Article Text

Abstract
Background Cross-presentation of exogenous antigens in HLA-class I molecules by professional antigen presenting cells (APCs) is crucial for CD8+ T cell function. Recent murine studies show that several non-professional APCs, including cancer-associated fibroblasts (CAFs) also possess this capacity. Whether human CAFs are able to cross-present exogenous antigen, which molecular pathways are involved in this process and how this ultimately affects tumor-specific CD8+ T cell function is unknown.
Methods In this study, we investigated the ability of human colorectal cancer (CRC)-derived CAFs to cross-present neoantigen-derived synthetic long peptides (SLPs), corresponding to tumor-derived mutant peptides, and how this affects tumor-specific T-cell function. Processing of the SLP was studied by targeting components of the cross-presentation machinery through CRISPR/Cas9 and siRNA-mediated genetic ablation to identify the key molecules involved in fibroblast-mediated cross-presentation. Multispectral flow cytometry and killing assays were performed to study the effect of fibroblast cross-presentation on T cell function.
Results Here, we show that human CRC-derived CAFs display an enhanced capacity to cross-present neoantigen-derived SLPs when compared with normal colonic fibroblasts. Cross-presentation of antigens by fibroblasts involved the lysosomal protease cathepsin S. Cathepsin S expression by CAFs was detected in situ in human CRC tissue, was upregulated in ex vivo cultured CRC-derived CAFs and showed increased expression in normal fibroblasts after exposure to CRC-conditioned medium. Cognate interaction between CD8+ T cells and cross-presenting CAFs suppressed T cell function, reflected by decreased cytotoxicity, reduced activation (CD137) and increased exhaustion (TIM3, LAG3 and CD39) marker expression.
Conclusion These data indicate that CAFs may directly suppress tumor-specific T cell function in an antigen-dependent fashion in human CRC.
- antigen presentation
- immunotherapy
- gastrointestinal neoplasms
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442-D450, doi:10.1093/nar/gky1106 (2019), with the dataset identifier PXD029507.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The majority of current immunotherapeutic modalities rely on the induction of a tumor-reactive cytotoxic CD8+ T cell response.1 Different cellular actors in the tumor-microenvironment (TME) can interfere with CD8+ T cell function, hampering an effective anti-tumor immune response. Cancer-associated fibroblasts (CAFs) are the most prominent cell type in the TME in a variety of malignancies, including colorectal cancer (CRC), in which their abundance is inversely correlated with patient survival.2 3 Since CD8+ T cells are often restricted to stromal zones and are thus spatially juxtaposed to CAFs in the TME, CAFs can directly interfere with CD8+ T cell function.4–6 Current known mechanisms for fibroblast-mediated T cell suppression in humans are all antigen-independent7 or involve antigen presentation to CD4+ (regulatory) T cells via specialized subsets of fibroblasts expressing major histocompatibility complex (MHC)-II molecules, that have been identified in the colon.8 9 However, the majority of CAFs do not express MHC-II, and it is unknown whether CAFs in human cancer can affect CD8+ tumor-specific T cell function directly via presentation of tumor-derived exogenous antigen on MHC-I.
Presentation of exogenous antigen by MHC-I molecules is a specialized form of antigen presentation, called cross-presentation, and thought to play an important role in cancer immunology.10 Professional antigen-presenting cells (APCs), including dendritic cells (DCs), macrophages and B cells, are able to cross-present antigens via two distinct routes; the cytosolic and vacuolar pathways.10–12 The cytosolic route entails the export of the acquired antigen from endosomes to the cytosol, followed by proteasomal degradation. The resulting peptides are translocated to the endoplasmic reticulum (ER) via the transporter-associated with antigen presentation (TAP), after which they can be loaded onto MHC-I molecules and transported to the cell surface. In the vacuolar route, disassembly of the acquired antigen takes place in the endolysosomal compartments via degradation by lysosomal proteases, including cathepsins, after which loading onto recycling MHC-I occurs. It is important to note that exceptions to these two pathways, for example, proteasome-dependent, TAP independent13 or proteasome-independent, TAP dependent14 cross-presentation, have been described. Professional APCs, especially DCs,15 are very efficient in cross-presenting tumor antigens. However, recent murine data show that CAF might also be capable of cross-presentation and thereby suppress tumor-specific T cell function in an antigen-specific fashion.6
In this study, we investigated the ability of human CRC-derived CAFs to cross-present synthetic long peptides (SLPs), corresponding to tumor-derived mutant peptides, and how this affects tumor-specific CD8+ T cell function. CRC-derived CAFs were used as a model system due to their abundance in the TME and reported immunosuppressive roles.8 9 Our results show that CRC CAFs display enhanced cross-presentation capacity compared with patient-matched, normal colonic fibroblasts. Mechanistically, enhanced cross-presentation in CAFs required the lysosomal protease cathepsin S, which was detected in CAFs in human CRC tissue. Interestingly, cathepsin S was upregulated in a subset of cultured CRC-derived CAFs and induced in normal fibroblasts after exposure to CRC-conditioned medium. Finally, we studied the role of fibroblast-mediated SLP cross-presentation on the induction of checkpoint molecule expression and the cytotoxic activity of tumor-specific T cells.
Collectively, our data show the ability of human CAFs to cross-present antigen via the cathepsin S dependent vacuolar pathway, to cytotoxic CD8+ T cells. CAF-mediated cross-presentation results in a diminished cytotoxic T cell response, showing that human CRC CAFs are potentially able to directly interfere with CD8+ T cell function by cathepsin-S dependent cross-presentation of tumor antigens. Combined with the already known mechanisms of antigen-independent suppression of T cell function, this identifies CAFs as an important next target to further enhance T cell-based immunotherapies in human CRC.
Materials and methods
Primary fibroblast isolation, cell culture and tumor-conditioned medium
Primary fibroblasts were derived from human CRC according to the Code of Conduct for Responsible Use of human tissues or after written informed consent was obtained. Adjacent normal colon or liver tissue was used for the isolation of matched, normal fibroblasts from primary and liver-metastasized CRC resection specimens, respectively. Tumor or normal tissue was minced into small fragments and subsequently digested with a mix (3:1 ratio) of collagenase (Gibco/Thermo Fisher Scientific, Leiden, The Netherlands) and dispase II (Roche, Basel, Switzerland) and incubated for 2 hours at 37°C. Cells were subsequently cultured and expanded. Prior to further use in experiments, verification of fibroblast marker expression via qPCR (vimentin/αSMA) and absence of markers of endothelial, immune or epithelial origin (CD31, CD45, Keratin20, respectively), was performed. Fibroblasts were used until passage 7 after initial isolation.
Primary fibroblasts and the immortalized Nijmegen Breakage Syndrome (NBS) fibroblast cell line16 were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Thermo Fisher Scientific) supplemented with 8% FCS, 100 IU/mL penicillin and 100 µg/mL streptomycin (all Thermo Fisher Scientific). CRC cell lines (HCT116, HT29, SW480, LS180) and melanoma cell lines (BLM, FM3, 518A2) were obtained from ATCC and early-passages of ATCC cell stocks were used. The Mel04.01 and Mel12.07 melanoma cell lines were generated before and have been described previously.17 All tumor cell lines were cultured in DMEM supplemented with 8% FCS, 100 IU/mL penicillin and 100 µg/mL streptomycin (all Thermo Fisher Scientific). Tumor-conditioned medium was harvested from subconfluent tumor cell lines that were incubated with serum-free DMEM for 5 days. After incubation, the conditioned medium was centrifuged to remove cellular debris, aliquoted and stored at −20°C until use.
T cells were cultured in Iscove’s Modified Dulbecco’s Medium with penicillin (100 IU/mL), streptomycin (100 µg/mL), L-glutamine (4 mM) (all from Life Technologies, Breda, The Netherlands), 7.5% heat inactivated pooled human serum (Sanquin, Amsterdam, The Netherlands) and supplemented with 150 IU/mL Interleukin-2 (IL-2, Aldesleukin, Novartis, Netherlands). Monocyte-derived DCs (MoDCs) and Epstein-Barr Virus (EBV)-immortalized B cells were generated and cultured as described previously.17
All cells were cultured at 37°C and 5% CO2. Mycoplasma tests were performed regularly by PCR and were negative throughout the duration of the experiments.
Constructs, lentiviral transduction, generation and validation of transgenic cell lines
Third-generation packaging vectors and HEK293T cells were used for the generation of lentiviral particles.18 Knockdown constructs for cathepsin S were acquired from the Mission TRC1 shRNA library (Sigma-Aldrich, Zwijndrecht, The Netherlands), with target sequences 5’-GCTATGGTGATCTTAATGGGA-3’ and 5’-CACAGTTGCATAAAGATCCTA-3’, respectively. The lentiviral cDNA expression vector expressing cathepsin S was generated through Gateway cloning, using the cathepsin S entry vector (pDNR223 backbone) from the human ORF library (Sigma) and pLex307 (addgene: #41392) as the destination vector. Cells were selected and cultured with 2 µg/mL of Puromycin (Sigma).
