Article Text

Statistics from Altmetric.com
Background
Radiotherapy is employed as a standard cancer therapy for more than 60% of patients with cancer.1 It is used for controlling the growth of local tumors by inducing DNA damage and promoting apoptosis in tumor cells.2 Additionally, local radiation therapy promotes the release of diverse tumor-associated antigens that can be engulfed by antigen presenting cells to stimulate the tumor-specific immune response and then presented to the CD8 T cells to eradicate tumor cells.2 3 However, radiotherapy can simultaneously recruit immunosuppressive cells such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) into the tumor microenvironment,4–6 which are strongly associated with the regrowth and radio-resistance of the tumor.7 Therefore, combined use of a therapy that targets the immunosuppressive tumor microenvironment is required to enhance the antitumor immunity after radiotherapy. Previous studies have demonstrated that the abscopal effect, defined as the induction of a systemic antitumor response after localized radiotherapy,8 is mediated by effector CD8 T cells and inhibition of immunosuppressive cells.9 10 Demaria et al first reported that immune checkpoint blockade enhances the abscopal effect of local irradiation in preclinical syngeneic mice models.11 Since the advent of immunotherapy, many preclinical and clinical studies have evaluated the abscopal effect after the combination treatment of radiation and immunotherapy during the past decade.3 However, the abscopal effect is still rarely observed in patients with cancer, and its underlying mechanisms are unclear.3 8
The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is essential for cell growth, motility, survival, metabolism, and survival.12 Activation of the PI3K/AKT/mTOR pathway contributes to the development of tumors and resistance to radiotherapy and chemotherapy.13 PI3Ks are classified into three different classes (class Ⅰ, Ⅱ and Ⅲ).12 Class I PI3Ks are heterodimeric molecules composed of a catalytic subunit (p110α, β, δ, and γ) and a regulatory subunit (p85).14 One of PI3K isoforms, PI3Kα is frequently mutated or amplified in the most common human cancers.12 Thus, class I PI3Kα are the clinically-validated targets for antitumor treatment or its combination with radiotherapy or chemotherapy.15 16 Notably, PI3Kα or β are expressed in all nucleated cells, but PI3Kδ or γ are explicitly expressed in leukocytes, suggesting the role of PI3Kδ/γ in the modulation of tumor microenvironment by regulating the immune cells.17 For instance, inactivation of PI3Kδ in Tregs activates CD8 T cell-mediated antitumor activity,18 and inhibition of PI3Kγ in myeloid cells enhances immunotherapy in preclinical tumor models.19 20 Therefore, these studies suggest that PI3Kδ/γ inhibition could restrain the immune-suppressive activity in tumor microenvironment following radiotherapy. However, the role of PI3Kδ/γ in radiation-induced antitumor immunity and the abscopal effect has not been studied yet.
Herein, we used a PI3Kδ/γ inhibitor, BR101801, a drug in phase I clinical trial, to understand the role of PI3Kδ/γ in inducing antitumor immunity after radiotherapy. For the first time, we show that PI3Kδ/γ inhibition along with local irradiation potentiates the effector CD8α+ T cell-mediated antitumor immunity in syngeneic mouse model.
Materials and methods
Mice and tumor implantation
BALB/c and C57BL/6 mice (6-week-old-female) were obtained from Orient Bio (Gyeonggi-do,Korea).
CT-26, MC38 and LL/2 (LLC1) cell-lines were purchased from American Type Culture Collection (VA, USA). All cell-lines were cultured in dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS, 35–015-CVR; Corning, New York, USA) and 1% penicillin/streptomycin (P/S, 30–002 CI; Corning). Once thawed, the cells were cultured in an incubator at 37 °C with 5% carbon dioxide and 95% air and passed twice prior to tumor implantation. 2×105 CT-26 cells, 5×105 MC38 cells, and 4×105 LL/2 cells in 50 µL of Dulbecco's phosphate buffered saline (DPBS) were subcutaneously injected into the right flank of the mice. For rechallenge and abscopal effect experiments, 2×105 CT-26 cells were subcutaneously injected into the left flank of the mice. Tumor growth was monitored by measuring the perpendicular diameters (width/length) every 2 or 3 days, and the tumor volume was calculated using the formula {(width2 × length)/2}.
