Article Text

Abstract
Background The combination of antiangiogenic agents with immune checkpoint inhibitors could potentially overcome immune suppression driven by tumor angiogenesis. We report results from a phase IB study of ziv-aflibercept plus pembrolizumab in patients with advanced solid tumors.
Methods This is a multicenter phase IB dose-escalation study of the combination of ziv-aflibercept (at 2–4 mg/kg) plus pembrolizumab (at 2 mg/kg) administered intravenously every 2 weeks with expansion cohorts in programmed cell death protein 1 (PD-1)/programmed death-ligand 1(PD-L1)-naïve melanoma, renal cell carcinoma (RCC), microsatellite stable colorectal cancer (CRC), and ovarian cancer. The primary objective was to determine maximum tolerated dose (MTD) and recommended dose of the combination. Secondary endpoints included overall response rate (ORR) and overall survival (OS). Exploratory objectives included correlation of clinical efficacy with tumor and peripheral immune population densities.
Results Overall, 33 patients were enrolled during dose escalation (n=3) and dose expansion (n=30). No dose-limiting toxicities were reported in the initial dose level. Ziv-aflibercept 4 mg/kg plus pembrolizumab 2 mg/kg every 2 weeks was established as the MTD. Grade ≥3 adverse events occurred in 19/33 patients (58%), the most common being hypertension (36%) and proteinuria (18%). ORR in the dose-expansion cohort was 16.7% (5/30, 90% CI 7% to 32%). Complete responses occurred in melanoma (n=2); partial responses occurred in RCC (n=1), mesothelioma (n=1), and melanoma (n=1). Median OS was as follows: melanoma, not reached (NR); RCC, 15.7 months (90% CI 2.5 to 15.7); CRC, 3.3 months (90% CI 0.6 to 3.4); ovarian, 12.5 months (90% CI 3.8 to 13.6); other solid tumors, NR. Activated tumor-infiltrating CD8 T cells at baseline (CD8+PD1+), high CD40L expression, and increased peripheral memory CD8 T cells correlated with clinical response.
Conclusion The combination of ziv-aflibercept and pembrolizumab demonstrated an acceptable safety profile with antitumor activity in solid tumors. The combination is currently being studied in sarcoma and anti-PD-1-resistant melanoma.
Trial registration number NCT02298959.
- immunotherapy
- clinical trials as topic
- programmed cell death 1 receptor
- drug therapy
- combination
- tumor microenvironment
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- clinical trials as topic
- programmed cell death 1 receptor
- drug therapy
- combination
- tumor microenvironment
Introduction
There is increasing evidence to support the role that angiogenic factors play in affecting immune regulation and immune effector cell trafficking into tumors.1 Angiogenic factors such as the vascular endothelial growth factor (VEGF) family are known drivers of immune suppression that directly inhibit antigen-presenting cells while augmenting the effects of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) to induce T cell exhaustion.2–6 We have previously shown that upregulated pretreatment soluble VEGF is a predictive biomarker for lack of clinical benefit to ipilimumab.7 We have also demonstrated that the combination of ipilimumab and bevacizumab (a monoclonal antibody against VEGF-A) in metastatic melanoma yields favorable response rates along with increased T cell infiltration.8 Given that immune suppression driven by tumor angiogenesis can lead to both primary and secondary resistance to immune checkpoint inhibitors (ICIs),9 the combination of angiogenesis inhibition with checkpoint blockade represents a promising strategy.
Ziv-aflibercept is a recombinant fusion protein consisting of extracellular VEGF-binding domains from human VEGF receptor VEGFR-1 and VEGFR-2 fused to the Fc domain of human IgG1. It functions as a soluble decoy receptor that ‘traps’ endogenous VEGF, thus inhibiting neovascularization and suppressing tumor growth.10 Unlike other monoclonal antibodies to VEGF, ziv-aflibercept features potential pharmacological advantages including a 100-fold increased binding affinity to VEGF-A compared with bevacizumab10 11 as well as binding to VEGF-B and placental growth factor, which have been implicated in cancer resistance to bevacizumab.12–14 VEGF blockade with aflibercept not only reduced tumor vascularization in mouse glioblastoma but also promoted dendritic cell (DC) maturation and reduced CD8 T cell exhaustion markers.15 The combination of ziv-aflibercept with CTLA-4 blockade demonstrated enhanced antitumor efficacy in two mouse colon cancer models.16 Furthermore, the combination of aflibercept, antiangiopoietin 2, and antiprogrammed cell death 1 (PD-1) blockade significantly improved survival in a mouse model of glioblastoma, showing that antiangiogenesis can overcome checkpoint blockade resistance in a traditionally non-T cell-inflamed cancer.17
Pembrolizumab, a humanized IgG4 monoclonal antibody that binds to PD-1 receptor, has been shown to effectively produce significant antitumor immune responses in advanced melanoma and renal cell carcinoma (RCC)18 19 but plays less of a role in tumors with low immune infiltrates such as colorectal cancer (CRC) and ovarian cancer.20 In contrast, antiangiogenesis agents have demonstrated activity in both CRC and ovarian cancer.21 22 We hypothesized that a novel combination of ziv-aflibercept and anti-PD-1 would be tolerable and lead to clinical benefits in both tumors that are and are not traditionally responsive to ICIs.
In this phase IB clinical trial, we assessed the safety and efficacy of ziv-aflibercept in combination with pembrolizumab. The primary objective was to determine the safety and the recommended phase II dose (RP2D) in patients with advanced solid tumors. Secondary objectives included preliminary estimates of response rate, progression and overall survival (OS) outcomes, and correlative analyses to elucidate the mechanism of action of this combination treatment.