A sgRNA, 5’-caccgTTACGGGCCGCCTCACTGAC-3’ (lowercase nucleotides are compatible with the restriction site) designed to generate a TAP1 KO was cloned into BsmBI-digested plentiCRISPRv2.19 Isolation of clonal NBS fibroblast cell lines containing the desired knockout was performed as described previously.20 Briefly, puromycin-resistant fibroblasts were sorted based on the expression of HLA class I. Fibroblasts with the lowest expression of HLA class I were seeded at single cell density and subsequently expanded to acquire clonal lines. Knockout verification of clones was performed via Western Blot and Sanger sequencing (Macrogen, Amsterdam, The Netherlands) of the targeted region (exon 2, TAP1). For sanger sequencing, genomic DNA was first extracted using the NucleoSpin DNA isolation kit (Macherey-Nagel) and the region of interest was amplified with PCR using DreamTaq Green PCR Master Mix (Thermo Fisher Scientific) and gel purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). The primers 5’-AGAGGTTCTGGGCTCCATTG-3’ and 5’-CAAACACCTCTCCCTGCAAGTG-3’ were used as forward and reverse primers, respectively. The same forward primer was also used in the Sanger sequencing reaction and chromatograms were visualized using FinchTV v1.4.0 (Geospiza, Inc., WA, USA). Cells were selected and cultured with 2 µg/mL of Puromycin (Sigma).
Fibroblast cell lines (NBS) transduced with HLA-A*03:01 and HLA-B*07:02 were generated and described previously,21 and were confirmed to express the desired transgene by FACS analysis.
Real-time quantitative PCR
Total RNA was isolated using the NucleoSpin RNA isolation kit (Macherey-Nagel, Düren, Germany) according to manufacturer’s instructions. cDNA was synthesized with the RevertAid First strand cDNA synthesis kit (Thermo Fisher Scientific) using 0.5–1.0 µg as RNA input. Real-time quantitative PCR (RT-qPCR) was performed with SYBR Green Master mix (Bio-Rad laboratories, Nazareth, Belgium) using the iCycler Thermal Cycler and iQ5 Multicolour RT-PCR Detection System (Bio-Rad). Target genes were amplified using specific primers (online supplemental table 1). Target gene expression levels were normalized to β-actin, determined to be the most stable reference gene out of a panel of five genes (β-actin, GAPDH, HPRT1, HMBS, β2M). The ΔCt or ΔΔCt method was applied to calculate the levels of gene expression, relative to the reference gene or a control condition, respectively.
Supplemental material
Western blot
Cells were lysed in RIPA buffer after which protein content was determined using a DC protein assay (Bio-rad) according to manufacturer’s instructions. Western blot analysis was performed as described before.22 Briefly, equal amounts of protein were separated with electrophoresis using 10%–12% SDS-polyacrylamide gels under reducing conditions. Proteins were transferred to PVDF membranes (Sigma) and non-specific binding was blocked with 5% milk powder in tris-buffered saline containing 0.05% Tween-20 (Merck, Darmstadt, Germany). Blots were incubated overnight with mouse anti-CTSS (Santa Cruz Biotechnology, Santa Cruz, USA), mouse anti-TAP1 hybridoma supernatant (clone mAb 148.3, E. Wiertz, Dept. of Medical Microbiology, UMC Utrecht, The Netherlands), rabbit anti-V5 tag (Abcam, Cambridge, UK) or mouse anti-β-actin (Santa Cruz Biotechnology). Detection was performed by horseradish peroxidase-conjugated secondary antibodies (all from Agilent, CA, USA) and chemiluminescence (Thermo Fisher Scientific) was used to visualize the target proteins.
Peptide synthesis
Peptides were synthesized as described previously23 by an in-house peptide facility or commercial vendor (Pepscan, Lelystad, The Netherlands) and sequences are shown in online supplemental table 2. The synthetic peptide sequences are named after their HLA-restriction and show the name of the protein and the position of the non-synonymous mutation within that protein. The integrity of all SLPs as well as the synthetic heavy A3-RPL2874-84(p76S>F) peptide was assessed by MALDI-TOF mass-spectrometry to exclude the presence of short epitopes or light peptide, respectively, that could directly bind to HLA-class I (HLA-I) molecules.
For the A3-RPL2861-91(p76S>F) 31-mer, different internally (rhodamine-dabcyl) quenched variants were synthesized that were protected either at the N-terminus and/or C-terminus by acetyl or carboxamide-groups, respectively (table 3online supplemental table 3). Synthesis was performed using preloaded Fmoc-Ser(tBu)-PEG-PS resin (Rapp Biopolymere GmbH) or H-Rink amide Chemmatrix resin (Aldrich) on 20 µmol scale. After coupling of Fmoc-Lys(Alloc)-OH on position 87, the Alloc was removed using Pd(PPh3)4, PhSiH in DCM for two times 15 min followed by coupling of di-Boc-Rhodamine. Subsequent automated solid phase peptide synthesis was performed on a MultiSyntech Syro II. Fmoc-protected amino acids were coupled using four-fold excess relative to the pre-loaded Fmoc amino acid trityl resin (0.2 mmol/g) using double couplings in N-Methyl-2-pyrrolidone (NMP) for 40 min followed by a second coupling of 60 min using PyBOP (four eq) and DiPEA (eight eq) at room temperature. After coupling each amino acid, Fmoc deprotection was performed in 20% piperidine in NMP for three times 4 min at room temperature. Fmoc-Lys(Dabcyl)-OH was incorporated on position 65. The resins were split in two portions and one portion was acetylated using acetic anhydride. Treatment of all the resins using TFA/ H2O/Phenol/TIS (90.5 : 5.0 : 2.5 : 2.0, v/v/v/v) for 2.5 hours liberated the peptides from the resin and removed all protective groups. Precipitation from Et2O/Pentane (1 : 1, v/v) followed by RP-HPLC purification and lyophilization of the appropriate fractions yielded the target compounds.
LC-MS measurements were performed on a Waters Acquity H-class UPLC using a Phenomenex Kinetex C18-column (2.1×50, 2.6 µm) and LCT ESI-Mass Spectrometer. Samples were run using two mobile phases: A=1% CH3CN, 0.1% formic acid in water and B=1% water and 0.1% formic acid in CH3CN. Flow rate=0.6 mL/min, runtime=3 min, column T=40°C. Gradient: 0%–95% B. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1.
Spinning-disk confocal microscopy
For live cell imaging of peptide cross-presentation, NBS fibroblasts were grown on 35 mm glass bottom dishes (MatTek Life Sciences, MA, USA) coated with poly-D-lysine. Hoechst (Cell signaling, Leiden, The Netherlands; 0.5 µg/mL) and Lysotracker deep red (Life Technologies; 0.1 µM added 15–30 min before imaging) were used to stain the nucleus and the lysosomes for detection by confocal microscopy. After addition of internally quenched peptide (20 µg/mL), time-lapses were collected on a Dragonfly spinning disk microscope (Oxford instruments, Abingdon, UK) adapted with a climate control chamber. Images were acquired using Hcx PL 63×1.32 oil objectives (Leica Camera, Solms, Germany), and data were analyzed using Fiji software.
Isolation of HLA Class I-presented peptides
To obtain anti-HLA class I A,B,C antibodies, the hybridoma cell line W6/32 (ECACC 84112003) was expanded in roller bottles using protein-free hybridoma medium supplemented with penicillin, streptomycin and L-glutamine. The antibodies were purified from the W6/32 supernatant using Prot-A sepharose beads. Antibodies were eluted with 0.1M glycine pH 2.5 and neutralized. The purified antibodies were covalently coupled to Prot-A sepharose beads (2.5 mg IgG/mL beads) using dimethylpimelimidate.24
Peptides associated with HLA–class I molecules were extracted from pellets of 1×109 PBS washed NBS fibroblasts, after 48 hours of incubation with the SLP, as described.24 In short, cells were lysed in lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 0.5% zwittergent 3–12 (pH 8.0) and Complete Protease Inhibitor cocktail (Roche). The total concentration of cells in lysis buffer was 1×108 cells/mL. After 2 hour incubation with tumbling at 4°C the lysate was centrifuged for 10 min at 3000xg and 4°C. The supernatant was transferred to a JA-25.50 tube and centrifuged for 35 min at 30 000 x g and 4°C. The supernatant was precleared on a 1 mL CL4B column and next subjected to a 1 mL anti-HLA immunoaffinity column at a flow rate of 0.5 mL/min. After washing successively with lysis buffer, TBS (20 mM Tris-HCl, 120 mM NaCl), TBS with 1 M NaCl and TBS, the bound HLA-class I/peptide complexes were dissociated and eluted from the column with 5 mL 10% acetic acid. Peptides were separated from the HLA-class I protein subunits by passage through a 10 kDa membrane (Microcon, Millipore). The filtrate was freeze dried, dissolved in 0.1% formic acid, and the peptides were further purified by C18 SPE (HLB Oasis, 100 µL bed volume, Waters), freeze dried again, and dissolved in 0.1% formic acid, containing 19 fmol/µL of the heavy labeled ATFYVRTTINK-peptide for analysis by parallel reaction monitoring (PRM).