Irradiation
Mice with a palpable sized (<150 mm3) tumor were selected. Tumors on the right flank of the mouse received 7.5 Gy of 200 kVp X-Ray local RT, using X-RAD 320 (Precision X-Ray, Connecticut, USA). Each anesthetized mouse was placed on a fixed shelf with an illuminator, indicating the irradiation site in X-RAD 320, to expose only the tumor and prevent the radiation from reaching any other part of the mouse.
PI3Kδ/γ inhibitor administration
The PI3Kδ/γ inhibitor BR101801 was prepared using the methods described in Korea patent 10-2016-0150006. Briefly, BR101801 was dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. Mice were intragastrically administered 50 mg/kg BR101801 (Boryung, Seoul, Korea) in a 200 µL compound mixture or with vehicle control {5% DMSO +55% polyethylene glycol 400 (Sigma-Aldrich, MO, USA)+40% deionized sterile water}. The compound mixture for drinking was filtered by passing through a 0.22 µm filter (Corning) and stored at 4°C. Once dissolved, diluted compounds were not used for over a week.
Flow cytometry
The excised tumor mass was soaked in media containing enzymes (Tumor dissociation kit; Miltenyi Biotec, Bergisch Gladbach, Germany) to facilitate tumor dissociation, according to the manufacturer’s recommendations. The dissected lymph nodes adjacent to the tumor were mechanically squished using a 70 µm nylon cell strainer (SPL Life sciences, Gyeonggi-do, Korea) to isolate single cells and washed under running buffer (0.5% bovine serum albumin in PBS). The spleen was homogenized on a 70 µm nylon cell strainer, and red blood cells were lysed with 1×RBC lysis buffer (Sigma-Aldrich, Missouri, USA), filtered through a 70 µm nylon cell strainer, and counted.
Single cells were stained using Fixable Viability Stain 780 (BD, New Jersey, USA), Fixable Viability Stain 620 (BD), or zombie NIR Fixable Viability (BioLegend, California, USA) for 15 min at room temperature (25°C–27°C) and incubated with anti-CD16/32 antibodies (Thermo Fischer Scientific, MA, USA) for 15 min at room temperature. Then, the cells were placed on ice and surface-labled with monoclonal antibodies (online supplemental table 1) for 30 min. All events were acquired and analyzed using a CytoFLEX flow cytometer (Beckman Coulter, California, USA). All flow cytometric data analyses were performed using Kaluza Analysis software (Beckman Coulter) and FlowJo software (BD).
Supplemental material
Intracellular staining
To examine the capacity to release IFN-γ and granzyme B, the cell suspensions were stimulated with 1 µM phorbol 12-myristate 13-acetate (PMA) (Sigma) and ionomycin (Sigma) at 37 °C for 2 hour. The cells were fixed and permeabilized using Forkhead box P3 (FoxP3)/Transcription Factor Staining Buffer Set (Thermo Fischer Scientific) following the manufacturer’s recommendations. Thereafter, cells were stained with monoclonal antibodies (online supplemental table 2) for 30 min on ice.
Immune cell depletion
Mice were treated weekly with 200 µg anti-CD4 antibodies (clone GK1.5, Bio X Cell, NH, USA), 200 µg anti-CD8α (clone 2.43, Bio X Cell), 10 µL anti-asialo GM1 antiserum (Wako Pure Chemical Industries, Osaka, Japan), or rat IgG2b isotype control (clone LT F-2, Bio X Cell) by intraperitoneal injection 3 days prior to irradiation. CD8+, CD4+, T cells, and NK cell depletion were confirmed by flow cytometry.