Patients and methods
Study design and treatment
This was a phase IB, multicenter, open-label, dose-escalation, and dose-expansion study investigating the efficacy and safety of the combination of ziv-aflibercept and pembrolizumab in patients with advanced solid tumors. This trial was sponsored by the Cancer Therapy Evaluation Program at the National Cancer Institute (NCI) and conducted at Dana-Farber Cancer Center (Boston, Massachusetts, USA), University Health Network–Princess Margaret Cancer Center (Toronto, Ontario, Canada), Moffit Cancer Center (Tampa, Florida, USA), and BC Cancer Agency–Vancouver Cancer Center (Vancouver, British Columbia, Canada).
The study was composed of a 3+3 dose-escalation design in patients with metastatic solid tumors (cohort 1) and dose expansion in PD-1 inhibitor-naïve metastatic melanoma, RCC, microsatellite stable (MSS) colon cancer and ovarian cancer (cohort 2). The dose-escalation phase in cohort 1 was open to enrollment for patients with any metastatic solid tumor fitting the eligibility criteria described further and included one patient with clear cell sarcoma, one patient with triple negative breast cancer (TNBC), and one patient with mesothelioma. Dose-expansion cohorts consisted of six patients per cancer type in addition to patients treated at the RP2D (figure 1). The study was amended in 2018 for additional dose-expansion cohorts (part 2) to include patients with PD-1 resistant melanoma, PD-1-resistant RCC, and sarcoma, and is still open to enrollment. Here we report on the first part of the study. The treatment regimen contained pembrolizumab 2 mg/kg in combination with ziv-aflibercept 2 mg/kg (dose level 1 (DL1)) or 4 mg/kg (dose level 2 (DL2)) every 2 weeks administered intravenously.

Study schema for dose escalation and dose expansion of pembrolizumab and ziv-aflibercept. Cohort 1 included the following tumors: clear cell sarcoma, TNBC, and mesothelioma. Other solid tumors in Cohort two were: epithelioid mesothelioma (2) and TNBC (1). TNBC, triple-negative breast cancer.
Patients
Key eligibility criteria included the following: patients with RCC must have had at least one prior line of treatment with a VEGF tyrosine kinase inhibitor. Patients with ovarian cancer must have developed resistance to platinum therapy within 6 months of treatment initiation and had no more than two prior platinum containing regimens. Patients with CRC must have progressed on at least one oxaliplatin-containing regimen. Other key eligibility criteria for all patients included Eastern Cooperative Oncology Group performance status of 0 or 1; ≤2 prior lines of systemic therapies for metastatic disease; expected life expectancy of >6 months; adequate hepatic, renal, and marrow function; measurable disease based on Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1; and available archival or newly obtained tumor biopsy sample.
Key exclusion criteria included the following: diagnosis of immunodeficiency; use of immunosuppressive therapy within 7 days; use of monoclonal antibody therapy within 4 weeks; additional progressing or actively treated malignancy; high risk of gastrointestinal or pulmonary bleeding; ulcerated skin lesions; active anticoagulation therapy with warfarin; blood pressure of >150/100; known progressing brain metastases; active autoimmune disease requiring systemic treatment; history of peptic ulcer disease, erosive esophagitis or gastritis, inflammatory bowel disease, diverticulitis, pulmonary embolism, or uncontrolled thromboembolic event within 3 months; and prior therapy with an anti-PD-1, anti-PD-L1, anti-PD-L2, or anti-CD137 agent, or ziv-aflibercept.
Safety and dose-limiting toxicity (DLT) assessment
The NCI Common Terminology Criteria for Adverse Events V.5.0 was used to assess adverse events (AEs). Patients were classified as having a DLT for any of the following occurring within the first 4 weeks of the study therapy: unexpected grade of ≥3 AE likely attributable to the study drugs, grade ≥3 AE that does not improve with or without intervention within 7 days of onset, eye pain of grade ≥2, grade 3 hypertension that does not improve with appropriate medical intervention within 14 days, urine protein to creatinine ratio of >3.5 g or >2 g protein on 24-hour urine collection, two delays of treatment (unrelated to scheduling non-adherence) each lasting >10 days within four cycles of the treatment, or arterial thromboembolic event.
Clinical assessments
All patients received a clinical assessment at baseline with CT of the chest, abdomen, and pelvis, and MRI of the head <35 days prior to start of therapy. Radiographic assessments were repeated every 12 weeks, and confirmatory scans were obtained 6–8 weeks following initial documentation of an objective response. Tumors were evaluated based on RECIST V.1.1.23
Statistical analysis
The primary objective was to determine the maximum tolerated dose (MTD) and recommended dose combination for ziv-aflibercept plus pembrolizumab. The secondary endpoints included overall response rate (ORR), progression-free survival (PFS), and OS.
For the expansion cohort (cohort 2), ORR was estimated and summarized with 90% CIs using exact binomial methods. Time-to-event endpoints (PFS and OS) were summarized using the product-limit method of Kaplan-Meier; 90% CIs were based on log (−log (outcome)) methodology. Six-month PFS and 12-month OS were summarized with 90% CIs.
For correlative analyses, pretreatment Luminex measurements were compared according to response or disease control defined as complete response (CR), partial response (PR), or stable disease (SD) using Wilcoxon rank-sum tests. SD was defined using the RECIST method and was not associated with any duration. Longitudinal models, to assess relationships over time, were fit to the log2 transform of marker expression; predictors in each model were response or disease control, time (Pre, time 1, time 2), and their interaction. Each model was fit using a linear mixed model with an autoregressive covariance structure. Estimates of differences according to response or disease control, or of changes over time were made using contrasts. Statistical analyses were performed using SAS V.9.4.