PRM-analysis
For PRM analysis, the samples were lyophilized and resuspended in buffer A. The equivalent of 100 million cells of HLA-eluate was injected together with 19 fmol of the heavy labeled ATFYVRTTINK peptide. The Orbitrap Fusion LUMOS mass spectrometer was operated in PRM-mode. The transitions and collision energies are in online supplemental table 4. The isolation width of Q1 was 0.7 Da. MS2 resolution was 30 000 at a custom AGC target value of 200% at a maximum fill time of 250 ms. PRM data analysis and data integration were performed in Skyline 3.6.0.10493. Peptide abundances were calculated by comparing the peak area of the eluted (light) and the peak area of the spiked-in heavy peptides.
T cell activity assays
Neoantigen-specific T cells were obtained previously and their antigen reactivity has been extensively characterized.17 For the reactivity assays, T cells were incubated with target cells (primary fibroblasts, NBS fibroblast cell line), verified to express crucial genes involved in cross-presentation, that were pre-loaded overnight with the desired SLP antigen. Tumor-conditioned target fibroblasts were exposed to tumor-conditioned medium for 48 hours prior to antigen loading. Inflammatory cytokine content of tumor-conditioned media was determined by Bio-Plex Pro Human Cytokine Th1/Th2 (Bio-Rad) Assay according to manufacturer’s instruction. Subsequently, T cells were added in a 1:1 effector:target (E:T) ratio and co-incubated for 24 hours. Reactivity of T cells was measured using both IFNγ ELISA (Sanquin and Mabtech, OH, USA), Th1/Th2 cytokine Luminex kit (Bio-rad) and/or expression of the activation marker CD137 by FACS analysis, as described previously.25 Four h26Cr-release cytotoxicity assays were performed, as described previously,27 with neoantigen-specific T cells that were pre-incubated for 24 hours with various target cells prior to the cytotoxicity assay.
Flow cytometry and fluorescence-assisted cell sorting
For cell surface staining, cells were harvested and washed twice with FACS buffer, consisting of PBS/0,5% BSA (Sigma)+0.05% sodium azide (Pharmacy LUMC, Leiden, The Netherlands). Subsequently, cells were incubated with directly conjugated primary antibodies (online supplemental table 5) or A3-RPL2874-84 (p76S>F)-specific tetramers (Manufactured in house, Jan-Willem Drijfhout, Dept. of Immunology, LUMC, The Netherlands) for 45 min. In multispectral flow cytometry experiments, primary antibodies were additionally mixed with Trustain FcX blocking solution (Biolegend) to prevent non-specific staining. After incubation, cells were washed twice with FACS buffer and directly used for flowcytometry or FACS sorting. For HLA-typing of primary fibroblasts, we made use of human anti-HLA hybridoma supernatant (A. Mulder, Dept. of Immunology, LUMC, The Netherlands) to determine the presence of HLA-A2 (IgG mAb SN607D8 and IgG mAb SN230G6), HLA-A3 (IgM mAb OK2F3) and/or HLA-B7 (IgG mAb VTM3A1) expression. PE-conjugated secondary antibodies were either anti-human IgG or anti-human IgM (Biolegend, Fell, Germany). All these FACS experiments were performed on a LSRII (BD Bioscience, CA, USA) and sorting was performed with an Aria FACS sorter (BD Biosciences). Data analyses were performed with FlowJo v10.6.1 (BD biosciences). Multispectral flow cytometry was performed on a Cytek Aurora cytometer (Cytek Biosciences, CA, USA) and data analysis performed using Cytosplore.28
Immunohistochemical and immunofluorescent staining
Four-micrometers sections were deparaffinized and processed for immunohistochemistry (IHC) or immunofluorescent (IF) staining. For IHC, sections were deparaffinized and blocked in 0.3% hydrogen peroxidase (H2O2, Merck) in methanol for 20 min. Next, for both IHC and IF, slides were rehydrated, and antigen retrieval was performed by boiling in 0.01M sodium citrate (pH 6.0) for 10 min. Slides were washed and incubated with primary mouse anti-cathepsin S (Santa Cruz, TX, USA) and/or rabbit anti-human vimentin (Cell signaling) antibodies diluted in 1% PBS/Bovine Serum Albumin overnight at room temperature in a humidified box. The next day, slides were incubated with appropriate biotinylated secondary antibodies (Agilent technologies, California, USA) or anti-mouse-alexa 488 (Thermo Fisher Scientific) and anti-rabbit-alexa 568 (Thermo Fisher Scientific). For IF staining, slides were mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific) including DAPI. For IHC, slides were incubated with Vectastain complex (Vector Laboratories, CA, USA) at room temperature for 30 min. Staining was visualized with the Dako Liquid DAB+ Substrate Chromogen kit (Agilent technologies) for 10 min. Nuclei were counterstained with Mayer’s Hematoxylin (Merck) and slides were rinsed in tap water, dehydrated, and mounted using Entellan (Merck). Images for IHC were obtained with an Olympus BX51 Light Microscope equipped with an Olympus DP25 camera. For IF, images were taken with a Leica DMi8 microscope (Leica). All images were analyzed using Fiji software.
Statistical analysis
Data are presented as means±SD from representative experiments of independent replicates. Unpaired Student t tests were used to compare two groups. Differences between more than two groups were measured using one-way or two-way analysis of variance and corrected for multiple testing (Dunnett’s multiple comparisons test). All analyses were performed using GraphPad Prism software (San Diego, California, USA). P values of 0.05 or less were considered statistically significant.
Results
Human CRC-derived CAFs can elicit a neoantigen-specific T cell response
To study whether human colorectal CAFs are able to cross-present, we tested their ability to elicit a T cell response on loading with exogenous antigen. Three different primary, CRC-derived CAFs with different HLA haplotypes (HLA A3-B7-, A3+B7- or A3-B7+) were loaded with SLPs that correspond to tumor specific neoantigens and require intracellular processing to generate the epitope that can be recognized by neoantigen-specific T cells.17 Subsequently, these CAFs were co-incubated with neoantigen-specific T cells and T-cell activation was determined. Incubation of CAFs with either the HLA-A3 restricted 31-mer A3-RPL2861-91(p76S>F) or the HLA-B7 restricted 31-mer B7-KIAA0020436-466(p451P>L), only led to IFNγ production in HLA-matched conditions where antigen corresponding to the neoantigen epitope was present (figure 1A). Incubation with the 11-mer A3-RPL2874-84(p76S>F) and 9-mer B7-KIAA0020447-455(p451P>L), short peptides that can directly bind to their respective HLA-I molecule without intracellular processing, were used as a positive control and elicited strong T-cell activation in HLA-matched conditions. In contrast, the SLPs corresponding to the wildtype sequence, which do not contain the epitope required for T cell activation, did not result in T cell activation. Combined, these data show that fibroblasts, matched for the right HLA-restriction element, can activate neoantigen-specific T cells.

Human CRC-derived CAFs induce neoantigen-specific T cell activation. (A) IFNγ production by neoantigen-specific T cells (T-cell bulk reactive to both the A3 and B7-epitopes) after 24 hours coincubation with antigen presenting human CRC-derived CAFs (20 µg/mL SLP, 2 µg/mL SSP). The different panels show different primary CRC-derived CAFs with differential HLA-haplotypes. Experimental conditions are compared with the T cell—fibroblast only condition with 1-way ANOVA *p<0.05, **p<0.01, ***p<0.001. (B) IFNγ production of neoantigen-specific T cells (T-cell bulk reactive to both the A3 and B7-epitopes) after 24 hours coincubation with the antigen presenting NBS fibroblast line (20 µg/mL SLP, 2 µg/mL SSP). The different panels show wildtype, HLA-mismatched fibroblasts and fibroblasts that have been transduced to express the correct HLA-molecule needed for presentation. Experimental conditions are compared with the T cell—fibroblast only condition with one-way ANOVA *p<0.05, **p<0.01, ***p<0.001. (C) TNFα and IL-2 production by neoantigen-specific T cells after 24 hours coincubation with the antigen presenting HLA-A3+ NBS fibroblast line. Concentration of peptide used for the mutant SLP is depicted. Concentration for wildtype SLP is 20 µg/mL. Experimental conditions are compared with the T cell—fibroblast only condition with Student’s t-test **=p≤0.001, *=p≤0.05. (D) Surface CD137-expression on neoantigen-specific T cells after coincubation with antigen presenting NBS fibroblasts determined by FACS. Concentration of peptide used for the mutant SLP is depicted. Concentration for wildtype SLP is 20 µg/mL. Data are plotted from representative experiments performed with one replicate per condition (n=3). ANOVA, analysis of variance; CAFs, cancer-associated fibroblasts; CRC, colorectal cancer; NBS, Nijmegen Breakage Syndrome; SLP, synthetic long peptides.