NanoString
Intratumoral CD45+ cells were isolated using the EasySep Mouse CD45+ Cell Isolation Kit (StemCell Technologies, Vancouver, Canada), following the manufacturer’s instructions. Briefly, single cells digested from the tumor mass were resuspended in PBS with 5% FBS and 1 mM EDTA (FACS buffer) and magnetically labeled in a two-step process. Cells were incubated for 8 min at room temperature with a CD45 positive selection cocktail and tetrameric antibody complexes in tubes. Thereafter, the tubes containing the mixture were placed into EasySep magnetic for 5 min, and the supernatant was discarded. Finally, magnetically labeled CD45+ cells were resuspended in FACS buffer.
RNA was isolated from the sorted intratumoral CD45+ cells using the MasterPure complete RNA Purification Kit (Lucigen, MA, USA). NanoString analysis was conducted using the nCounter Analysis System (NanoString Technologies, Washington, USA). The nCounter Mouse Immunology Kit (NanoString) was used to analyze 561 immunology-related mouse genes.
ELISpot
CD8+ T cells from tumor-draining lymph nodes (TDLNs) were isolated using the EasySep Mouse CD8+ T Cell Isolation Kit (StemCell Technologies). Isolated CD8+ T cells (1×105 cells/well) and irradiated (30 Gy) CT-26 cells (1×105 cells/well) were co-cultured on anti-IFN-γ mAb coated strip plates (Mabtech, Stockholm, Sweden) for 48 hours. ELISpot assay was performed according to the manufacturer’s instructions. The cytokine spots of IFN-γ were counted using the EliSpot Reader System (AID iSPOT ELR07IFL, Germany).
T cell proliferation and Treg functional assay
Splenic Tregs from tumor-bearing mice were collected 5 days after irradiation, and the CD3+CD4+CD25+CD127- cells were sorted on a flow cytometer (FACSAria III, BD). Naive splenic CD8+ T cells were isolated using the EasySep Mouse CD8+ T Cell Isolation Kit (StemCell Technologies). Naive splenic CD4+ T cells and Treg cells were isolated using the EasySep Mouse CD4+CD25+ Regulatory T Cell Isolation Kit II (StemCell Technologies).
For the Treg suppression assay, naive splenic CD8+ T cells were incubated with 5 µM CellTrace CFSE (Thermo Fischer Scientific) for 15 min. Sorted 1×105 Tregs and labeled 1×105 CD8+ T cells were co-cultured in complete RPMI1640 medium containing 10% FBS, 1% P/S, 1 mM sodium pyruvate (Corning), 10 mM HEPES buffer (Corning), 1×MEM NEAA (Gibco, MA, USA), and 1× β-mercaptoethanol (Gibco) at a ratio of effector to responder (1:1). Labeled CD8+ T cells were stimulated in a flat-bottom 96-well plate coated with 2 µg/mL anti-CD3 (clone 145–2 C11, Bio X Cell) and soluble 1 µg/mL anti-CD28 (clone 37.51, Bio X Cell). After 4 days, the cells were harvested and analyzed for CFSE dilution by flow cytometry.
To investigate naive T cell proliferation, isolated naive CD8+ T cells, and Tregs were stained with 5 µM CellTrace CFSE and then cultured with PI3Kδ/γ inhibitor (BR101801) in serial dilutions, starting from 1 µM. Additionally, the cells were stimulated with DynabeadsTM Mouse T-Activator CD3/CD28 (Gibco) at a 1:1 cell-to-bead ratio in a V-bottom 96-well plate. Tregs were cultured in the presence of rhIL-2 (300 U/mL, Proleukin; Novartis, Basel, Switzerland) and TGF-β1 (5 ng/mL, Peprotech, NJ, USA). After 3 days, the cells were analyzed for CFSE dilution and counted using precision count beads (BioLegend).
Quantitative RT-PCR
Total RNA was isolated from the tumor mass using the MasterPureTM complete RNA Purification Kit (Lucigen) following the manufacturer’s instructions and reverse transcribed using the ImProm-II reverse transcription system (Promega, Wisconsin, USA). Quantitative RT-PCR was performed on a Chromo 4 Cycler (Bio-Rad, California, USA) using Power SYBR green PCR master mix (Applied Biosystems, Massachusett, USA) with PCR primers (online supplemental table 3).