Analysis of biomarkers and correlatives
Correlative analysis was performed on available peripheral blood mononuclear cell (PBMC) and plasma samples from 23 patients and tumors from 27 patients.
For cytokine analysis, serum samples from 23 patients were available from pretreatment, 1 month, and 2 months after the start of the study. A panel of 29 cytokines and chemokines, including VEGF, interleukin (IL)-1Rα, IL-6, IL-7, IL-8, IL-10, CD40L, CCL4, CXCL5, CXCL10, and TNF-a was analyzed using the Bio-Techne Luminex Assay Kit (Bio-Techne, Minneapolis, Minnesota).
Flow cytometry was performed on PBMC samples at three time points (pre-treatment, 1 month, and 2 months) to investigate immunological changes during treatment. Four panels were applied to study T cells, B cells, DCs, monocytes, natural killer cells, memory T cells, Tregs, monocytic MDSCs, and checkpoint and activation markers. Additional methods on flow cytometry procedures may be found in the online supplemental material.
Supplemental material
Tissue samples were analyzed through immunohistochemistry (IHC) and multiplex immunofluorescence (mIF). Tumor specimens were collected from metastatic deposits of solid tumors or pretreatment archived specimens if no fresh tumor could be obtained prior to study treatment initiation. IHC was used to analyze vessel content and morphology through staining with anti-CD31 antibody. Vessel density was categorized by two pathologists as ‘low,’ ‘moderate’, or ‘high,’ while endothelial phenotype was assessed as ‘skinny’ (non-activated) or ‘plump,’ as described previously.8 Tumor and tumor–stroma interface was analyzed through an mIF panel of five markers: SOX10 (tumor marker), PD-1 (exhausted T cell marker), CD31 (endothelial marker), CD8 (cytotoxic T cells), and PD-L1 (tumor cells and immune cells). Analysis of expression of markers was assessed using inForm V.2.2 image analysis software (PerkinElmer, Waltham, Massachusetts, USA). Three to five regions were selected by two pathologists and were annotated. Additional methods on IHC and mIF procedures may be found in the online supplemental material.
Results
Patients
In both cohorts of this study, patients who received at least one dose of ziv-aflibercept and pembrolizumab were considered evaluable for the safety analysis. Between April 2015 and March 2017, 33 patients were enrolled at four centers in the USA and Canada for dose escalation and dose expansion as highlighted in figure 1. Median follow-up was 8.2 months.
Patient demographics are summarized in table 1. Out of 33 patients enrolled in this study, 6 patients had melanoma (18%); 8 patients had ovarian cancer (27%); 7 patients had RCC (21%); and 6 patients had CRC (18%). Six patients had other solid tumors (18%); three patients in cohort 1 had clear cell sarcoma, TNBC, or mesothelioma, and three in cohort 2 had epithelioid mesothelioma (n=2) and TNBC (n=1).
Baseline patient characteristics overall and by cohort
The median age was 64 (range: 36–79) years and 58% of patients were male. All patients had prior surgery for their tumors; most patients (26/33, 79%) had at least one previous line of chemotherapy; 12 patients (36%) received prior antiangiogenic agents; 11 patients (33%) had prior radiation therapy; 5 patients (15%) had targeted therapy; and 2 patients (6%) had prior immunotherapy (one patient with melanoma and one patient with ovarian cancer in cohort 2 had ipilimumab monotherapy). Prior lines of therapy for each tumor type are shown in online supplemental table 1. At the time of data cut-off (January 21, 2021), 33 patients (100%) had discontinued treatment. Twenty-one patients (64%) discontinued treatment due to disease progression on study and 8 patients (24%) discontinued due to treatment-related adverse events (TRAEs). Two patients (6%) refused further treatment; one patient (3%) died on study; and one patient (3%) completed the follow-up period.
Safety
No DLTs were reported in the dose-escalation part of the study, which included three patients in cohort 1 who received 2 mg/kg of pembrolizumab plus 2 mg/kg of ziv-aflibercept every 2 weeks (DL1) and three patients in cohort 2 who received 2 mg/kg of pembrolizumab plus ziv-aflibercept at 4 mg/kg (DL2), which was established as the MTD and recommended dose in expansion cohorts. TRAEs of any grade were reported in all 33 patients and are summarized in table 2.
Summary of treatment-related AEs overall and by dose level (any-grade frequency ≥10%, grade 3 frequency of ≥5%, and all grade 4/5)
The most common TRAEs of any grade across all cohorts were proteinuria (20/33, 61%), hypertension (18/33, 55%), diarrhea (13/33, 39%), fatigue (10/33, 30%), and hoarseness (9/33, 27%). Grade ≥3 TRAEs were reported in 19/33 patients (58%), with the most common being hypertension (12/33, 36%) and proteinuria (6/33, 18%). In total, three deaths occurred in the study, of which one was deemed treatment-related due to cardiac arrest. This patient was a 70-year-old white man with baseline hypertension and CRC. After undergoing one course of study therapy on DL2, he experienced grade 1 vomiting and diarrhea 10 days after treatment. He additionally developed six new AEs that were possibly related to treatment 14 days after treatment including limb edema (grade 1), thrombocytopenia (grade 1), proteinuria (grade 2), weight gain (grade 1), lymphocytopenia (grade 1), and dehydration (grade 1). Finally, 18 days after undergoing study treatment, he developed cardiac arrest considered to be possibly related to therapy.
