. Importantly, this includes soluble CD137 isoforms that circulate. In these cases, the CD137 agonist agent might retain sCD137 in the circulation delaying renal elimination, in addition to enhancing production. Monitoring of sCD137 in several models allowed us to conclude that it is a powerful candidate biomarker to monitor CD137-based immunotherapies. Moreover, sCD137 increased in the blood of a group of patients intratumorally treated with Urelumab and sCD137 was readily produced by human chimeric antigen receptor (CAR) T cells whose CAR sequence encompasses CD137’s cytoplasmic tail.
Material and methods
Mice
C57BL/6 and Balb/c mice were purchased from Envigo RMS Spain (Barcelona, Spain). C;129S4-Rag2tm1.1Flv Il2rγtm1.1Flv/J (Rag2−/− IL2Rγ−/−) mice were bred at the animal facility of CIMA-Universidad de Navara. Female mice were used at 6–9 weeks of age and maintained under specific pathogen-free conditions.
Cell lines
MC38 and CT26 mouse colon carcinoma cell lines were kindly gifted by Dr Karl E Hellström (University of Washington, Seattle, Washington, USA) and by Mario Colombo (IRCCS Istituto Nazionale dei Tumori, Milano, Lombardia, Italia), respectively. Lewis lung carcinoma (LLC) and B16F10 cell lines were purchased for ATCC. Cells were grown in RPMI 1640 media+GlutaMAX (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 µM 2-mercaptoethanol, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2. HEK293T cell line were maintained with DMEM high glucose+GlutaMAX (Gibco) supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (complete DMEM media) at 37°C with 5% CO2.
Isolation of primary mouse and human T cells
PBMCs were density gradient separated (Ficoll–paque plus, GE healthcare) from blood of healthy donors (n=13). Remaining erythrocytes were lysed by ACK buffer. CD8 T cells were freshly isolated by a negative magnetic selection kit (human CD8 T cell isolation kit, Miltenyi Biotech) according to the manufacturer’s instructions.
Spleens from naïve C57BL/6 mice were mechanically processed and ACK lysed. Mouse CD8 T cells were obtained from splenocytes by the negative mouse CD8 T cell isolation kit (Miltenyi Biotech).
Microbead coating and preparation
Agonist anti-CD3ε (OKT3 and 17A2), anti-CD137 (6B4 and 3H3), mIgG1 isotype control (MOPC-21), RatIgG2a isotype control (RTK2758) mAbs and human Mesothelin-Fc protein (ACROBiosystems) were covalently coupled to Dynabeads M-450 Tosylactivated (ThermoFisher) according to the manufacturer’s instructions. OKT3 tagged with AlexaFluor647 and 6B4 tagged with AlexaFluor488 were used to visualize by Flow Cytometry the graded density of anti-CD137 antibody coupled to the beads. Non-fluorescent irrelevant MOPC-1 mAb was added to adjust for the total amount of mAb in the coupling reaction.37 For fluorescence-dye labeling of the antibodies, Alexa Fluor 488/647 Antibody Labeling Kits (ThermoFisher) were used according to manufacturer’s instructions.
CD8 T cell activation
Isolated human CD8 T lymphocytes were preactivated with anti-CD3ε-coated (OKT3, 1 µg/mL) plates for 24 hours and rested for 24 hours before stimulation with plate-bound agonist antibodies for 48 hours. The mAbs used were anti-CD3ε (OKT3, 1 µg/mL), anti-CD137 (6B4, 5 µg/mL), anti-CD28 (CD28.2, 2 µg/mL), anti-OX40 (MOXR0916, 2.5 µg/mL), hIgG1 isotype control and mIgG1 isotype control (MOPC-21).
In the indicated experiments, CD8 T cells were stimulated with anti-CD3ε or anti-CD3ε+anti-CD137 coated microbeads for the duration of cultures. Recombinant IL2 was used at 50 U/mL (Proleukine, Novartis).
Similarly, isolated mouse CD8 T cells from splenocytes were activated for 72 hours in plates coated with the mAbs anti-CD3ε (17A2, 0.5 µg/mL), anti-CD137 (3H3, 10 µg/mL), anti-CD28 (37.51, 5 µg/mL) and/or RatIgG2a isotype control (RTK2758).
Anti-OX40 mAb and its isotype control hIgG1 were kindly provided by Bristol-Myers Squibb. Anti-hCD137 (6B4) is the laboratory’s own hybridoma.38 OKT3 is produced and purified from the hybridoma.38 3H3 and MOPC-1 antibodies were purchased from BioXCell, and the rest of antibodies for in vitro stimulations were from Biolegend. Isotype control mAbs were used in all experiments.
Blockade of CD137 costimulation using recombinant soluble CD137
Isolated human and mouse CD8 T lymphocytes were cultivated with anti-CD3ε+anti-CD137 coated microbeads in absence or presence of increasing concentrations of soluble recombinant human or mouse CD137 (purchased CELLSCIENCE and R&D systems, respectively).
Plasmids
The anti-mesothelin M11 scFv was generated from a human phage display library. M11-based CARs contain the CD8α hinge, CD8α (M11-BBz) or CD28 (M11-28z) transmembrane domains and the 4-1BB or CD28 intracellular domains in tandem with CD3z. cDNA sequences containing the M11-28z or the M11-BBz were custom synthesized (GenScript) and cloned into the third-generation lentiviral vector pCCL, under the control of EF1α promoter.
Lentiviral production and CAR gene transfer
Lentiviral particles were generated as previously described with slight modifications.39 10×106 HEK293T cells were plated 24 hours before transfection in 15 cm dishes. At the time of transfection, DNA-PEI complexes (18 µg CAR vector, 15 µg pMDLg/pRRE, 15 µg pRSV-Rev and 7 µg pMD2.G, 94.5 µL PEI) were added onto the cells, and media was replaced 4 hours later. Viral supernatants were collected 48 and 72 hours later and clarified by centrifugation and filtration. Viral supernatants were concentrated using ultracentrifugation and vector-containing pellets were resuspended and stored at −80°C until use. The number of transducing units per milliliter was determined by the limiting dilution method in Jurkat cells.