Statistical analysis
A two-tailed Student’s t-test was performed to analyze the statistical differences between the groups. Tumor growth statistics were calculated using two-way ANOVA with Tukey’s analysis using GraphPad Prism (GraphPad software, California, USA). One-way ANOVA with Tukey’s post hoc test was used to compare data between multiple groups. Differences were considered statistically significant at p<0.05. All data are presented as mean±SEM.
Results
BR101801 combined with irradiation synergistically increased the antitumor efficacy in a syngeneic mouse model of colorectal carcinoma
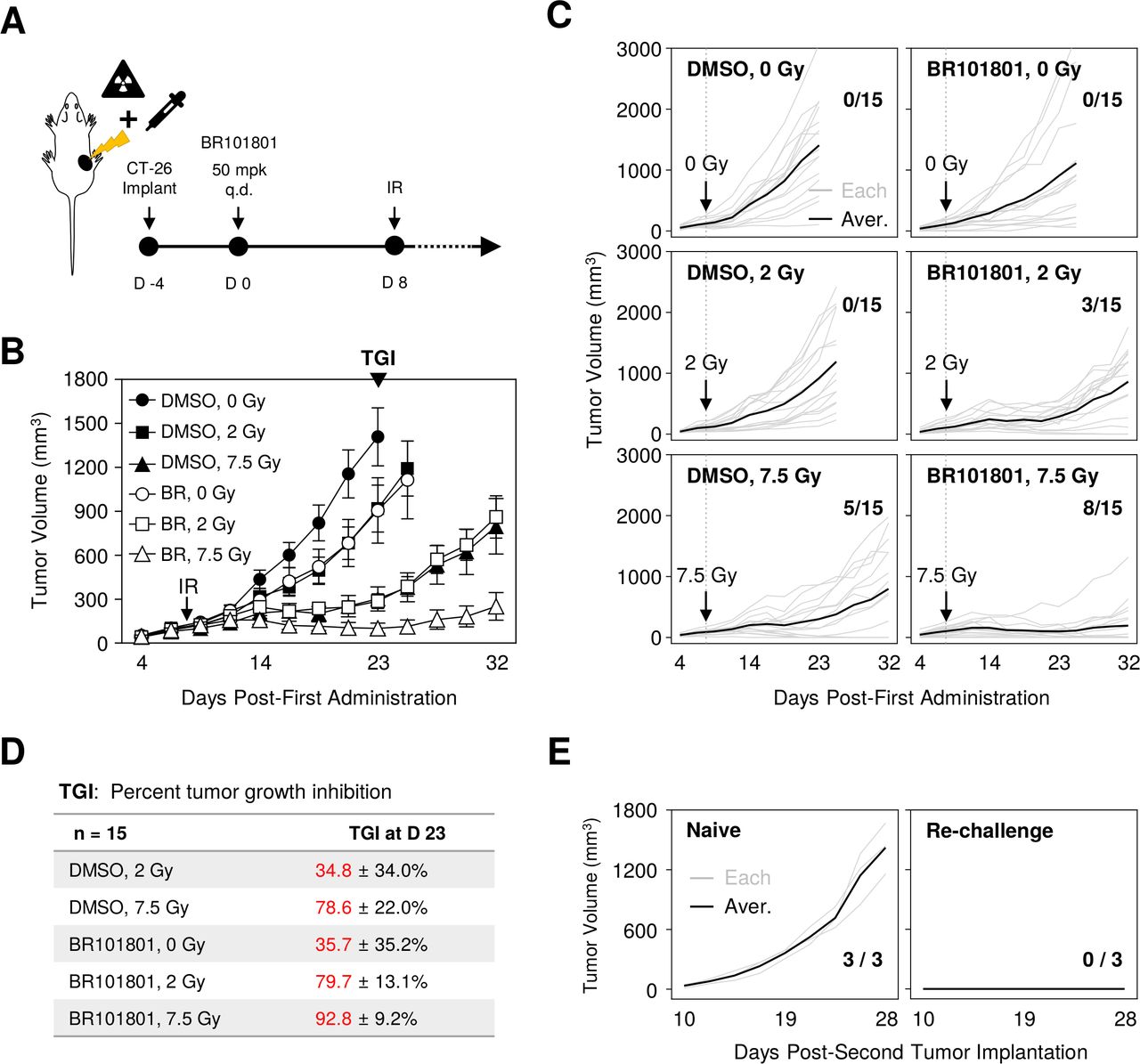
We first examined whether PI3Kδ/γ inhibition could synergistically enhance the antitumor efficacy after irradiation in solid tumors. In this study, we used, BR101801 [4-[[(1S)−1-(4,8-Dichloro-1-oxo-2-phenyl-3-isoquinolyl)ethyil]amino]−8Hpyrido[2,3-d]pyrimidin-5-one], a new PI3Kδ/γ inhibitor (online supplemental figure 1A), which shows highly selective and potent inhibitory effects against PI3Kγ and δ with IC50 values of 15 and 2 nM, respectively (online supplemental figure 1B); it is currently in phase I clinical trials for lymphoma (NCT04018248).21 22 Additionally, a CT-26 (colorectal cancer cell line derived from BALB/c mouse) tumor model, which has a highly immunogenic profile,23 was used to determine the antitumor effect. Mice with palpable tumors were administered BR101801 (50 mg/kg) daily, and the tumor mass was locally irradiated (figure 1A). The optimal irradiation dose was selected as 7.5 Gy showing the most stable and constant CT-26 tumor growth tendency with a mean tumor growth inhibition (TGI) rate of 78.2%±8.0% (online supplemental figure 2). Our data indicated that BR101801 combined with irradiation significantly reduced the tumor growth relative to BR101801 or