Efficacy
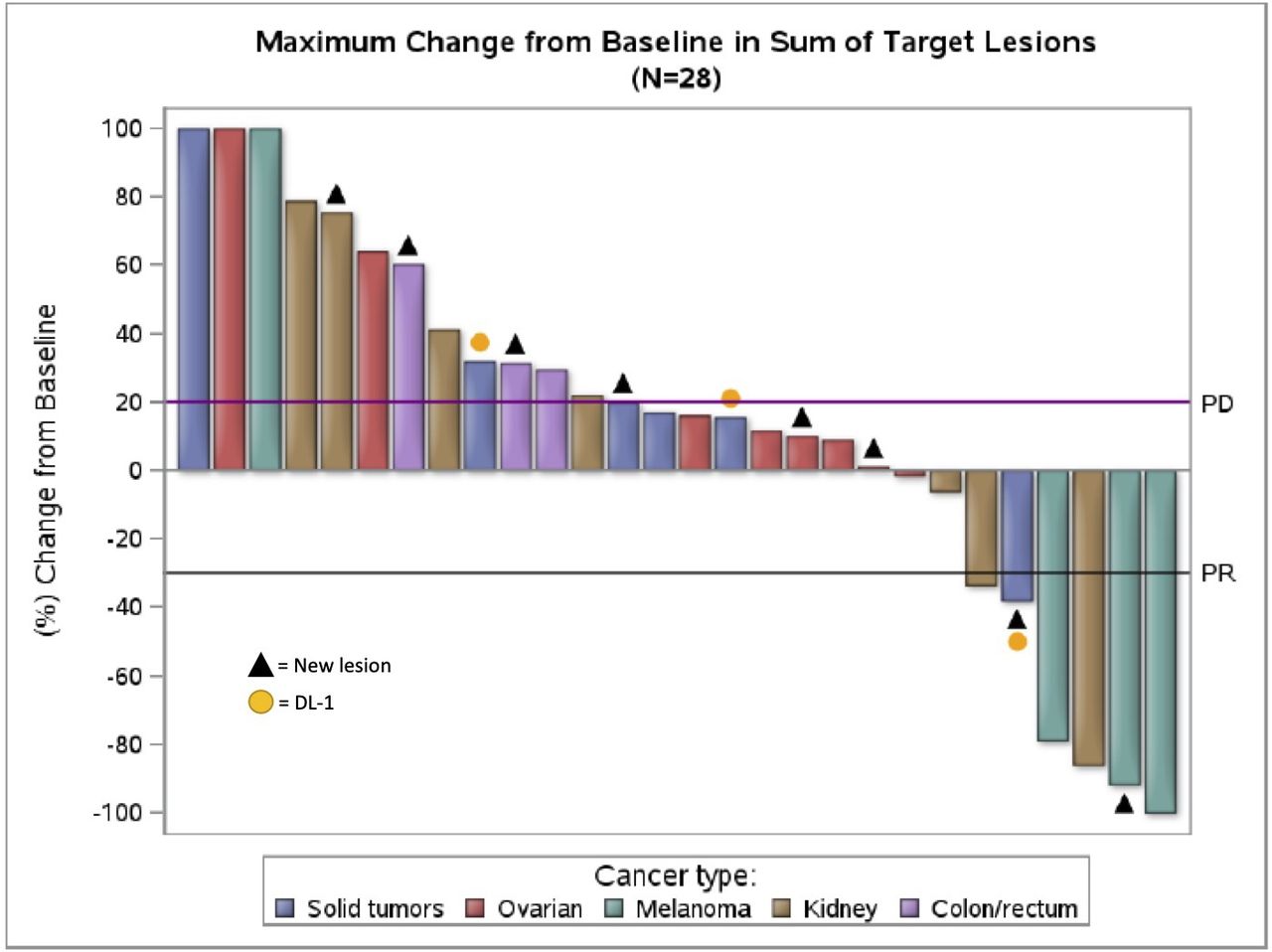
Efficacy outcomes by dose level and tumor type are summarized in table 3. A waterfall plot of RECIST responses in 28 patients with tumor measurements over time is shown in figure 2; five patients are not shown due to lack of follow-up scans. The response rate in 30 patients treated at DL2 was 16.7% (5/30, 90% CI 7% to 32%). Of the five patients who responded, two patients with melanoma had CR; one patient with melanoma had PR; one patient with RCC had PR; and one patient with mesothelioma had PR. The disease-control rate (DCR) (best overall response of CR, PR, or SD) was 40% (12/30, 90% CI 25% to 57%).

Waterfall plot of best Response Evaluation Criteria in Solid Tumors response. Waterfall plot of maximum change from baseline in sum of target lesions for 28 patients with tumor measurements over time. Plot is color-coded by tumor type. Triangles indicate patients who developed new lesions; yellow circles indicate the three patients who received DL1. Five patients from this study were not included due to lack of follow-up scans: one patient withdrew consent; one patient died on study; and three patients experienced treatment-related adverse events and withdrew. DL1, dose level 1.
Efficacy outcomes by dose level and tumor type
Response rates (CR and PR) by tumor type were as follows: 50% in melanoma (3/6, 90% CI 15% to 85%), 14% in RCC (1/7, 90% CI 1% to 52%), 17% in other solid tumors (1/6, 90% CI 1% to 48%), 0% in CRC (0/6, 90% CI 0% to 39%), and 0% in ovarian cancer (0/8, 90% CI 0% to 31%).
Median PFS was 2.6 months (90% CI 1.0 to not reached (NR)) for patients receiving DL1 and 3.3 months (90% CI 2.7 to 5.7) for patients in receiving DL2. OS was NR for patients receiving DL1 and 13.6 months (90% CI 8.7 to NR) for patients receiving DL2.