CAR transduction and stimulation
Isolated human CD8 T cells were activated with Dynabeads human T-Activator CD3/CD28 (ThermoFisher) at 1:3 cell-to-bead ratio in complete RPMI Media plus IL2 (50 U/mL). 24 hours later, lentivirus-containing supernatant was added to the activated human CD8 T cells. On day 3, cells were debeaded, and their population was controlled so as not to exceed 2×106 cell/mL confluency each day. Mesothelin-Fc-bead stimulation was carried out at day 8 post-gene transfer at an effector-to-target (E:T) ratio of 1:3 for 72 hours. Percentages of CAR+ T cells determined by Flow Cytometry were taken into account to normalize the amount of cells in these experiments.
Mouse models
To evaluate the potential biomarker role of sCD137, 5×105 MC38, CT26, LLC or B16F10 tumor cells were subcutaneously inoculated into the right flanks of syngeneic mice. When tumors reached 16–25 mm2, the mice were randomized into two groups. CD137 agonist therapy was provided by 5 µg of anti-CD137 (clone 3H3, BioXcell) intratumorally administered three times with a 3-day interval. Control mice received intratumoral injections of Rat IgG. Blood samples were collected in tubes containing 30 µL of Heparin (Hospira) before treatment and 2 or 3 days post-therapy in the MC38 and CT26 experiments, respectively. Tumor-free mice were treated intraperitoneally according to the same treatment scheme, and their blood samples were collected at the indicated time points.
To study human sCD137 kinetics in a model without circulating anti-CD137 mAbs, 100–120×106 PBMCs from a healthy donor were cocultivated in vitro with microbeads with mIgG1, OKT3+mIgG1 or OKT3+6B4 mAbs covalently coupled at an E:T ratio of 1:3 for 24 hours. Peripheral blood mononuclear cells (PBMCs) together with beads were washed with PBS and injected embedded in ice-cold Matrigel matrix into both flanks of Rag2−/− IL2Rγ−/− mice. Blood was drawn 24 and 48 hours after plug inoculation, and the Matrigel plugs (containing cells and beads) were recovered at 48 hours.
Data for hCD137 knock-in mice transgenic for human CD137 in the endogenous CD137 locus will be reported elsewhere (Fan et al manuscript in preparation).
CD137 expression by RT-qPCR
Total RNA was extracted from CD8 and PBMCs using a Maxwell RSC simplyRNA tissue kit (Promega) in a Maxwell RSC 48 instrument. M-MLV reverse transcriptase (Invitrogen) and random primers (Invitrogen) were used to obtain cDNA from total RNA. Quantitative PCR was carried out with iQ SYBR Green Supermix (Bio-Rad) in a CFX Connect Real-Time PCR Detection System (Bio-Rad) following the program: 95°C 3’; [95°C 15”, 60°C 15”, 72°C 25”, 76°C 5”, 78°C 5”, 80°C 5”] x42; 64°C 5”, 95°C 50”, 22°C 30” (SYBR fluorescence was captured in the underlined steps). The primer strategy for qPCR was based on previous research by this group40: human total CD137 (Fw: 5′CACTCTGTTGCTGGTCCTCA-3′, Rv: 5′-CACAGGTCCTTTGTCCACCT-3′), human transmembrane CD137 (Fw: 5′-GAAGGAGAGGGACGTGGTCT-3′, Rv: 5′-GCGCAAGAAAGAAGGAGATG-3′), human β-Actin (Fw: 5′-AGCCTCGCCTTTGCCGA-3′, Rv: 5′-CTGGTGCCTGGGGCG-3′); mouse total CD137 (Fw: 5′-AACATCTGCAGAGTGTGTGC-3′, Rv: 5′-AGACCTTCCGTCTAGAGAGC-3′), mouse transmembrane CD137 (Fw: 5′-AGAAGGACGTGGTGTGTGG-3′, Rv: 5′-TAAGGACCTGCAAGGAGTGC-3′) and mouse β-Actin (Fw: 5′-CGCGTCCACCCGCGAG-3′, RV:5′- CCTGGTGCCTAGGGCG-3′).
Flow Cytometry
Human cells were stained with the following fluorochrome-labeled antibodies: anti-CD45-PECy7 (Biolegend), anti-CD8-BV510 (Biolegend), anti-CD25-BV421 (Biolegend), anti-CD69-PerCPCy5.5 (Biolegend), anti-CD137-Biot (5D1, the laboratory’s own hybridoma), anti-Ki67-AF488 (Biolegend) and anti-Granzyme-AF647 (Biolegend). Streptavidin-PE was added to detect 5D1-Biot Ab. Isotype control mixes was prepared with mIgG1-BV421 (Biolegend), mIgG1-PerCPCy5.5 (Biolegend), mIgG1-AF488 (Biolegend), mIgG1-AF647 (Biolegend). In the case of 5D1-Biot, FMO was performed. M11 CAR expression was verified with anti-mIgG(H+L)-AF647 (Invitrogen) just before stimulation with mesothelin-Fc-beads.
Mouse T cells were stained with: anti-CD8-PECy7 (Biolegend), anti-CD25-APC (Biolegend), anti-CD69-BV510 (Biolegend), anti-CD137-PE (Biolegend), anti-PD1-PerCPCy5.5 (Biolegend), and anti-Ki67-AF488 (BD Bioscience). Rat IgG1-APC (Biolegend), Arm Hamster-BV510 (Biolegend), Syr Hamster-PE (Biolegend), Rat IgG2a-PerCPCy5.5 (Biolegend) and mIgG1-AF488 (Biolegend) antibodies were used as isotype-matched negative controls.