irradiation (two or 7.5 Gy) alone group (BR101801 plus 7.5 Gy; TGI, 92.8±9.2% vs. BR101801 alone; TGI: 35.7±35.2% or 7.5 Gy; TGI: 78.6%±22.0% at day 23) (figure 1B–D). Additionally, the synergistic antitumor effect was observed in both the MC38 and LL/2 syngeneic tumor models (online supplemental figure 3). Although the CT-26 tumor mass of the control group reached a mean of 1408 mm3 at the end point, mice treated with BR101801 and 7.5 Gy irradiation had a tumor burden with a mean of 101 mm3 at day 23 (figure 1B). Notably, additional 9 days later, a complete response was observed for eight of the fifteen mice treated with BR101801 plus 7.5 Gy irradiation at day 32 (figure 1C), suggesting that PI3Kδ/γ inhibition and irradiation synergistically increases the antitumor effect in CT-26 tumor model.
Supplemental material

Combining BR101801 and irradiation synergistically increases the antitumor efficacy in CT-26 syngeneic mouse model. (A) Schematic representation of PI3Kδ/γ inhibitor (BR101801) schedules and local irradiation. BALB/c mice-bearing CT-26 colon cancer orally administrated (q.d.) with 50 mg/kg BR101801 for 32 days. The tumor mass was locally irradiated 8 days after the first dose of BR101801. (B) Mean tumor volume of subcutaneous (CT-26) implants in BR101801 and irradiation treated mice (n=15 mice per group). (C) Individual CT-26 tumor growth curves with the number of mice showing complete antitumor response at the end point (black line indicates the mean tumor volume in each group; gray line indicates individual tumor growth). (D) Percentages of tumor growth inhibitor in each group. (E) Tumor growth following CT-26 cells rechallenge to naive mice (left) and complete responder mice (right) with the number of mice showing tumor mass (n=3 mice in the naive and re-implanted group). DMSO, dimethyl sulfoxide.