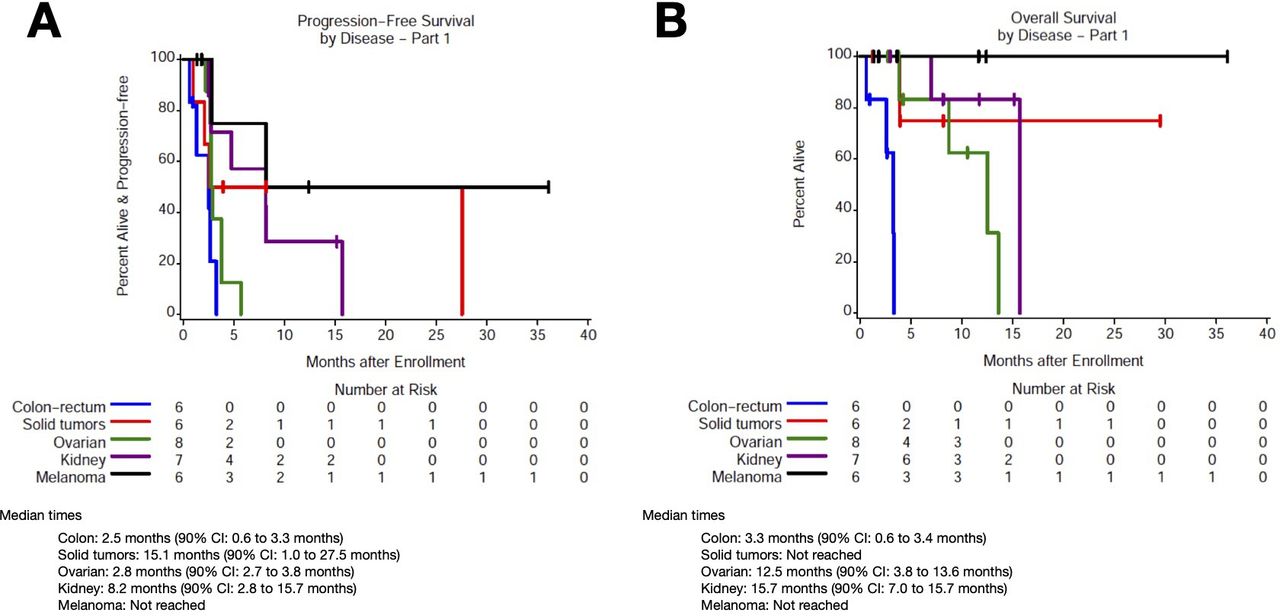
Among patients with CRC (n=6, cohort 2), median PFS was 2.5 months (90% CI 0.6 to 3.3) and median OS was 3.3 months (90% CI 0.6 to 3.4; figure 3A,B). Among patients with RCC (n=7, cohort 2), median PFS was 8.1 months (90% CI 2.5 to 15.7) and median OS was 15.7 months (90% CI 7.0 to 15.7). Among patients with melanoma (n=6, cohort 2), median PFS and OS were NR. Among patients with ovarian cancer (n=8), median PFS was 2.8 months (90% CI 2.7 to 3.8) and median OS was 12.5 months (90% CI 3.8 to 13.6). Among patients with other solid tumors (n=6; 3 in cohort 1, 3 in cohort 2), median PFS was 15.1 months (90% CI 1.0 to 27.5) and median OS was NR.
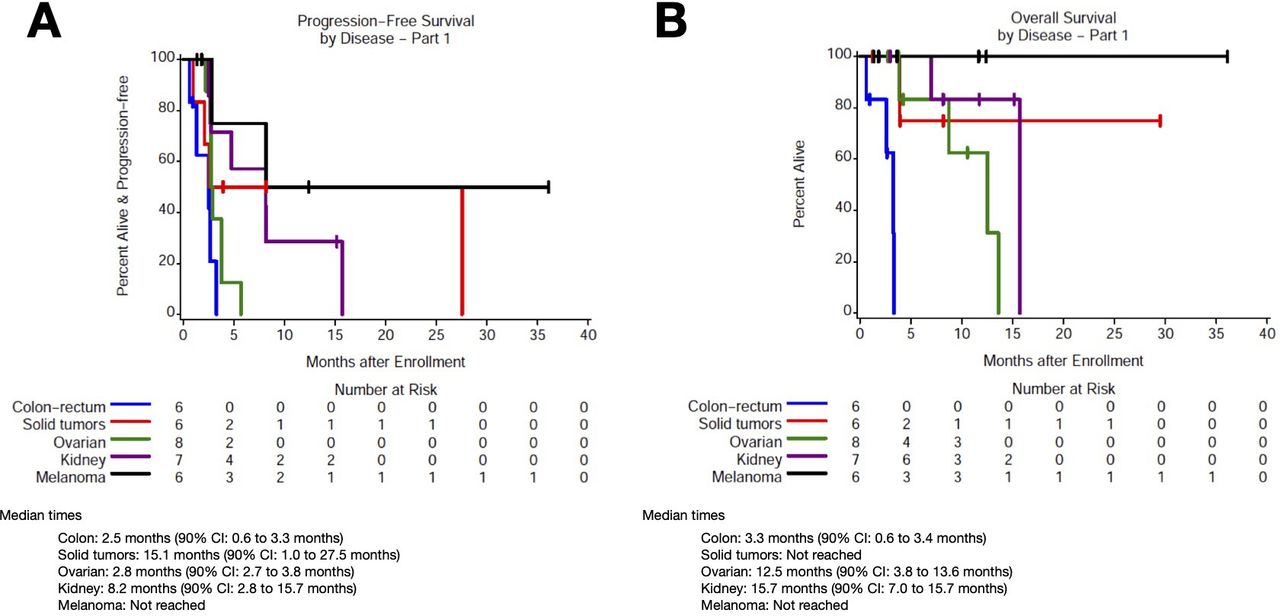
Progression and OS based on tumor type. Kaplan-Meier curves for (A) progression-free survival and (B) OS based on tumor type. OS, overall survival.