Zombie NiR (Biolegend) was used to exclude cell death. Samples were acquired on a BD Canto II (BD Biosciences) and CytoFlex S systems (Beckman Coulter). Analyses were performed using FlowJo (Tree Star) and CytExpert software (Beckman Coulter).
sCD137 protein levels analysis
Concentrations of sCD137 in the supernatant of cell cultures and plasma of MC38 and CT26 in vivo experiments were measured by ELISA from R&D (human and mouse 4-1BB/TNFRSF9 DuoSet ELISA kit).
To measure the concentrations of sCD137 coupled to 3H3 in peripheral blood, anti-Rat IgG (Biolegend) was coated on 96-well ELISA plates (Nunc) at 5 µg/mL overnight at room temperature (RT). Plates were blocked with PBS+1% BSA for 1 hour at RT. Then, plasma from mice bearing CT26 tumors in vivo (day +9) was incubated for 2 hour at RT. A dilution curve of 3H3+CD137 recombinant protein (ratio 5:1) was used as a standard. The sCD137complexed by 3H3 was detected using anti-4-1BB biotinylated mAb, followed by Streptavidin-HRP. CD137 recombinant protein, anti-4-1BB biotinylated mAb and Streptavidin-HRP were taken from the mouse 4-1BB/TNFRSF9 DuoSet ELISA kit (online supplemental figure S2). In parallel, a total sCD137 concentrations were quantified by conventional sandwich ELISA with antibodies that do not compete with 3H3 for 4-1BB binding.
Supplemental material
Human sCD137 concentrations in mouse plasma from the Matrigel matrix costimulation experiment were quantified with a predesigned human sCD137 ProcartaPlex kit (ThermoFisher Scientific Inc, Waltham, Massachusetts, USA) in a Luminex MAGPIX Instrument System (ThermoFisher Scientific Inc) according to the manufacturer’s instructions. The Matrigel plugs were resolubilized on ice in a known volume of cold PBS to measure sCD137 concentrations using the R&D human sCD137 kit according to the manufacturer instructions. sCD137 levels in the plasma of patients in the INTRUST clinical trial and melanoma patients treated with nivolumab+ipilimumab at our hospital (Clinica Universidad de Navarra) were determined with the TNFRSF9 Human ELISA Kit (Invitrogen) or the human sCD137 ProcartaPlex kit, as indicated.
All samples were centrifuged at high speed (12 100 g, 5 min) to clean particles. Concentrations below the detection limit of any assay were reported as 0.
Statistical analysis
GraphPad Prism V.8 (LA Jolla, California, USA) was used for appropriate statistical analysis as indicated in figure legends. Significance is marked on figures as *(p<0.05), **(p<0.01) and ****(p<0.0001).
Results
CD137 ligation costimulates expression of soluble and transmembrane CD137 in mouse and human T lymphocytes
CD137 costimulation can be mimicked by stimulation with anti-CD3ε mAbs attached to the culture plate together with an agonist anti-CD137 mAb. Many CD8 T-lymphocyte functional effects are costimulated in these culture settings. Interestingly, transcripts encoding CD137 itself are upregulated more readily by CD137-mediated costimulation in comparison with similarly provided CD28 costimulation (figure 1A–D). For those experiments, CD8 T cells were immunomagnetically sorted, preactivated for 24 hours with plate-bound anti-CD3ε to ensure acquisition of CD137 expression and then these primed lymphocytes were plated on plates with plastic-absorbed anti-CD3ε mAb plus the costimulatory mAbs. Moreover, in these CD8 T cells isolated from the peripheral blood of a panel of donors, both mRNA encoding the transmembrane and the soluble forms of CD13740 were further increased in the presence of CD137 costimulation (figure 1A). Accordingly, sCD137 was detected in the tissue culture supernatant (figure 1B) and surface CD137 was detected on the plasma membrane at higher levels (figure 1C). Of note, CD28 is much less active at causing CD137 upregulation in this setting and concomitant CD28+CD137 dual costimulation only marginally increased the expression of the CD137 isoforms in human CD8 T lymphocytes when compared with CD137 single costimulation. As expected, both CD137 and CD28 enhanced CD25, CD69, Ki67 and Granzyme B expression (figure 1D).