Since BR101801 marginally modulated radiosensitivity of CT-26 cells in vitro (online supplemental figure 4), we assumed that the antitumor effect of the combined treatment could be associated with antitumor immunity. Next, to determine whether BR101801 and irradiation together induce tumor-specific memory response, naïve and complete responder mice were reimplanted with identical amounts of CT-26 tumors on the opposite flank, and the tumor volume was monitored regularly (figure 1E). Interestingly, all three complete responder mice rejected tumor formation, contrary to naive mice-bearing tumor burden (figure 1E). Therefore, our data suggest that BR101801 combined with irradiation synergistically enhances antitumor efficacy with tumor-specific memory response in CT-26 tumors.
CD8α+ T cells are essential for maintaining the antitumor effect of BR101801 plus irradiation
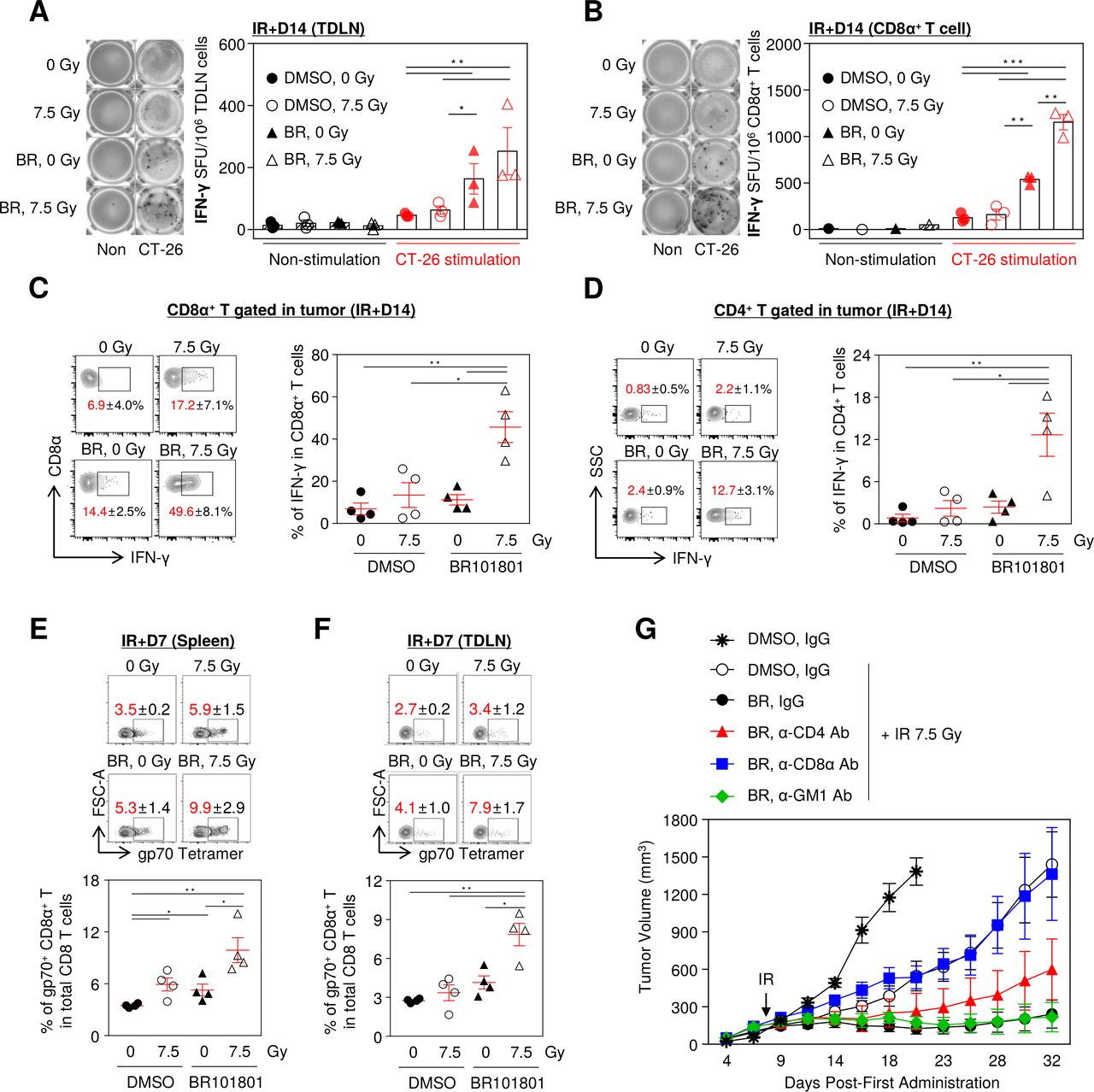
Given that complete responder mice showed rejection of CT-26 tumor re-implantation, we hypothesized that the tumor-specific T cell responses are required for antitumor activity. To explore effector T cell-mediated responses, we performed an ELISpot assay to analyze the IFN-γ secretion from TDLNs in tumor-bearing mice. TDLNs were isolated 14 days after 7.5 Gy irradiation and 30 Gy irradiated CT-26 cells were used as a source of tumor antigen stimulation. Owing to the combined therapy, the tumor-specific IFN-γ-secreting cells increased significantly in all TDLN cells (figure 