To study whether normal fibroblasts are also capable of cross-presentation, similar experiments were performed with three different primary normal skin fibroblasts with the same differential haplotypes (HLA A3-B7-, A3+B7- or A3-B7+). Also in this case, HLA-restricted, T cell activation was observed (online supplemental figure 1A), indicating that fibroblasts under physiological conditions already possess antigen-presenting capacities. As expected, T cell reactivity against a mutant SLP-loaded HLA-mismatched fibroblast cell line could only be elicited after retroviral transduction with the appropriate HLA-restriction element (figure 1B, online supplemental figure 1B). The possibility that extracellular degradation of the SLP by proteases present in serum and/or direct binding of (fragmented) SLP to HLA-I on T cells resulted in T cell activation was excluded by incubation of neoantigen-specific T cells with SLPs only (online supplemental figure 1C). CAF-mediated stimulation of neoantigen-specific T cells also induced production of TNFα and IL-2 and the increased expression of CD137 by CD8+ T cells (figure 1C,D). Comparison of the antigen-presenting capacities of normal fibroblasts with professional APCs (allogeneic Mo-DCs and autologous EBV-immortalized B cells (EBV-B cells) showed similar T cell activation after presentation by fibroblasts, indicating similar cross-presentation capacity under physiological, non-cancerous conditions (online supplemental figure 1D). Collectively, these data show the ability of (cancer-associated) fibroblasts to activate neoantigen-specific T cells via intracellular processing of exogenous antigen.
Supplemental material
Fibroblasts cross-present SLPs through a lysosomal endopeptidase
To formally establish that cross-presentation occurs in fibroblasts, a mass-spectrometry-based approach was chosen to demonstrate the processing of SLP and presentation of the cognate peptide epitope in HLA-class I. The limited availability and lifespan of HLA-matched, CRC-derived fibroblasts necessitated the use of the NBS fibroblast cell line as a model system for these and subsequent experiments. NBS fibroblasts were incubated with the 31-mer A3-RPL2861-91(p76S>F) for 48 hours, to resemble the time point at which T cell activation is assessed in all other experiments, after which protein lysate was collected and affinity purified for HLA class I/peptide complexes. We previously identified the 11-mer epitope A3-RPL2874-84(p76S>F), embedded within the 31-mer SLP, to be the cognate peptide epitope recognized by the neoantigen-specific T cells.17 Elution of the fibroblast HLA-I peptidome was performed and targeted mass-spectrometry, using heavy-isotope labeled A3-RPL2874-84(p76S>F) as a reference, revealed that the A3-RPL2874-84(p76S>F) peptide was indeed present in the fibroblast HLA-class I eluate (figure 2A). Although exact determination of the amount of endogenous peptide present is limited by technical factors,29 30 the amount of A3-RPL2874-84(p76S>F) peptide detected by PRM was determined to be equivalent to 3.67 fmol/million cells. This amount surpasses concentrations found in the endogenous MHC-I ligandome of tumor cells, suggesting that this epitope is present at levels that should be readily capable of eliciting T cell activation.31 These data show that intracellular processing of the SLP antigen (31-mer) indeed occurs in fibroblasts and results in the formation of the exact previously identified cognate 11-mer epitope17 that can be eluted from their MHC-I ligandome.

Cross-presentation of the A3-RPL2861-91(p76S>F) SLP relies on a lysosomal endopeptidase. (A) Parallel reaction monitoring (PRM)-analysis shows the detection of the A3-RPL2874-84(p76S>F) epitope (blue peak) in HLA-eluate of HLA-A3+ NBS that were co-incubated with A3-RPL2861-91(p76S>F) SLP for 48 hours. Heavy-isotope labeled A3-RPL2874-84(p76S>F) was spiked-in as a reference and showed the same retention time. (B) Internally quenched A3-RPL2861-91(p76S>F) was loaded on HLA-A3+ NBS fibroblasts and fluorescent spectroscopy was performed to analyze peptide processing. (C) FACS analysis of internally quenched A3-RPL2861-91(p76S>F) variants, chemically protected at either the N- and/or C-termini (orange label), after co-incubation with NBS fibroblasts (18 hours). (D) Spinning confocal microscopy shows colocalization of the internally quenched A3-RPL2861-91(p76S>F) SLP (green) and the lysosome compartment (red). Dashed line indicates the outlines of the fibroblast. Closed line indicates the location of the nucleus. Arrowheads indicate areas of colocalization. Lysotr=lysotracker. NBS, Nijmegen Breakage Syndrome; SLP, synthetic long peptide.
Next, we investigated the route of antigen processing and presentation. Distinct endo- and exopeptidases that mediate proteolytic cleavage are involved in antigen processing.32 To establish the role of these endopeptidases and exopeptidases in SLP processing, internally quenched fluorescent peptides of the 31-mer A3-RPL2861-91(p76S>F) were synthesized that were unmodified or chemically protected at the N-terminus and/or C-terminus by acetyl or carboxamide groups, respectively. This chemical protection of the N-terminus and/or C-terminus allows the differentiation between processing by two classes of exopeptidases, aminopeptidases and carboxypeptidases, respectively. Proteolytic cleavage of the native (unmodified N-and C-terminus) peptide resulted in substantial fluorescent emission exclusively when the peptide was incubated with fibroblasts. No substantial spontaneous fluorescence was detected, showing the feasibility of our approach, and maximum signal intensity within 1 hour (figure 2B). Next, incubation of fibroblasts with the different chemically protected SLPs showed high fluorescent signal in all conditions, indicating efficient cleavage of the peptide regardless of chemical constitution of the N-termini and C-termini (figure 2C). This pattern is compatible with proteolytic cleavage by an endopeptidase, since their function is not hampered by chemical protection of the N-termini and C-termini,33 as would be the case for cleavage by exopeptidases. Finally, we visualized the subcellular compartment in which SLP processing occurs by loading fibroblasts with unmodified, internally quenched A3-RPL2861-91(p76S>F) and a dye that labels the lysosomal compartment. Spinning disk confocal microscopy revealed co-localization of peptide and the lysosomal tracker (figure 2D, online supplemental video 1), indicating that peptide cleavage indeed occurs inside the lysosomes. A clear size increase of the peptide containing vesicles could be observed (online supplemental video 1), which is consistent with SLP uptake by means of pinocytosis. To study whether the pathway utilized by fibroblasts is cell-specific, we also performed these experiments with professional APCs (Mo-DCs) using the same internally quenched SLP. These experiments showed that the vacuolar route is also employed by professional APCs, as illustrated by colocalization of the lysosomal tracker and the SLP. This indicates that the same pathway, that is, the vacuolar pathway, is used for processing of this SLP in both professional APCs (Mo-DCs) as well as non-professional (fibroblasts) APCs (online supplemental figure 2). Consistent with the results from fluorescence spectroscopy, SLP processing in the lysosomes occurred very rapidly (<1 hour). These results convincingly show that SLPs are cross-presented by fibroblasts and that this occurs via a lysosomal endopeptidase, consistent with the vacuolar route of cross-presentation.
Supplementary video
Cross-presentation of SLP by fibroblasts is TAP1 independent
To further study the routing of antigen presentation in fibroblasts, we looked at the role of a central protein in cross-presentation, the heterodimeric TAP complex. TAP serves both as a transporter for antigen into lysosomes as well as the ER and thereby also partially controls the efflux of HLA-I-peptide complexes to be available for antigen loading on HLA-I when processed via the vacuolar route.14 First, transient blockade of TAP with a soluble TAP-inhibitor (sCPXV012),34 which selectively blocks TAP-mediated transport of antigen to the ER without affecting HLA-I surface expression,34 was performed but showed little effect on T cell activation (figure 3A). This shows that TAP-mediated antigen transport to the ER is not required for efficient presentation of the A3-RPL2874-84(p76S>F) epitope. To further investigate the role of TAP in fibroblast mediated antigen cross presentation, two clonal fibroblast cell lines harboring a TAP1 KO were generated using CRISPR/Cas9 (figure 3B, online supplemental figure 3A-E). As expected, total surface HLA-I was lower in the KO clones as compared with the vector control (figure 3C) since TAP controls the efflux of HLA-I/peptide complexes from the ER and expression on the cell surface. TAP1 KO also results in the presentation of an alternative pool of TAP-independent peptides, some of which are immunogenic, and known as T cell epitopes associated with impaired peptide processing (TEIPP).35 To establish the functional KO of TAP1 in the clonal fibroblast cell lines, a TEIPP-specific T cell bulk that recognizes a TEIPP-epitope from the endogenous protein LRPAP1 was used.20 Expression of LRPAP1 was similar between vector control and TAP1KO clones (online supplemental figure 4), but recognition of LRPAP1 only occurred in the TAP1KO clones (figure 3D), indicating that TAP1 is indeed functionally knocked out and TEIPP epitopes are now presented in HLA-I and recognized by TEIPP-specific T cells. As a control, a short synthetic peptide (SSP) corresponding to the LRPAP1 epitope (A2-LRPAP122–25 27–31 was loaded directly on fibroblasts and this elicited T cell activation in both control and TAP1KO fibroblasts. Having established the functional knock-out of TAP1, we next loaded A3-RPL2861-91(p76S>F) on these fibroblasts and co-incubated them with neoantigen-specific T cells to reveal TAP1-dependency for cross-presentation by fibroblasts. In contrast to the results with the chemical TAP inhibition, T cell reactivity towards A3-RPL2861-91(p76S>F) was strongly decreased in TAP1 KO conditions (figure 3E). Interestingly, reactivity towards two A2-restricted epitopes, A2-EML150-80(p64R>W) and A2-MP1 Flu46-75, was not hampered (figure 3F), suggesting that TAP1-dependency for cross-presentation differed between the A2 and A3-restricted epitopes. Since TAP deficiency can affect the cell surface expression of different HLA-class I molecules to variable degrees,36 we determined the expression of HLA-A2 and HLA-A3, separately. HLA-A3 surface expression was strongly reduced, while HLA-A2 expression was only slightly decreased (online supplemental figure 5) in TAP1 KO clones. Based on these observations, we concluded that the impaired presentation of A3-RPL2861-91(p76S>F) was due to a decreased efflux of HLA-A3-peptide complexes from the ER to the cell surface, rather than the result of blockade of TAP-mediated antigen transport to the ER, as this would limit available HLA-A3 for binding to the SLP-derived epitope generated through lysosomal processing. In summary, SLP cross-presentation by fibroblasts does not rely on the TAP complex for transport of the epitope to the ER but on its function to ensure enough ER efflux of HLA-I class molecules to allow binding of antigen derived through lysosomal processing before they are shuttled to the cell surface.