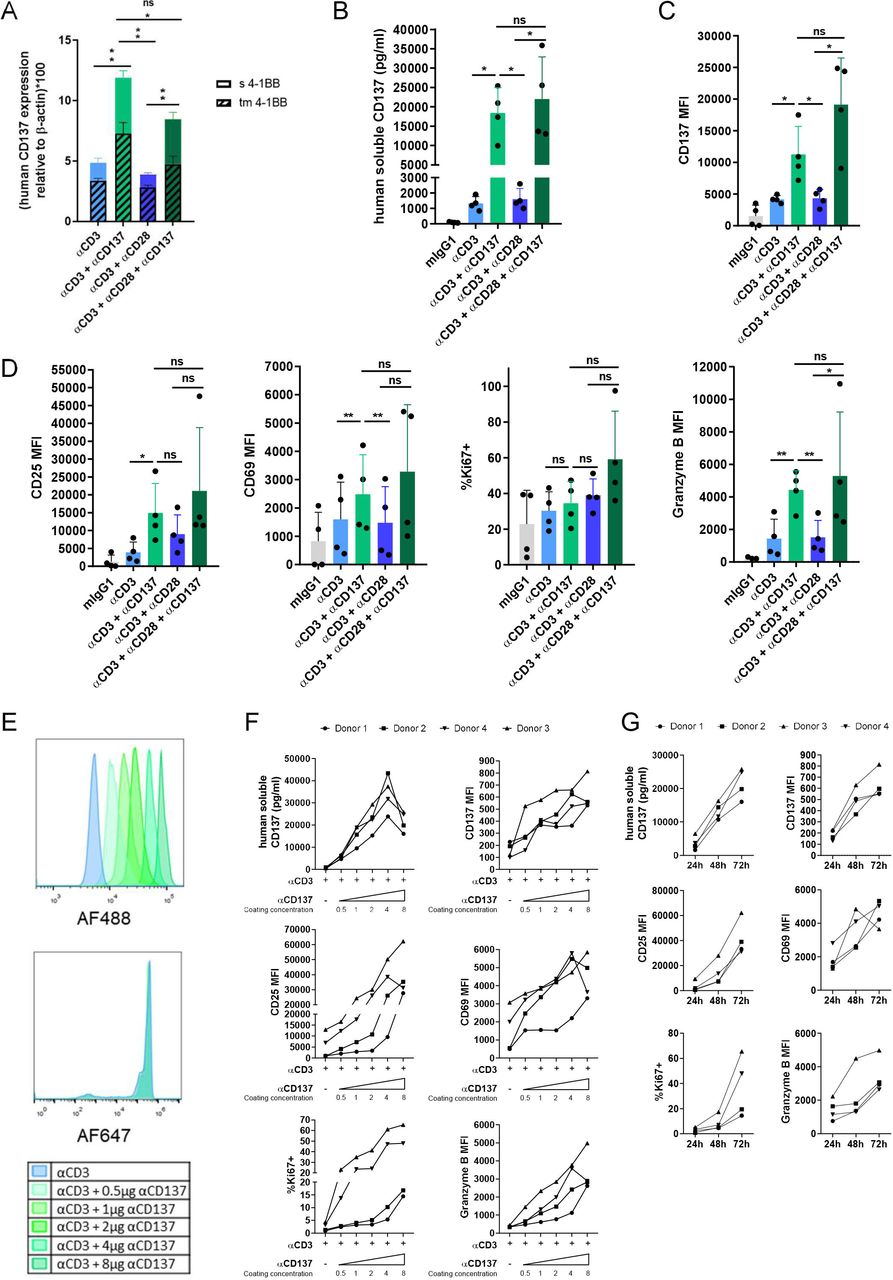
Soluble and transmembrane CD137 are upregulated by CD137 costimulation. (A) Quantitative RT-PCR measurements of mRNAs encoding transmembrane and sCD137 in isolated human CD8 T cells from three different donors stimulated by anti-CD3ε mAb bound to the culture plate in conjunction or not with anti-CD137 or anti-CD28 as indicated. (B) Measurement of the sCD137 concentration in the 48 hours tissue culture supernatants. (C) Surface expression levels of CD137 on CD8 T lymphocytes stimulated as in part A. (D) Surface expression levels of CD25 and CD69 as well as intracellular staining levels of Ki67 and Granzyme B in experiments as in part B. (E) Microbeads were covalently cocoated with fixed concentrations of anti-CD3ε tagged with AlexaFluor647 and serially increasing concentrations of anti-CD137 mAb tagged with AlexaFluor488 (0.5–8 µg of mAb per 50 µL of beads). Flow cytometry was used to show levels of microbead coating by relative fluorescence intensity units. F shows stimulation of CD8 T cells isolated from four healthy donors cocultured with the microbeads as indicated measuring sCD137 in culture supernatants or the intensity of surface expression of CD137, as well as other markers of T cell activation as indicated. Results were assessed at 72 hours of culture. G shows a time-course of these parameters in the cultures using the beads coated with the highest content of anti-CD137. Bars indicate mean±SEM. Experiments were performed with four independent donors. Statistical significance was assessed by Student’s paired t-test in parts A, B, C and D, and Wilcoxon test in part D (CD25 MFI). *p<0.05, **p<0.01. MFI, mean fluorescence intensity; ns, not significant.
To quantitatively assess these phenomena, we covalently coated microbeads with a fixed amount of anti-CD3ε and graded amounts of anti-CD137 mAb that were fluorescently labeled in a manner that Flow Cytometry permitted to estimate the gradual coating of the beads (figure 1E).37 Using such microbeads as stimulators for human CD8 T cells, we could observe a dose (figure 1F) and time dependent (figure 1G) induction of soluble and membrane bound CD137 in addition to other activation markers such as CD25, CD69 Ki67 and granzyme B. These results are further indicative of the value of CD137 to quantitatively estimate the intensity of CD137 costimulation.
A close costimulatory relative of CD137 in the TNFR family is OX40 (CD134, TNFRSF4).41 42 Interestingly, costimulation of human CD8 T cells with an agonist anti-OX40 mAb showed increased release of sCD137 into the culture supernatant and increased levels of membrane immunostaining for CD137, indicating that CD137 upregulation is a function shared by other members of this TNFR costimulatory subfamily (online supplemental figure 1A,B). Again, side-by-side comparisons favored OX40 over CD28 for CD137 induction as well as to some extent for CD25 and Ki67 upregulation (figure 1A–D). Of note, recombinant IL-2 also readily enhanced CD137 expression both as a membrane bound and as a soluble moiety (online supplemental figure 1C,D). IL-2 also augmented the surface expression of CD25 and CD69 as expected (online supplemental figure 1D). Therefore, CD137 as a marker denotes activation of T cells by a variety of stimuli and not solely by CD137 agonists.
Similarly, mouse CD8 T cells derived from splenocytes were costimulated by plate-bound anti-CD137 agonist mAb to upregulate transcription of CD137 (figure 2A). In these conditions, more sCD137 was measured released into the culture supernatants (figure 2B) and CD137 increased on the surface of such costimulated T lymphocytes (figure 2C). In the case of mice, anti-CD28 mAbs also readily induced comparable levels of CD137 suggesting some species dissimilarities.10 Again, both CD28 and CD137 ligation gave rise to upregulation of CD25, CD69, Ki67 and PD1 expression (figure 2D).