There was no correlation between grade≥3 TRAEs and treatment response. In addition, 4/18 patients with treatment-related hypertension responded to therapy compared with 1/15 patients without hypertension. Overall, treatment-related hypertension (any grade) had non-significantly increased odds of response compared with patients who did not report hypertension (OR 4.2, 90% CI 0.7 to 25.9, p=0.19). Similarly, 5/20 patients with treatment-related proteinuria had response compared with 0/13 patients without proteinuria, but overall proteinuria was not statistically significantly associated with treatment response (OR 6.1, 90% CI 0.5 to 82.0, p=0.25).
Association between tumor pathology and clinical response
Tumor biopsy samples were available from 27 patients, of which 18 patients had pre-treatment samples only, and 9 patients had both pre-treatment and post-treatment samples. Based on anti-CD31 staining analysis of vessel density, 16/27 (59%) had high vascularity, 7/27 (26%) had moderate vascularity, and 4/27 (15%) had low vascularity. In addition, 19 patients had a plump (activated endothelium) phenotype and 8 patients had a skinny (non-activated) phenotype.
Of the two responders (one melanoma with CR and one RCC with PR) included in the pretreatment pathology analysis, one had high baseline vascularity and the other had low vascularity. Of the eight patients with disease control (CR, PR, or SD), four patients had high baseline vascularity; two had moderate vascularity; and two had low vascularity. In addition, both responders had a plump phenotype at baseline, while the eight patients with disease control were evenly divided between plump (n=4) and skinny phenotypes (n=4). There were no statistical relationships between baseline vascularization and response (p=0.67) or disease control (p=0.99). There were also no statistical relationships between baseline vascularization phenotype and response (p=0.47) or disease control (p=0.47).
In addition, when comparing vascularity in nine tumor samples before and after treatment, one patient changed from high to moderate vascularity; and one patient changed from low to moderate vascularity, while seven patients remained the same. Three patients changed phenotype from plump to skinny, and three patients changed from skinny to plump as a result of treatment, while three patients remained the same phenotype.
Correlation between tissue biomarkers and clinical response
mIF was performed for 22 patients, of which 21 patients had pretreatment samples, 5 patients had both pretreatment and post-treatment samples, and 1 patient had only post-treatment samples. Of the 21 patients with pretreatment data, 2 had PR (1 melanoma and 1 RCC) and 5 had SD (2 RCC, 1 ovarian, 1 TNBC, and 1 mesothelioma) as best response to treatment. A representative mIF analysis with CD8, PD-1, PD-L1, CD31, and SOX10 staining for a patient with melanoma is shown in online supplemental figure 1.
Responders (CR and PR) had statistically significantly higher levels of pretreatment CD8+/PD1+ in the tumor than non-responders (mean 83.5 vs 21.2 cells/mm2, p=0.02). There were no other statistically significant differences noted for response. Similarly, there were no statistically significant relationships between mIF biomarker expression and disease control.
Association between cytokine changes and clinical response
A Luminex panel of 29 cytokines and chemokines was performed to compare differences in inflammatory mediators between patients with disease response (‘responders’, defined as CR or PR) or non-responders, as well as between patients with disease control (defined as CR, PR, or SD) or no disease control. Serum samples were available from 23 patients, of which 2 had CR, 3 had PR, 7 had SD, and 10 had PD, and 1 was not assessable due to mortality and was included in the non-responder and no DCR analysis.
Pretreatment cytokine measurements were compared between 5 responders and 18 non-responders, as well as between 12 patients with disease control and 11 with no disease control. Notably, responders had significantly lower pretreatment levels of IL-6 compared with non-responders (mean 3.2 vs 16.1 pg/mL, p=0.009), as well as a trend towards higher pretreatment levels of CD40L (3033.5 vs 2462.8 pg/mL, p=0.06; online supplemental figure 2A). There were also significant pretreatment cytokine level differences between patients with disease control versus no disease control. Patients with disease control had significantly higher baseline levels of CXCL2 (1679.0 vs 903.1 pg/mL, p=0.006), CXCL5 (574.1 vs 317.2 pg/mL, p=0.02), and CD40L (3404.8 vs 1694.6 pg/mL, p=0.001; online supplemental figure 2B). There was no correlation between baseline levels of VEGF and treatment response or disease control.
The longitudinal expression of cytokines was also compared between patients with disease control versus no disease control based on changes between pretreatment, 1-month, and 2-month levels. Of note, the average expression of CD40L was higher for patients with disease control at all time points, and significantly higher at baseline (p=0.03) compared with patients with no disease control. In addition, patients with disease control exhibited significant increases in IL-6 between baseline and 1 month (p=0.04) as well as baseline and 2 months (p=0.04), whereas patients with no disease control did not demonstrate significant changes over time. There is a trend towards lower levels of IL-6 across all time points for patients with disease control compared with patients with no disease control (p=0.08). There were no other statistically significant differences in longitudinal expression of cytokines between responders and non-responders.
Association between flow cytometry markers and clinical response
Flow cytometry was performed on PBMC samples at three time points (pre-treatment, 1 month, 2 months), and four panels were applied to study T cell, regulatory T cell (Treg), and monocyte populations as well as checkpoint markers. Of 23 patients included in this analysis, 5 patients were responders (CR or PR) and 12 patients had disease control (CR, PR, or SD). In addition, 15 patients in this analysis had grade ≥3 TRAEs.