Role of TAP1 in SLP cross-presentation by fibroblasts. (A) NBS fibroblasts were preincubated with sCPVX012 at indicated concentrations and subsequently loaded with A3-RPL2861-91(p76S>F) (20 µg/mL). After 24 hours coincubation with neoantigen-specific T cells, IFNγ production was determined and plotted as a percentage decrease compared with the uninhibited condition. Means with SD are plotted from representative experiments (n=2). *p≤0.001 as determined by one-way ANOVA with correction for multiple testing. (B) TAP1 expression in TAP1KO fibroblast clones and vector control as determined by Western blot. (C) Total (left panel) and relative (right panel) HLA-I expression on TAP1KO fibroblast clones and vector control determined by flow cytometry. (D) IFNγ production of LRPAP1-specific T cell bulk after coincubation with empty or LRPAP-1 synthetic short peptide (SSP)-loaded TAP1KO fibroblast clones after 24 hours (left panel). Right panel shows the expression of CD137 on LRPAP1-specific T cells after coincubation with empty TAP1KO fibroblast clones or vector control. Representative figures are plotted from independent replicates (n=2). (E) IFNγ production by neoantigen-specific T cells after 24 hours coincubation with antigen presenting HLA-A3+ NBS fibroblast (gray, vector control; blue, TAP1KO clones) loaded with varying concentrations of the A3-RPL2861-91(p76S>F) SLP. Means with SD are plotted from representative experiments (n=3). (F) IFNγ production by antigen-specific T cells after 24 hours co-incubation with antigen presenting HLA-A2 +NBS fibroblast (gray, vector control; blue, TAP1KO clones) loaded with varying concentrations of either the A2-EML150-80(p64R>W) or A2-MP1 Flu46-75 SLP. Means with SD are plotted from representative experiments (n=2). ANOVA, analysis of variance; DMSO, dimethylsulfoxide; NBS, Nijmegen Breakage Syndrome; SLP, synthetic long peptide.
CRC CAFs and CRC-conditioned fibroblasts show enhanced cross-presentation capacity compared with normal fibroblasts
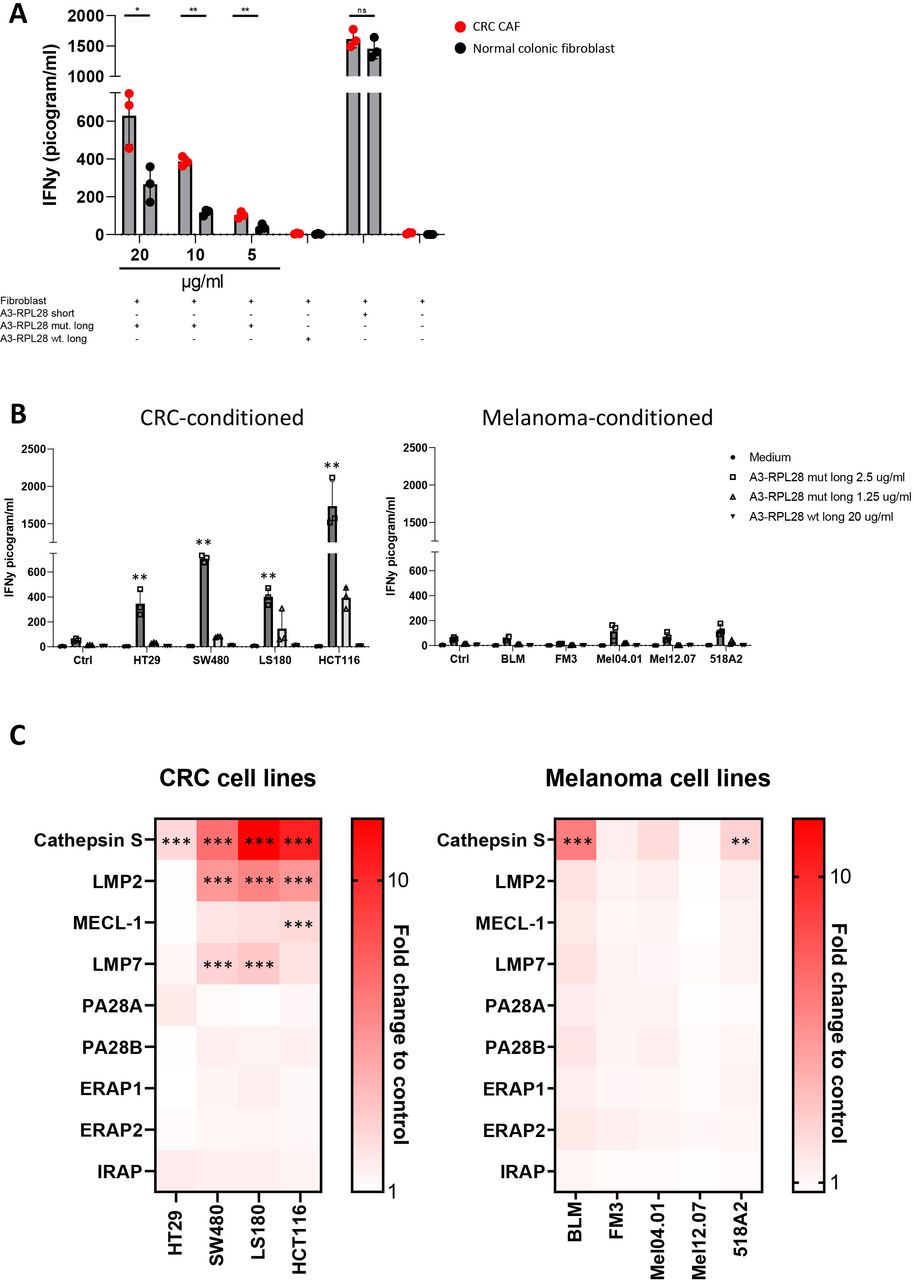
Having established the route of antigen cross-presentation in human fibroblasts, we next asked whether this process was different in human CAFs compared with normal fibroblasts, as was previously shown to be the case in mice.6 Therefore, we investigated T cell activation by antigen-cross-presenting human colon derived CAFs and normal colonic fibroblasts in an HLA-matched setting. HLA-A3+ primary CRC CAFs induced stronger activation of neoantigen-specific T-cells than their matched normal tissue derived counterparts after loading with the A3-RPL2861-91(p76S>F) SLP (figure 4A). The IFNγ response on loading with the short peptide A3-RPL2874-84(p76S>F), that can directly bind to HLA-A3, was similar between CAFs and normal fibroblasts, indicating that only cross-presentation is enhanced in CAFs. Since CAF phenotype and function can be strongly influenced by cues from adjacent tumor cells,37 38 we hypothesized that the enhanced cross-presentation phenotype observed in CAFs might be mimicked by exposing normal fibroblasts to tumor-conditioned medium. Exposure of NBS fibroblasts to CRC-conditioned medium led to upregulation of fibroblast activation protein alpha (FAPα) and vimentin (online supplemental figure 6A,B), indicating a CAF resembling phenotype. Indeed, loading of the A3-RPL2861-91(p76S>F) SLP on CRC-conditioned fibroblasts led to strongly enhanced T-cell reactivity as compared with Ag presentation under physiological conditions by control fibroblasts and professional APCs (allogeneic Mo-DCs and autologous EBV-B cells) (figure 4B; online supplemental figure 1D). This difference was not only observed for the A3-RPL2861-91(p76S>F) SLP but increased T cell activity was also observed after B7-KIAA0020436-466(p451P>L) cross-presentation by CRC-conditioned fibroblasts (online supplemental figure 6C). Interestingly, preconditioning of fibroblasts with conditioned medium from melanoma cells, did not endow fibroblasts with this enhanced cross-presentation phenotype (figure 4B), indicating a CRC-specific mechanism. To study the molecular pathways involved in this enhanced cross-presentation observed in CRC-conditioned fibroblasts, we performed a targeted qPCR-screen to look at the expression levels of genes that have been established to play a role in cross-presentation, both in vitro and in vivo (figure 4C). Strikingly, stimulation of fibroblasts with CRC-derived conditioned media led to strong upregulation of cathepsin S (4/4 tumor cell lines≥2.5 FC, 3/4 tumor cell lines≥5 FC), a member of the group of lysosomal endopeptidases that we show to be critically involved in the cross-presentation route used by fibroblasts. This strong upregulation of cathepsin S (≥5 fold change) was not observed after stimulation with conditioned media from 5 of the six melanoma cell lines investigated. Minor upregulation of the immunoproteasomal subunits LMP2 and LMP7 was also observed after stimulation with CRC-derived conditioned media. Since both cathepsin S and immunoproteasomal subunits are regulated by IFNγ,39 40 we investigated the presence of this and other inflammatory cytokines in both CRC and melanoma-derived conditioned media (online supplemental figure 6D). No IFNγ expression was detected, and none of the other inflammatory cytokines differed between melanoma or CRC-derived conditioned medium, indicating that another, as yet unknown soluble factor is responsible for the observed upregulation. These data indicate that CRC tumor cell-fibroblast cross-talk leads to enhanced cross-presentation capacity and is accompanied by the specific upregulation of cathepsin S, which matches the finding that a lysosomal endopeptidase is responsible for the cross-presentation of exogenous SLP (figure 2D).