CD137 transcription and protein expression are costimulated by CD137 on mouse CD8 T cells. (A) Mouse CD8 splenocytes were cultured with plate-bound anti-CD3ε mAb in conjunction or not with anti-CD28 or anti-CD137 mAbs and the levels of soluble and transmembrane-encoding mRNAs were measured by quantitative RT-PCR. (B) sCD137 concentrations in the tissue culture supernatants and (C) surface expression of CD137 on the corresponding T cells. (D) As a control of the cell costimulation, the immunostaining levels of CD25, CD69 as well as the % of positive cells for Ki67 and PD1 assessed by immunostaining and flow cytometry are provided. Bars indicate mean±SEM. Statistical significance was assessed by Student’s standard and ratio paired t-test in parts A, B, C, and D, as appropriate. *p<0.05. These experiments were repeated three times. mAb, monoclonal antibodiy; MFI, mean fluorescence intensity; ns, not significant.
sCD137 concentrations are raised in tumor-bearing mice undergoing immunotherapy with agonist anti-CD137 mAb
Agonist anti-CD137 is an efficacious immunotherapy against transplanted mouse tumors such as those derived from the MC38 and CT26 colon carcinoma cell lines.40 43 In the case of MC38-derived tumors, a three-dose regimen of intratumoral anti-CD137 mAb or control antibody starting on day +4 following tumor-cell inoculation was administered (figure 3A). Four out of 13 mice experienced complete tumor regression, while most other treated tumors were transiently delayed in their progression as compared with IgG control groups (figure 3B,C). Interestingly, in sequential plasma samples drawn from such mice, abundant levels of circulating sCD137 were measured over the course of treatment and these remained high (figure 3D). Interestingly, when correlating plasma sCD137 on day +9 and tumor size on day +20, an association was found between higher CD137 concentrations and smaller or cured tumors (figure 3E), suggesting some value of sCD137 as a predictive biomarker while early on treatment.

sCD137 increases in peripheral blood of tumor-bearing mice undergoing treatment with agonist anti-CD137 mAb. (A) Experimental treatment and blood sample collection in mice bearing MC38-derived tumors. (B) Tumor size follow-up and fractions of complete rejections attained in mice (n=13 per group) treated with anti-CD137 mAb or control antibody and the corresponding overall survival (C) are shown. (D) Plasma sCD137 concentrations (mean±SD) in sequential follow-up overtime measured by ELISA in the groups of mice treated as indicated. (E) Plotted statistical correlation of plasma sCD137 on day +9 since tumor cell inoculation and the size of the MC38-derived tumors on day +20. (F) Similar experimental design in Balb/c mice bearing syngeneic CT26 tumors. (G) Individual tumor size follow-up and fractions of mice that completely rejected their tumors. (H) Overall survival of such mice. (I) Plasma sCD137 concentrations (mean±SD) in sequential follow-up overtime of the mice treated in part F. Statistical significance was assessed by log-rank (Mantel-Cox) test in figure parts C and H, and two-way ANOVA in figure parts D and I. *p<0.05, **p<0.01, ****p<0.0001. ANOVA, analysis of variance; ns, not significant.
In the CT26 model (figure 3F) in Balb/c mice, efficacy was more marked with every mouse treated since day +6 completely rejecting their tumors (figure 3G,H). In this experimental setting, high concentrations of sCD137 were measured in plasma on day +9 that fell on day +12 when most tumors had already become undetectable (figure 3I).
Two mechanisms could account for increased sCD137 plasma concentrations in such mice, namely, increased production by T cells and retention in the circulation by the treating mAb. In support of this double mechanism, increases of sCD137 were detectable in mice treated with agonist anti-CD137 mAb, which were bearing tumors such as LLC and B16F10 (online supplemental figure 2A) considered to have a very low degree immunogenicity and which did not respond at all to the CD137-agonist therapy (online supplemental figure 2B). Furthermore, in tumor-free mice, it was possible to detect increases in circulating sCD137 although clearly lower than those observed in MC38 tumor-bearing mice (online supplemental figure 2C). Hence, both retention in blood and higher pharmacodynamic production by activated T lymphocytes account for such increased sCD137 circulating concentrations. Indeed, an ELISA analysis was set up to quantify total sCD137 and CD137 captured by the antibody directed to CD137 used as a therapeutic agent (online supplemental figure 3A). As can be seen in online supplemental figure 3B, both free and mAb-bound sCD137 could be detected in similar quantities. sCD137 concentrations may serve a number of functions such as a negative feed-back mechanism partially blocking CD137L, as previously reported.40 In keeping with this, sCD137 can also act as a decoy for the activity of anti-CD137 mAb. In this sense, we tested increasing quantities of recombinant human or mouse sCD137 to inhibit costimulation as provided by anti-CD137 mAb either coated in microbeads or plate bound. As can be seen in online supplemental figure 3C,D, such soluble CD137 form was indeed capable of neutralizing part of the anti-CD137 mAb costimulatory activity.
sCD137 increases in response to CD137 costimulation in human systems
To humanize the findings in mouse models, we implanted Matrigel plugs under the skin of Rag2−/− IL2Rγ−/− mice containing a suspension of human PBMC together with beads coated with anti-CD3ε mAb (OKT3) plus control antibody or agonist anti-CD137 mAb (6B4). This experimental system is free of circulating anti-CD137 mAbs that would be able to mediate capture and retention. In this experimental setting (figure 4A–C), we were able to detect a clear increase in human sCD137 in the plasma of the mice contingent on CD137 costimulation by the antibodycoated microbeads (figure 4D). In the excised and resolubilized Matrigel plugs, concentrations of human sCD137 were much higher and were clearly dependent on CD137 costimulation (figure 4E). In this regard, higher CD137 surface expression was also detectable on the CD8 T cells recovered from the Matrigel plugs. Online supplemental figure 4 shows this costimulation effect for CD137 surface expression, while CD25 levels were comparable with those without CD137 costimulation from the beads (online supplemental figure 3A). A comparable coating of the beads was verified by immunostaining and Flow Cytometry (online supplemental figure 4B).