2A). Also, the highest number of IFN-γ-secreting CD8α+ T cells isolated from TDLN were observed in the combination group (figure 2B). Moreover, a considerable amount of IFN-γ was released from intratumoral CD8α+ and CD4+ T cells in the combination treatment group following PMA and ionomycin stimulation (figure 2C,D). Further, we measured the distribution of tumor antigen-specific CD8α+ T cells which influence antitumor immunity in lymphoid organs including the spleen and TDLN. As expected, the frequency of gp70-specific CD8α+ T cells was the highest in the combination treatment (figure 2E,F). Further, to determine which immune cells are primarily responsible for maximal efficacy, we depleted CD4+, CD8α+ T cells, and NK cells with specific antibodies. Notably, the depletion of CD8α+ T cells significantly eliminated the antitumor activity of the combination treatment, indicating that the antitumor effect was mediated by effector CD8α+ T cells. In contrast, CD4+ T cell depletion marginally attenuated the antitumor activity, and depletion of NK cells did not affect the antitumor activity (figure 2G and online supplemental figure 5). Thus, our data suggest that IFN-γ secreting tumor antigen-specific CD8α+ T cells are the key players in enhancing the antitumor efficacy of BR101801 in combination with irradiation.
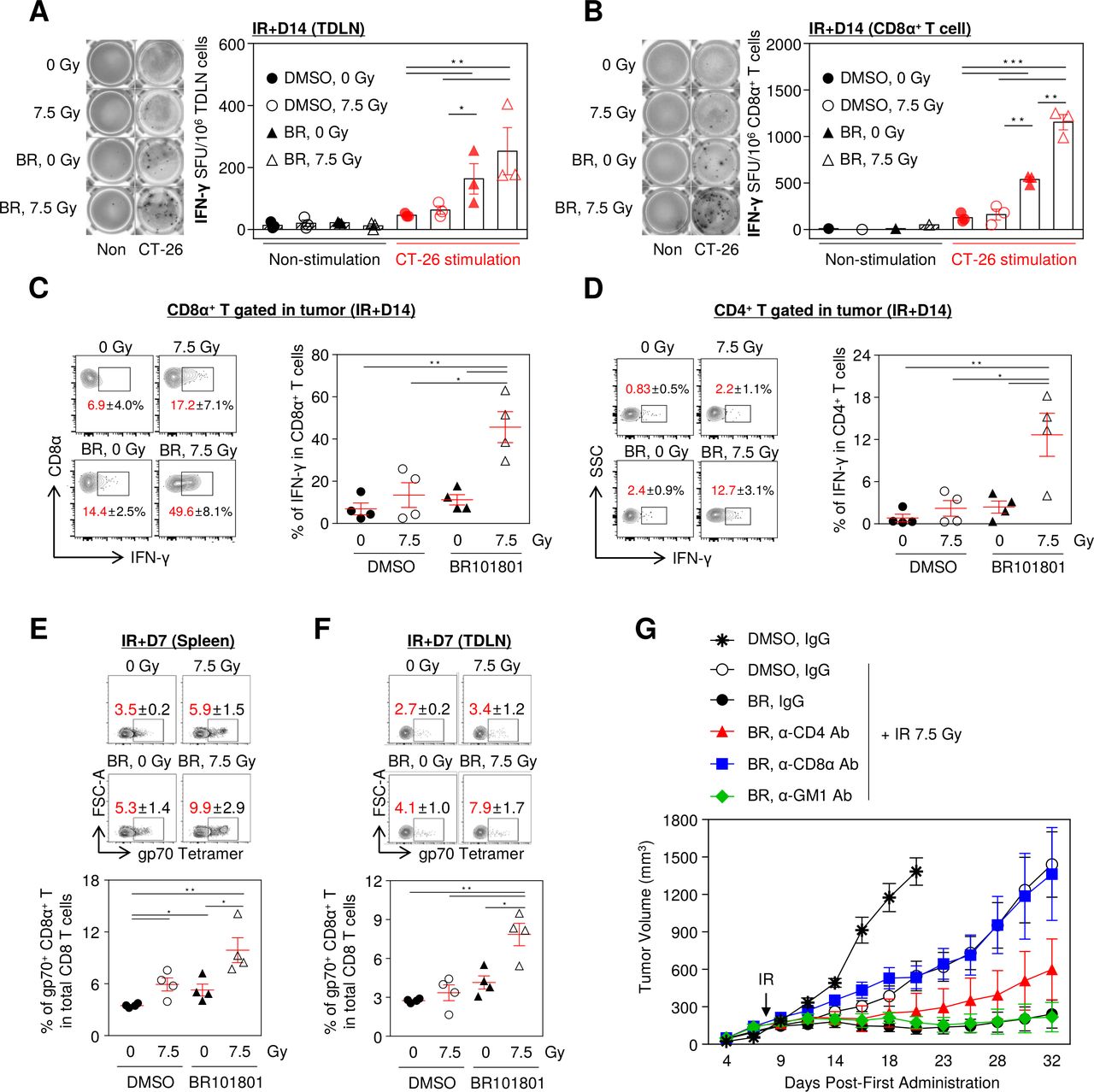