Enhanced cross-presentation in CRC-derived CAFs and CRC-conditioned fibroblasts. (A) IFNγ production by neoantigen-specific T cells after 24 hours coincubation with antigen presenting human CRC-derived CAFs (red) and matched, normal colonic fibroblasts (black). Concentration of peptide used for the mutant SLP is depicted. Concentration for wildtype SLP is 20 µg/mL. Means with SD are plotted from representative experiments (n=2). Student’s t-test *p≤0.05, **p≤0.01. (B) IFNγ production by neoantigen-specific T cells after 24 hours coincubation with CRC or melanoma-conditioned fibroblasts that are loaded with A3-RPL2861-91(p76S>F) at 2.5 µg/mL (dark gray), 1.25 µg/mL (light gray) or the control wildtype SLP (20 µg/mL). Data from representative experiments are shown (n=3). Experimental conditions are compared with the unstimulated (Ctrl) condition with one-way ANOVA, **p≤0.01. (C) Fold change to control fibroblast (non-conditioned) in mRNA expression of indicated genes in NBS fibroblasts conditioned with CRC or melanoma-derived conditioned medium for 6 hours. Mean from independent replicates are plotted in the heatmap (n=3). Significance was calculated for conditions with a >2.5 FC in expression as compared with control. Two-tailed ANOVA, **p≤0.01, ***p≤0.001. ANOVA, analysis of variance; CAF, cancer-associated fibroblasts; CRC, colorectal cancer; NBS, Nijmegen Breakage Syndrome; ns, not significant; SLP, synthetic long peptides.
Cross-presentation of SLP is dependent on the lysosomal endopeptidase cathepsin S
In the vacuolar route in DCs, cathepsin S is known to be a lysosomal protease that is involved in generating peptides for cross-presentation both in vitro and in vivo.41 To establish the role of cathepsin S in cross-presentation of the A3-RPL2861-91(p76S>F) SLP in fibroblasts, we generated two, fibroblast cell lines with cathepsin S overexpression (figure 5A,B). As expected, T cell activation was observed in both wildtype, vector control and cathepsin S-overexpressing fibroblasts at high concentrations (10–20 µg/mL) of A3-RPL2861-91(p76S>F) SLP. However, in the lower peptide ranges, only cathepsin S overexpressing fibroblasts are able to activate T cells supporting their increased cross-presenting capacity compared with wildtype fibroblasts and vector control cells (figure 5C). Conversely, shRNA-mediated knockdown of cathepsin S expression (figure 5D) abrogated T-cell reactivity in CRC-conditioned fibroblasts (figure 5E). Combined, these data show that enhanced cross-presentation of the A3-RPL2861-91(p76S>F) SLP in CRC-conditioned fibroblasts is regulated by the lysosomal endopeptidase cathepsin S. Finally, to study whether cathepsin S also mediates cross-presentation of other SLPs, we studied cross-presentation of the A2-EML150-80(p64R>W) SLP by fibroblasts that overexpressed cathepsin S. In contrast to the A3-restricted epitope, processing of this A2-restricted SLP resulted in strongly decreased T cell activity (online supplemental figure 7). The observation that cathepsin S overexpression affects cross-presentation and subsequent T cell activation indicates that this SLP is also processed by the vacuolar pathway, since cathepsin S is not involved in the proteasomal processing route. The fact that cathepsin S overexpression in this case decreases T cell activity suggests that cathepsin S can also destroy neoepitopes during antigen processing. Interestingly, cathepsin S-mediated processing may thus act in two ways (increased or decreased presentation) depending on the neoepitope studied. Altogether, these data show that cathepsin S is crucially involved in fibroblast-mediated cross-presentation of SLPs and the activity of this protease actively contributes to the shape of the MHC-I ligandome that is ultimately presented at the cell surface.

Cathepsin S enhances cross-presentation in fibroblasts. (A) Relative expression of cathepsin S (CTSS) in NBS wildtype (gray bar), vector control (gray bar) and two CTSS overexpressing NBS fibroblasts (#1 and #2, red bars) normalized for expression of β-actin. Student’s t-test ***p≤0.0001 (B) Western blot analysis of cathepsin S expression in NBS wildtype, vector control and CTSS overexpressing fibroblasts. The CTSS/β-actin ration is shown for quantification. (C) IFNγ production by neoantigen-specific T cells after 24 hours coincubation with wildtype (gray bar), vector control (gray bar) and CTSS overexpressing NBS fibroblasts (red bars). Means with SD are plotted from representative experiments (n=4). Two-tailed ANOVA with correction for multiple testing *p≤0.05, **p≤0.01. (D) Expression of cathepsin S in CRC-conditioned fibroblasts with shRNA-mediated cathepsin S knockdown. Student’s t test *p≤0.05. (E) IFNγ production by neoantigen-specific T cells after 24 hours coincubation with cathepsin-S KD (blue bars) and vector control CRC-conditioned NBS fibroblasts (gray bar). Means with SD are plotted from representative experiments (n=2). Two-tailed ANOVA with correction for multiple testing *p≤0.05. ANOVA, analysis of variance; CRC, colorectal cancer; NBS, Nijmegen breakage syndrome.
Cathepsin S is expressed by CRC CAFs in vivo and upregulated in a subset of ex vivo cultured human CAFs
Cathepsin S is expressed by professional APCs in humans,42 but it is unknown whether it is also expressed by human CRC-derived CAFs. Examination of cathepsin S expression in a matching set of primary CRC derived CAFs and CRC-liver-metastasis CAFs, including their matched normal fibroblast counterparts, showed expression in all fibroblasts. However, cathepsin S upregulation was found in a subset of both primary CRC (4 out of 8) and liver-metastasis derived CAFs (4 out of 6) (figure 6A), including the HLA-A3+ CAF that showed enhanced cross-presentation (figure 4A, patient 10). Second, we investigated the expression of cathepsin S in CRC surgical resection specimens. IHC revealed cathepsin S expression in spindle-shaped cells in stromal-rich regions of the tumor (figure 6B). Moreover, CD8 and cathepsin S staining on sequential slides revealed close proximity of CD8+ T cells to cathepsin S-expressing CAFs (online supplemental figure 8). IF double-staining was finally used to show cathepsin S expression in vimentin-positive, spindle-shaped cells (figure 6C, n=3 patients). Altogether, our data demonstrate the high and selective expression of cathepsin S in CAFs in human CRC specimens.

Detection of cathepsin S expression in colorectal cancer CAFs. (A) Relative expression of cathepsin S in primary tumor and liver-metastasis-derived CRC CAFs compared with matched normal fibroblasts. Mean and SD are plotted of a technical triplicate. Student’s t test *p≤0.05. (B) IHC staining of primary CRC tissue for cathepsin S. Black arrows point to cathepsin S positive spindle-shaped cells. (C) IF staining of primary CRC tissue for vimentin (red), DAPI (green) and cathepsin S (pink). CAFs, cancer-associated fibroblasts; CRC, colorectal cancer; IHC, immunohistochemistry; IF, immunohistochemistry.