sCD137 is produced by human T cells transiently engrafted in immunodeficient mice. (A) Experimental setting implanting subcutaneous Matrigel plugs under the skin of Rag2−/− IL2Rγ−/− mice and subsequent excision/resolubilization of the plugs as well as serial plasma collection. (B) Human PBMCs were mixed with microbeads coated with mIgG1, anti-CD3+mIgG1 or anti-CD3+anti-CD137. (C) H&E staining of sections of the Matrigel plugs showing the lymphocytes and microbeads indicated in the rectangle area that is shown under higher power magnification. (D) Concentrations of sCD137 in plasma from individual mice and in the resolubilized Matrigel plugs (E). Bars indicate mean±SEM. Statistical significance was assessed by two-way ANOVA in D and Welch’s test in part E. *p<0.05, **p<0.01. ANOVA, analysis of variance; ns, not significant.
To provide further proof of this concept of increased sCD137 production in vivo, we used human CD137 knock-in mice (Fan et al manuscript in preparation). Such mice were engrafted with MC38-derived syngeneic tumors and were treated as indicated in figure 5A with an intraperitoneal course of the anti-CD137 mAb Urelumab or control antibody. As can be seen in figure 5B, the treatment induced detectable circulating levels of human sCD137.
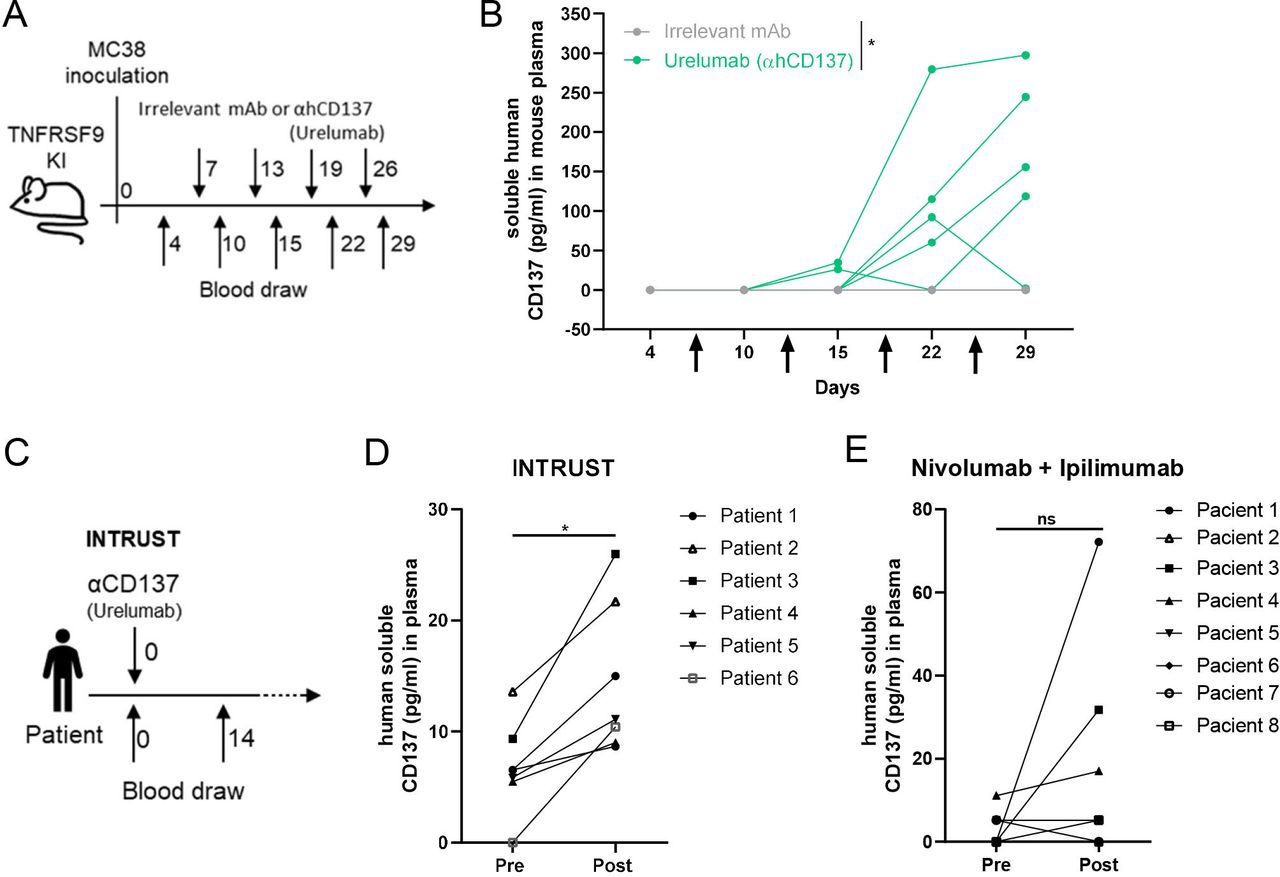
Circulating human sCD137 is induced both in hCD137 knock-in mice and in cancer patients treated with Urelumab. (A) Scheme of treatment and plasma collections from knock-in human CD137 mice in C57BL/6 background treated intratumorally with control mAb or Urelumab. (B) Sequential follow-up of circulating concentrations of human sCD137 in these mice intraperitoneally treated with Urelumab or control antibody. (C) Scheme of patients undergoing intratumoral injections on 8 mg of Urelumab inside accessible tumor lesions, as performed in the six first patients treated in the INTRUST clinical trial (NCT03792724). (D) sCD137 was determined in samples drawn on day +14 per protocol, showing increases over baseline of sCD137 that were measured by ELISA. (E) determinations sCD137 in the plasma of patients treated with ipilimumab+nivolumab determined baseline and on day +14 following treatment administration as assessed by luminex. Bars indicate mean±SEM. Statistical significance was assessed by two-way ANOVA in part B and Student’s paired t-test in parts D and E, as appropriate. *p<0.05. mAb, monoclonal antibody; ns, not significant.