For T cell population analysis, patients who responded to treatment appeared to demonstrate higher CD4+ cells compared with non-responders at baseline (p=0.08), 1 month (p=0.11), and 2 months (p=0.09, figure 4A). In contrast, responders appeared to demonstrate lower CD8+ cells compared with non-responders at baseline (p=0.08), 1 month (p=0.06), and 2 month (p=0.11, figure 4B). Among patients who developed grade≥3 TRAEs, there was a trend towards significantly lower CD8+ TCRab at baseline (p=0.09), 1 month (p=0.06) and 2 months (p=0.009). There was no correlation between grade ≥3 TRAEs and treatment response.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Flow cytometry analysis of T cell and monocyte. Flow cytometry analysis comparing T cell populations and monocytes between patients with CR (or PR, n=5) and non-responders (n=18) and patients with disease control (CR, PR, or SD, n=12) and no disease control (n=11). (A) CD4+ populations were higher in responders versus non-responders at all time points. (B) CD8+ populations were lower in responders versus non-responders at all time points. (C) Treg CD4+/CD25+/FoxP3+ populations were lower at baseline in responders. (D) Treg CD4+/CD25+/FoxP3+ populations were lower at baseline and 1 month in patients with disease control versus no disease control. (E) Central memory T cells (TCM) CD8+/CD45RO+/CCR7+ was higher at all time points in responders versus non-responders. (F)Effector memory T cells (TEMRA) CD8+/CD45RO−/CCR7− was lower in responders at baseline. (G) Non-classical monocytes expressing TIE2 were higher at baseline in non-responders. (H) Classical monocytes were higher at all time points in non-responders. CR, clinical response; PR, partial response; Treg, regulatory T cell.
In addition, patients who responded to treatment appeared to have lower baseline levels of CD4+/CD25+FoxP3+ Treg cells compared with non-responders at pretreatment (p=0.09, figure 4C), although there were no differences at later time points. Similarly, patients with disease control had significantly lower levels of CD4+/CD25+FoxP3+ Treg cells compared with patients with no disease control at both pretreatment (p=0.03) and 1 month (p=0.03, figure 4D). On longitudinal fold-change analysis, non-responders exhibited a significant reduction in CD4+/CD25+FoxP3+ Treg cells between baseline and the 2-month time point (p=0.02), as did patients with no disease control (p=0.047).
We also performed flow cytometry analysis of memory phenotype markers. Responders had significantly higher measurements of TCM CD8+/CD45RO+/CCR7+ at all time points compared with non-responders at baseline (p=0.03), 1 month (p=0.02), and 2 months (p=0.02, figure 4E). In addition, responders had significantly lower levels of TEMRA CD8+/CD45RO−/CCR7− than non-responders at baseline only (p=0.04, figure 4F). Non-classical monocytes expressing TIE2 were higher at baseline in non-responders compared with responders (p=0.06, figure 4G). Classical monocytes were higher at all time points in non-responders compared with responders at baseline (p=0.04), 1 month (p=0.048), and 2 months (p=0.02, figure 4H).
Patients with disease control had significantly lower levels of CD4+/OX40+ (p=0.04) and CD8+/OX40+ (p=0.02) compared with patients with no disease control at the 2-month time point. In addition, for patients with disease control, measurements of CD4+/41BB+ were lower at the 2-month time point compared with patients with no disease control (p=0.01). Longitudinal analysis showed a significant increase in CD4+/41BB+ cells in patients with no disease control (p=0.007), but this change was not found in patients with disease control. Finally, patients with disease control had significantly higher levels of CD8+/41BB+ at the 2-month time point compared with patients without disease control (p=0.02). Longitudinal analysis showed a significant increase in CD8+/41BB+ cells in patients with no disease control (p=0.007), but this change was not found in patients with disease control.
Analysis of monocyte populations was delineated between classical CD86+, intermediate, and non-classical monocytes. Responders had significantly lower levels of classical CD86+ monocytes compared with non-responders at baseline (p=0.04), 1 month (p=0.048), and 2 months (p=0.02). Similarly, patients with disease control had lower levels of classical monocytes compared with patients with no disease control but only at baseline (p=0.04). Interestingly, responders had higher measurements of non-classical monocytes compared with non-responders at baseline (p=0.07), 1 month (p=0.07), and 2 months(p=0.05). Patients with disease control had a significantly higher measurement of non-classical monocytes at the 1-month time point only (p=0.02). There was no statistical relationship between classical MDSC populations and clinical response or disease control.
Patients with disease control had a significantly higher baseline intermediate TIE-2 compared with patients with no disease control (p=0.01). There was a trend towards lower non-classical monocytes expressing TIE-2 in responders compared with non-responders (p=0.06). Finally, responders had significantly higher levels of parent monocyte-derived dendritic cells (mDC) at the 2-month time point only (p=0.04), as did patients with disease control (p=0.008). There was no statistical relationship between monocyte populations and grade ≥3 TRAEs.
Discussion
The combination of ICIs and antiangiogenic agents has recently emerged as an effective strategy in tumors that traditionally respond to ICIs. Here, we sought to investigate the combination of ziv-aflibercept (VEGFR-1/2 inhibitor) and PD-1 blockade in both traditionally immune responsive tumors (melanoma and RCC) and non-immune responsive tumors (MSS CRC, ovarian, and other solid tumors). The combination was found to be safe and there was no DLT observed during dose escalation. The responses we observed occurred only in the tumors that are known to be immunogenic (two melanoma and one RCC); however, the small sample size precluded any meaningful conclusions in immune-excluded tumors including MSS CRC, and ovarian cancer.