Fibroblast-mediated antigen cross-presentation results in decreased T cell cytotoxicity
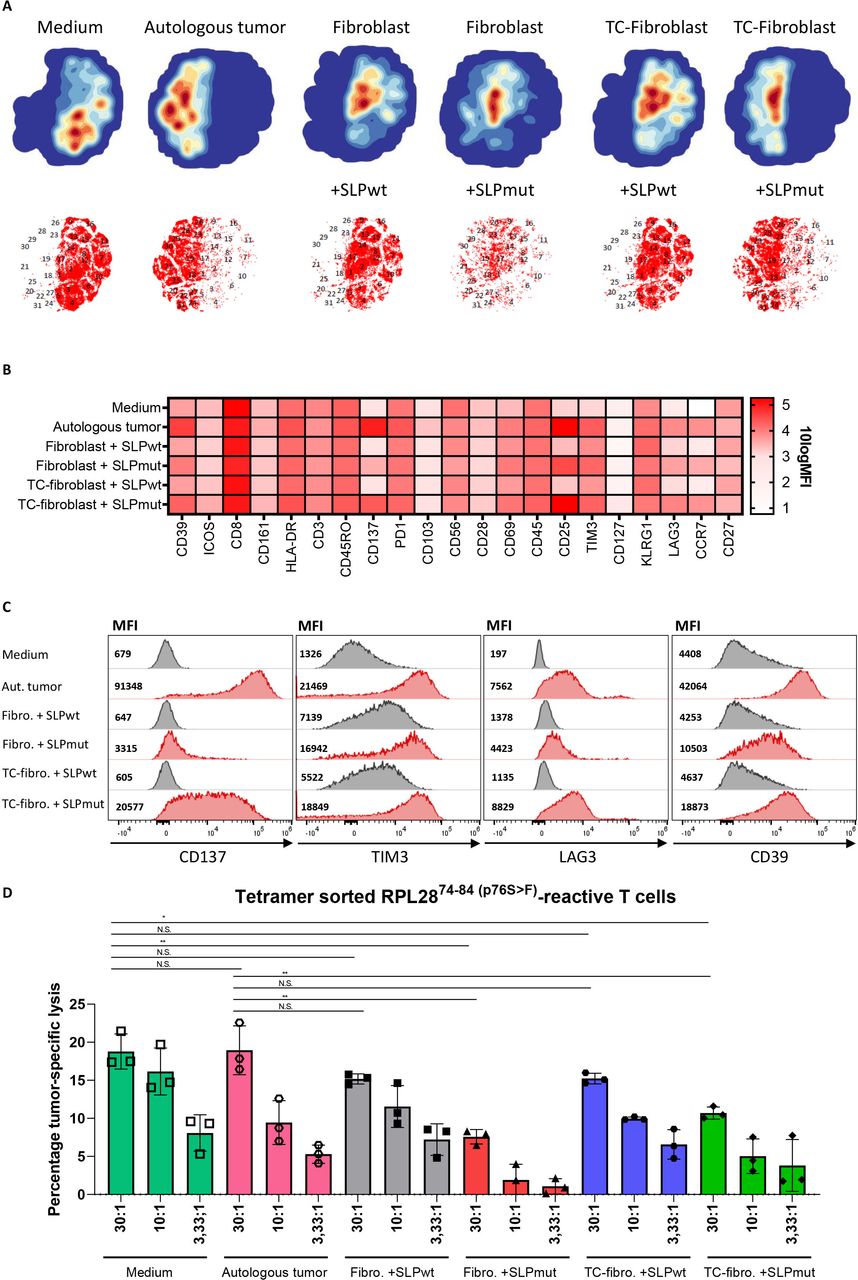
Having established the mechanism and route of antigen cross-presentation in CAFs, we investigated the effect of fibroblast-mediated antigen presentation on T cell function using a multispectral flow cytometry panel consisting of activating and inhibitory molecules. A3-RPL2874-84(p76S>F)-tetramer specific T cells were sorted from a neoantigen-specific T cell bulk to have a pure antigen-restricted T cell population (online supplemental figure 9). Tetramer-positive T cells were expanded and subsequently incubated with medium, autologous tumor cells or CRC-conditioned fibroblasts loaded with either the neoantigen or wildtype A3-RPL28 SLP. Hierarchical clustering of the multispectral flow cytometry data of all conditions revealed a cluster partitioning consisting of 31 clusters (figure 7A, online supplemental figure 10A). The tSNE plots of individual conditions show a striking difference between conditions with antigen presence or absence, most notably with regard to the expression of CD137, T-cell immunoglobulin and mucin-domain containing-3 (TIM3), lymphocyte-activation gene 3 (LAG3) and CD39 (figure 7B,C; online supplemental figure 10B-D). Interestingly, T cells that are exposed to fibroblasts that cross-presented the neoantigen-based SLP, showed upregulation of both activating and inhibitory markers and this effect was strongest in the CRC-conditioned fibroblasts. However, the expression of the activating marker CD137 in the conditions where fibroblast presented the SLP remained lower than that elicited by presentation of the endogenous neoantigen by autologous tumor cells, indicating suboptimal T cell activation in conditions where fibroblasts present the antigen (figure 7C). Notably, expression of TIM3, LAG3 and CD39, known inhibitory checkpoint molecules, was upregulated in T cells exposed to antigen-presenting, CRC-conditioned fibroblasts (figure 7C). Both LAG3 and TIM-3 also showed an antigen-independent, fibroblast-dependent upregulation while CD39 was upregulated in both an antigen-dependent and fibroblast-dependent fashion. In comparison to the autologous tumor condition, antigen-presenting, tumor-conditioned fibroblast showed less activation (CD137, 4.5-fold decrease) while the inhibitory markers TIM3 and LAG3 showed a similar expression. Although T cell activation depends on a complex interplay and balance of antigen-specific and co-inhibitory and stimulatory interactions, these results may imply that the net result of T cell activation by fibroblasts could be skewed to a more exhausted effector function. Both fibroblast as well as tumor cells do not express co-stimulatory molecules (CD80/CD86) and differ in this respect from professional APCs. To study costimulatory-independent effects of fibroblast-mediated antigen presentation on T cells, we investigated the tumor-cell killing capacity of T cells that were pre-exposed to Ag cross-presenting CAFs or autologous tumor cells (endogenously expressing the neoantigen) and compared that to the tumor-cell killing capacity of T cells that were precultured in medium only. Interestingly, we observed that pre-exposure of specific T cells with autologous tumor cells did not reduce their subsequent tumor-cell cytotoxicity. This shows that the recovery time of the pre-exposed T cells is sufficient to test the impact of pre-exposure to fibroblasts. Pre-exposure of T cells to both normal or CRC-conditioned antigen-presenting fibroblasts led to a significant decrease in tumor-cell killing by neoantigen-specific CD8+ T cells (figure 7D). However, no apparent difference was found between normal and CRC-conditioned fibroblast conditions, indicating that antigen cross-presentation by fibroblasts in itself is sufficient to cause CD8+ T cell dysfunction. A previous study in mice6 showed that CD8+ tumor-specific T cell function was abrogated due to fibroblast-mediated killing of T cells that bound to HLA-I/peptide complexes on the fibroblast cell surface via interaction with both PD-L2 and FAS-L. However, we found no evidence for fibroblast-induced T cell death, since T cell numbers did not decrease after fibroblast-mediated SLP cross-presentation (online supplemental figure 11A). Moreover, we could not detect FAS-L expression in our primary, CRC-derived CAFs (online supplemental figure 11B). In conclusion, we show that cross-presentation of antigen by human fibroblasts impairs CD8+ T cell effector function and is accompanied by a decrease in activating (CD137) and increase in inhibitory (TIM3, LAG3 and CD39) checkpoint molecule expression when compared with cognate interactions between tumor cells and T cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fibroblast-mediated antigen cross-presentation results in decreased T cell cytotoxicity. (A) tSNE plots of the expression of activating and inhibitory surface receptors on A3-RPL2874-84(p76S>F) sorted T-cells incubated for 24 hours with medium, autologous tumor cells and (CRC-conditioned) fibroblasts loaded with either A3-RPL2861-91(wildtype) or A3-RPL2861-91(p76S>F). (B) Heatmap showing the 10logMFI of the separate T cell surface markers in the different experimental conditions. (C) Histograms and MFI of CD137, TIM3, LAG3 and CD39 in the different experimental conditions. (D) Percentage specific lysis of autologous tumor cells, determined by26Cr-release assay, by A3-RPL2874-84(p76S>F) sorted T cells after preincubation with the indicated target cell conditions. Means with SD for the 30:1 ratio are plotted from representative experiments (n=2). *P<0.05, **p<0.01 as determined by one-way ANOVA with correction for multiple testing (Dunnett’s test). ANOVA, analysis of variance; tSNE, t-distributed stochastic neighbor embedding CRC, colorectal cancer; MFI, mean fluorescent intensity; ns, not sognificant; TC, tumor conditioned.
Discussion
The ability of fibroblasts to interfere with tumor-specific CD8+ T cell function is thought to be mainly reliant on antigen-independent mechanisms, including T cell exclusion and hampering of DC maturation.5 7 43–48 In this study, we show the ability of human CRC-derived CAFs to process and cross-present an exogenous, SLP-derived epitope to neoantigen-specific T cells and thereby directly influence T cell activation status, resulting in a decrease of cytotoxic CD8+ T cell function.