Moreover, in our institution, we are running an investigator initiated clinical trial (NCT03792724; INTRUST) in which patients first received an intratumoral dose of 8 mg of Urelumab. In the first six consecutive patients, we were able to detect by ELISA a clear increase in sCD137 in their plasma in comparison with pretreatment samples (figure 5C,D). By contrast, in a series of sera from melanoma patients treated with nivolumab+ipilimumab drawn 2 weeks after treatment onset, only two out eight patients experienced an increase of sCD137 as detected by a Luminex assay (figure 5E).
CARs containing CD137 signaling sequences induce the release of sCD137 on antigen recognition
CARs in the clinic encompass CD137 signaling domains in conjunction with CD3ζ cytoplasmic sequences.44 The mesothelin recognizing second-generation CARs created by the group of Carl June have two versions: one encompassing the CD137 signaling tail, while the other has that of CD28 instead. Human isolated CD8 T cells were lentivirally transduced with such CARs and then transduced T cells were stimulated with microbeads coated with recombinant mesothelin (figure 6A). In this experimental setting, it was observed that the CD137-containing and the CD28-containing versions in the absence of antigen released some sCD137 into the culture supernatant, probably due to tonic signaling as previously described.45 On antigen stimulation, much larger quantities of sCD137 were released into the tissue culture supernatant 24 and 48 hours after stimulation (figure 6B). The measured concentrations were comparable in the CD8 T cells expressing either the CD137-containing CAR or in those expressing the CD28-containing CAR (figure 6B). Furthermore, comparable increased levels of surface CD137 and secretion of IFNγ to the supernatant were observed on ligation of the two CARs (figure 6C,D). The appropriate transduction of the CARs to activated human T cells (approximately 20%–40% expression) and the proper coating of the microbeads were checked by immunostaining and Flow Cytometry (online supplemental figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD137 encompassing CARs induce the release of sCD137 on activation. (A) Antimesothelin second-generation CARs encoding cytoplasmic sequences of either CD137 or CD28 were lentivirally transduced to activated human CD8 T lymphocytes. Such resulting T cell cultures could be activated by microbeads decorated with chemically linked recombinant mesothelin-Fc. (B) sCD137 concentration in the culture of the corresponding CAR T cells stimulated or not with mesothelin-coated microbeads 24 and 48 hours following of exposure to the beads. (C) CD137 surface expression in the corresponding lymphocyte cultures. (D) IFNγ concentrations in the supernatants. Individual results from three or four independent donors whose individual values are linked by dotted lines are shown and the color-coded bars indicate the value of the means. CAR, chimeric antigen receptor.
According to these results, sCD137 and induction of surface CD137 can be proposed as dynamic markers for recent CAR T cell engagement with antigen including those encompassing the intracellular CD137 signaling domain.
Discussion
CD137 is commonly used in the laboratory as a marker of recent or ongoing T cell activation for a number of purposes. For example, it is used to select those tumor-infiltrating T lymphocytes (TILs) that recognize antigen.46–48 Once CD137 expression is induced, CD137 agonist antibodies in the TIL cultures enhance expression and protect T cells from apoptosis, giving rise to higher culture yields.49 50
In cancer-bearing mice and humans, agonist CD137-targeted agents aim to costimulate T cells as a result of crosslinking this surface receptor and thereby enforcing signaling.51 One of the most interesting features of this immunotherapy approach is that the engaged T cells would be precisely those involved in ongoing immune responses.46
With the advent of novel CD137 agonists and CD137-based CARs and their increasing use in clinical settings, there is a clear need to identify and validate suitable biomarkers that would show whether CD137 has been functionally engaged.52 Such pharmacodynamic biomarkers that can be sequentially monitored would be of use in that they would permit adjustments and refinements in doses and schedules. In addition, they will conceivably show early on-treatment who are the patients likely to benefit from such immunotherapies.
Our findings indicate that expression of CD137 itself is a prominent target of CD137 costimulation in culture and in vivo. Very conveniently, CD137 has soluble forms as the result of alternative splicing53 54 and shedding from the lymphocyte surface.55 Hence, sCD137 becomes detectable and measurable in the circulation56 as previously reported in autoimmune diseases.57 In that regard, the 16 kDa molecular weight of sCD137 is below the kidney filtration threshold and this suggests fast elimination kinetics. However, if a CD137 binding agent is in the circulation, this would result in sCD137 retention in plasma, making sCD137 kinetics easier to detect. It is of interest that in conditions with limiting or none circulating anti-CD137 mAb, such as the beads in the Matrigel plugs or the intraperitoneal injection in knock-in mice, we still found increases in circulating sCD137, thus showing the relevance of enhanced biosynthesis. It remains to be studied whether sCD137 is released from T cells in the tumor microenvironment or tumor-draining lymph nodes and how it gains access to the circulation. In our experimentation, we found that even in tumor-free mice sCD137 concentrations raised in plasma on treatment with agonist CD137 mAb, and it remains to be seen what is the T cell source of sCD137 in such conditions. In tumor-bearing mice circulating sCD137 increases were much more prominent and correlated early with eventual decreases of tumor sizes and tumor rejections.