Not surprisingly, activated tumor-infiltrating CD8 T cells at baseline (CD8+PD1+) and high CD40L, which is expressed on activated T cells, and increased memory CD8 T cells in the periphery correlated with response to the combination therapy, consistent with the effect of PD-1 blockade. Tumor-infiltrating CD8+PD1+ T cells have been previously described to be an indicator of an immunologically active tumor microenvironment and a predictor of response to anti-PD-1 therapy,24–26 although other studies have pointed to PD-1 expression as a marker for exhaustion.27 28 In addition, the relationship between high non-classical monocytes at baseline before decreasing in responders is potentially an indication of the on-target effect of VEGF inhibitors on myeloid cells, although this conclusion is difficult to confirm based on small sample sizes and warrants further investigation. This is also consistent with the decrease in IL-6 in patients with disease control. On the other hand, patients with favorable innate immune profile at baseline with high mDC were more likely to derive clinical benefits from this combination. Interestingly, patients with disease control exhibited higher baseline levels of CXCL2, CXCL5, and CD40L, which are all cytokines associated with promoting angiogenesis.29 30
The immune modulation effect of ICIs combined with antiangiogenesis has been reported in many studies. Most of those studies were conducted in patients with RCC31 32 and HCC.33 The addition of bevacizumab to atezolizumab improved outcomes in patients with RCC, particularly in tumors with immune-suppressed myeloid signatures, thus further emphasizing the potential effect of antiangiogenesis to overcome the immune-suppressive microenvironment.34 Interestingly, patients in this study exhibited high incidence of hypertension and proteinuria at rates similar to other combination therapies of ICIs and antiangiogenesis agents.32 33 35 For example, in the CLEAR study of lenvatinib and pembrolizumab for advanced RCC, 55.4% of patients developed hypertension and 29.5% developed proteinuria. While hypertension has been found to be associated with response to other VEGF inhibitors, our study did not show a statistically significant association between hypertension and proteinuria with treatment response.
This study is limited by its small sample size, the lack of a control arm of a single-agent PD-1 inhibitor, and limited availability of paired biopsies. While other combinations of ICIs and antiangiogenesis agents have been shown to cause morphological vessel alteration,8 36 including ‘plumpness’ indicating endothelial activation in high endothelial venules that enable lymphocyte extravasation,37 38 the limited number of biopsy samples in this study precluded this analysis. The ongoing cohort of patients with melanoma and RCC who progressed post-PD-1 blockade could help to answer the question of whether the addition of ziv-aflibercept to pembrolizumab could overcome the resistance to PD-1 inhibitor. Nonetheless, this study provided a proof of concept for the safety and feasibility of ziv-aflibercept combination with pembrolizumab and established the RP2D for future larger studies.
Supplemental material
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved for clinical trial by institutional review boards at all sites. Participants provided written informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @OsamaRahma2
Contributors OR helped conceive the study and was involved in patients’ treatment, data interpretation, supervision on the study, writing, editing, and reviewing the manuscript. OR acted as guarantor and accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish. KT was involved in data interpretation and editing and reviewing of the manuscript. AG-H had access to and verified the raw data and was involved in data interpretation. ASB, PLB, and DJR were involved in patients’ treatment, data interpretation, and supervision on the study. ES and HS were involved in supporting and supervising the study. EH, RC, MM, MS, and SR were involved in data analysis, data collection and curation, and data interpretation. FSH helped conceive the study and was involved in patients’ treatment, data interpretation, supervision of the study, writing, editing, and reviewing the manuscript. All other authors were involved in investigation; each author read and approved the final work.
Funding This trial was supported by the National Cancer Institute (NCI) and by Merck, Sharpe, and Dohme and Sanofi via Cooperative Research and Development Agreements with the NCI. PLB was supported by NCI UM1 Grant CA186644.
Competing interests OR has research support from Merck; educational grants from BMS and Merck; consulting agreements with Merck, Celgene, Five Prime Therapeutics, GSK, GFK, Defined Health INC, Roche/Genentech, Puretech, Leerink, and PRMA Consulting; and a pending patent, ‘Methods of Using Pembrolizumab and Trebananib’. ASB reports serving as a consultant or advisor for Bayer, Deciphera, EMD Serono, and PierianDx. PLB reports consulting or advisory role for Seattle Genetics, Lilly, Amgen, Merck, BMS, and Pfizer, and reports research funding from Bristol-Myers Squibb, Sanofi, AstraZeneca, Genentech/Roche, SERVIER, GlaxoSmithKline, Novartis, PTC Therapeutics, Nektar, Merck, Seattle Genetics, Mersana, Immunomedics, Lilly, Amgen, and Bicara. SR reports receiving commercial research grants from Bristol-Myers Squibb, Merck, and KITE/Gilead. SH reports the following: grants from BMS and Novartis; personal fees from BMS, Merck, Serono, Novartis, Takeda, Surface Pharmaceuticals, Genentech/Roche, Compass Therapeutics, Apricity, Bayer, Aduro, Partners Therapeutics, Sanofi, Pfizer, Pionyr Immunotherapeutics, 7 Hills Pharma, Verastem Oncology, Rheos Medicines, and Kairos Therapeutics; equity in Torque Therapeutics; and patents #20100111973 and #7250291 issued as well as #20170248603, #20160340407, #20160046716, #20140004112, #20170022275, and #20170008962, and ‘Methods of Using Pembrolizumab and Trebananib’ pending.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.