Our data show that fibroblasts can process peptides in a cathepsin S dependent manner and present the resulting epitope on HLA-I via the vacuolar route.11 Mechanistically, we show that SLPs are processed by cathepsin S and that this ultimately results in the trimming of the model antigen in our study (31-mer SLP) to an epitope (11-mer) which is then presented in an HLA-I-peptide complex on the cell surface of fibroblasts. Alternatively, cathepsin S can also lead to destruction of immunogenic epitopes and in this way shape the antigen repertoire presented by cross-presenting fibroblasts. Cathepsin S is considered to be a key protease involved in processing peptides for both the MHC-class II route and cross-presentation in professional APCs.41 49 Cathepsin S expression is mostly confined to these bone-marrow derived professional APCs, but also shown to be expressed and play an important role in antigen processing in another non-professional APC, intestinal epithelial cells.50 Expression of cathepsin S has been reported in lung fibroblasts51 and we show here that cathepsin S expression was also detected in tissue of CRC patients and cultured CAFs from both primary CRC and liver-metastasized CRC, pointing to an until now unappreciated role of this protease in human CAFs in vivo. This indicates that CAFs in CRC have the potential to serve as non-professional APCs. In this regard, it is interesting to note that certain colon fibroblasts8 have been reported to express MHC-II and that cathepsin S might therefore also be involved in generating peptides for MHC-II restricted antigens in this fibroblast subset.
Presentation of the SLP epitope partially relied on the heterodimeric TAP-complex for sufficient efflux of HLA-I molecules from the ER to which antigen formed through lysosomal processing could subsequently bind. Of note, in the tumor cell that harbors the neoantigen on which the SLP is based, endogenous processing results in exactly the same A3-RPL2874-84(p76S>F) epitope and it may well be that TAP translocation of the processed epitope to the ER is required in this setting. However, the same epitope can also be formed in fibroblasts through processing in the vacuolar pathway and TAP appears not to be required for translocation of the epitope in the ER. This paradoxal TAP-dependency has been previously observed in cross-presentation of SLPs in DCs,14 showing similarity between the route of SLP cross-presentation in fibroblasts and professional APCs. Moreover, previous studies have shown that fibroblasts can be engineered into a DC-like state by overexpression of BATF3, IRF8 and PU.1 and become equally proficient in antigen presentation and induction of cytotoxic T cell responses as professional APCs.26 52 It would be of interest to study the role of these transcription factors in the enhanced cross-presentation phenotype observed in CRC-derived CAFs and CRC-conditioned fibroblasts. However, the tumor-derived factor that drives this enhanced cross-presentation phenotype remains to be elucidated but was specifically present in CRC tumor-conditioned and not melanoma-conditioned medium, indicating that it is a tumor-type specific phenomenon. Enhanced capacity for cross-presentation in CAFs has previously been observed in the B16-F10 murine melanoma model.6 In this study, similar to our findings, cross-presentation of the OVA-antigen was reliant on endolysosomal processing and more efficient in CAFs as compared with normal fibroblasts, showing that the mode of antigen cross-presentation shows similarities between SLPs and full proteins. Intriguingly, our data show that exposure of fibroblasts to melanoma conditioned-medium does not enhance the cross-presentation capacity in human fibroblasts, indicating that the factors driving enhanced cross-presentation in that model might not be tumor cell-derived. In this study, the cross-presenting abilities of fibroblasts compared with professional APCs were found to be similar under physiological conditions but became more efficient in tumor-conditioned fibroblasts, similar to findings from other studies,53 indicating that the capacity to cross-present is context-dependent and clearly enhanced in the tumor setting.
Finally, we show that antigen cross-presentation by tumor-conditioned fibroblasts results in upregulation of both activating (CD137) and inhibitory (TIM3, LAG3 and CD39) receptors on neoantigen-specific T cells and that fibroblast-mediated antigen presentation results in diminished cytotoxic T cell function. However, upregulation of the activation marker CD137 was strongly increased after T cells were exposed to autologous tumor cells as compared with exposure to the antigen-presenting (tumor-conditioned) fibroblasts while the expression of TIM3, LAG3 and CD39 was almost similar. This suggests that cross-presentation of antigen by fibroblasts results in suboptimal activation of T cells. This could be an explanation for the observed decrease in cytotoxicity of T cells that are pre-exposed to antigen-presenting fibroblasts as compared with autologous tumor cells, in which there was no decrease in their subsequent ability to kill tumor cells as has been shown previously.54 Surprisingly, the extent to which fibroblasts decreased CD8+ T cell cytotoxicity did not differ between normal fibroblasts and CRC-conditioned fibroblast. However, it is important to note that in our experiments, antigen cross-presentation was observed at far lower concentrations of antigen in CAFs, tumor-conditioned fibroblasts and cathepsin S overexpressing fibroblasts as compared with normal fibroblasts. Thus, although there are no differences in the net effect on CD8+ T cell function, the antigen load at which cross-presentation can occur might be more physiologically relevant in CAFs as compared with normal fibroblasts. Diminished cytotoxic T cell function after pre-conditioning with antigen-loaded CAFs was also observed in the B16-F10 murine model6 and shown to be dependent on PD-L2 and FAS-L engagement, resulting in T cell apoptosis. However, we did not observe apoptosis induction in neoantigen-specific T cells that were exposed to antigen-presenting fibroblasts. Moreover, none of the human CRC-derived CAFs used in our study showed mRNA expression of FAS-L indicating that another mechanism might be involved in the reduction of cytotoxic effector function. In this regard, the upregulation of TIM-3, LAG3 and CD39 observed after T cell incubation with antigen-loaded tumor-conditioned fibroblasts might be of particular interest to investigate further, since antagonistic antibodies are under clinical development and might be able to partially relieve the fibroblast-imposed CD8+ T cell dysfunction.55–57 It would be of considerable interest to phenotypically characterize CD8+ T cells that are adjacent to CAFs in vivo, to see whether the pattern of activating and inhibitory receptor expression is similar to what we observe in vitro. Moreover, since T cell migratory capacity is linked to activation status58 it is tempting to speculate that CAFs might not only form a physical barrier that hampers T cell migration but that CD8+ T cells might also become ‘trapped’ in the stroma due to continuous antigen stimulation by cross-presenting fibroblasts. Finally, if cross-presentation by CAFs occurs in vivo this would also imply that there might be competition between professional APCs and non-professional APCs with regards to the available antigen. Stroma-rich tumors, such as CRC, that contain substantial amounts of CAFs might thus prevent adequate uptake of tumor-associated antigen by professional APCs, thereby also hampering an effective CD8+ T cell response indirectly.
Due to the unavailability of sufficient CRC-derived antigen-specific T cells, we used T cells specific for multiple neoantigens previously detected in melanoma patients.17 This model system allowed us to study clinically relevant tumor-specific T cells in a completely human setting. Where possible we used primary colonic CAFs, complemented by mechanistic studies in the NBS fibroblast cell line to investigate the mode of fibroblast-mediated cross-presentation and its subsequent effect on tumor-specific T cell function. Whether the enhanced cross-presenting capacity of SLPs by CAFs and CRC-conditioned fibroblasts also holds true for full proteins and other particulate antigens has not been studied here but similar to uptake of SLPs, pinocytosis has been described as an uptake route for this class of antigens suggesting similarities in the mode of antigen processing.
In conclusion, our findings show the capability of human CAFs to cross-present antigen. This process is enhanced in human CAFs and CRC-conditioned fibroblasts and requires the expression of the lysosomal endopeptidase cathepsin S. Fibroblast-mediated antigen presentation results in the upregulation of both activating and inhibitory receptors, of which the net result is a decrease in cytotoxic CD8+ T cell function. Our results demonstrate that CAFs, next to a plethora of indirect mechanisms, can also directly interfere with CD8+ T cell function and that this mechanism might serve as a potential target for the improvement of T cell-based immunotherapy in human CRC.
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442-D450, doi:10.1093/nar/gky1106 (2019), with the dataset identifier PXD029507.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
We thank Noel de Miranda (Department of Pathology, LUMC, The Netherlands) for providing critical feedback. We thank the Flow cytometry Core Facility (FCF) of Leiden University Medical Center (LUMC) in Leiden, the Netherlands, for technical support and cell sorting assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LJH and EMV contributed equally.
Contributors TH planned and performed majority of experiments and associated data analysis; MV, LdB and LP provided technical assistance; AM assisted in HLA-typing of primary fibroblasts; PAVV planned and performed mass-spectrometry experiments; GvdHvN, MJ and JN designed and synthesized peptides and assisted in spinning disk confocal microscopy. MMH and EW were involved in generating and verifying KO lines; LG and TVH isolated and characterized TEIPP-specific T cells; SJS co-performed the multispectral flow cytometry experiment; JH, SHVdB, LH and EV cosupervised and performed data interpretation. TH, LH and EV cowrote the paper. EV accepts full responsibility for the work and/or the conduct of the study. All authors contributed to editing of manuscript and critical review.
Funding TH is sponsored by a personal PhD grant from the Leiden University Medical Center. EV is sponsored by a grant from the Dutch Cancer Society-KWF grant 2017-10815. GvdHvN is supported by NWO (VIDI grant 192.011). This work was supported by the Dutch Research Council (NWO) Medium Investment Grant 91116004 (partly financed by ZonMw) to PAVV.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.