Inducibility of CD137 transcription by CD137 costimulation does not come as a surprise given the prominent roles o NF-κB and AP1 consensus sequences in the tnfrsf9 promoter.58 The control of the alternative splicing forms of the molecule remains to be studied. Furthermore, part of the released sCD137 could be the result of shedding following metalloprotease cleavage on the plasma membrane.55 We are currently studying the relative contribution of both processes to the overall sCD137 quantities present in culture supernatants under different stimulation conditions.
Of note, it has been reported that once agonist agents interact with the receptor, CD137 becomes internalized to an endosomal compartment.38 This might complicate the use of surface CD137 as a biomarker due to this first step of internalization.
The function of sCD137 in vivo remains to be completely elucidated, but we have previously shown that it could bind to CD137L and thereby attenuate endogenous costimulation.40 It is important to consider that in the presence of pharmacological agonists based on antibodies or on CD137L such a function would be exceeded with much greater amounts of the artificial than by the natural CD137-agonist ligand. In this sense, we were able to ascertain that recombinant sCD137 could neutralize costimulatory anti-CD137 mAb in costimulation cultures. In the case of therapeutic agonist anti-CD137 antibodies, raising doses should bypass the problem of neutralization by sCD137. In addition, sCD137 could potentially be involved in eliciting CD137L reverse signaling59 with potential functional outcomes that would be different depending on whether the CD137 agonist permits or not the interaction with CD137L.
As a biomarker, sCD137 increases measured in plasma will be the consequence of biosynthesis, release and retention in the circulation. These three features are considered advantageous to monitor the activity of agonist agents on the CD137 target. Fluctuation in the levels of CD137 expression overtime are proposed to be relevant to track the activity of CD137 agonists, and these properties seem to be largely conserved between mice and humans. For instance, when tumors are rejected and antigens are cleared from the system, circulating sCD137 concentrations decrease.
The relevance of sCD137 as a biomarker can be potentially extended to other members of the TNFR family targeted for immunotherapy purposes such as OX40 (CD134). In the case of patients treated with intratumoral doses of urelumab (INTRUST clinical trial), the increases of sCD137 in plasma were remarkably observed in six out of six patients. However, the biomarker should not be considered specific since it was also induced in culture by recombinant IL-2 and in two out of eight patients with cancer on treatment with nivolumab+ipilimumab. Towards clinical development of the biomarker, it will also be important to consider assays that detect antibody-bound or antibody-free sCD137.
According to our results, increased sCD137 following CD137 costimulation quantitatively monitors the intensity of CD137 costimulation during treatment. This has been observed in tumor-bearing mice on anti-CD137 mAb treatment. This might also be useful in CAR T-cell adoptive therapy.60 61 Indeed, we have observed the increased sCD137 phenomenon in six out of six patients intratumorally given low doses of Urelumab, and increased levels of circulating sCD137 have been reported in the plasma of patients with cancer receiving systemic treatment with the anti-CD137 mAb Utomilumab27 or with the new CD137 agonists such as the bispecific construct FAP-4-1BBL62 and the PDL1-CD137 bispecific antibody GEN1046.63 In the case of CARs, sCD137 becomes a biomarker candidate to pharmacodynamically monitor both CD137 and CD28 encompassing CARs since both of them readily costimulated the expression of soluble and surface CD137.
All in all, sCD137 is a biomarker candidate linked to the biology of the target, and we propose that its measurement will have clinical utility using it in conjunction with other parameters. Our work provides the basis for the use of sCD137 and CD137 surface expression levels on T cells as surrogate pharmacodynamics biomarkers not only of antigen recognition but more importantly of CD137 costimulation as artificially provided by immunotherapy agents already in or approaching the clinic.30
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by IRB from the local government of Navarra. All experiments involving animals were approved by the Ethics Committee of Animal Experimentation at the University of Navarra (R-030-19GN). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We are grateful to D Alignani from the Flow Cytometry facility and to Eneko Elizalde and Elena Ciordia from the animal facility for their excellent work. Drs Hervas-Stubbs, Aranda and Berraondo for helpful discussions. English editing by Paul Miller and project managing by E Guirado and B Palencia are also acknowledged.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @SoniaGuedanPhD
JG-V and AA contributed equally.
Correction notice This article has been amended since it was first published online. Miguel F Sanmamed was incorrectly listed as Miguel F Sanamed.
Contributors JG-V performed experiments analyzed results and wrote the manuscript. AA, IO, AC, AT, MCO, MA, IE-S, CL-R and MER-R performed experiments, analyzed results and edited final versions of the manuscript. XN, SG and LC provided experimental models and key reagents and edited the manuscript. MFS and JLPG provided samples from the intrust clinical trial and edited the manuscript. IM analyzed and takes responsibility for the integrity of the results, attained funding, and wrote the manuscript.
Funding This project has been supported by PID2020-112892RB funded by MICIN/AEI /10.13039/501100011033 and SAF2017-83267-C2-1-R funded by MICIN/AEI /10.13039/501100011033/ and by FEDER 'Una manera de hacer Europa' to IM. This project has also received funding from the European Union Horizon 2020 research and innovation program (grant agreement n° 635122 - PROCROP), Fundación de la Asociación Española Contra el Cáncer (AECC) (HR21-00083), Fundación La Caixa and Fundación BBVA.
Competing interests IM acknowledges grants from Roche, Alligator, Genmab, BMS, AstraZeneca, Pharmamar and Bioncotech, as well as consultancy fees from BMS, Roche, Genmab, Numab, Gossamer, Alligator, AstraZeneca and Pharmamar. MFS receives a grant from Roche.
Patient and public involvement statement Provided in the informed consent to INTRUST trial.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.