Combining BR101801 and irradiation induces CD8α+ T cell-mediated antitumor immunity. BALB/c mice-bearing CT-26 colon cancer were orally administrated (q.d.) with 50 mg/kg BR101801 till the sacrifice time point (duration of BR101801 treatment; IR+1: for 9 days, IR+3: for 11 days, IR+7: for 15 days, IR+14: for 22 days). (A) Representative images of IFN-γ ELISpot assay (left) and a graph (right) indicating the number of spots for tumor-draining lymph nodes (TDLNs) induced by BR101801 and irradiation with or without additional stimulated CT-26 cells (n=3 mice per group) (black: non-stimulation, red: CT-26 stimulation). (B) Representative images of IFN-γ ELISpot assay (left) and a graph (right) indicating the number of spots for CD8α+ T cells isolated from TDLNs (n=3 mice per group). (C, D) IFN-γ releasing CD8α+ T cell population in the tumor. cells were stimulated with PMA and ionomycin for 2 hour and accumulated IFN-γ was detected in (C) CD8α+ T cells and (D) CD4+ T cells (n=4 mice per group). (E) Representative contour plots of systemic distribution of tumor antigen (gp70)-specific CD8α+ T cells in spleen (top) and quantification of the percentages of gp70+ CD8α+ T cells in total CD8 T cells (bottom) (n=4 mice per group). (F) Representative contour plots of systemic distribution of gp70-specific CD8α+ T cells in tumor-draining lymph nodes (TDLNs) (top) and quantification of the percentages of gp70+ CD8α+ T cells in total CD8 T cells (bottom) (n=4 mice per group). (G) Antibody-mediated depletion of CD4, CD8 T, and NK cells in BALB/c mice treated with BR101801 for 32 days and irradiation. Each mouse was intraperitoneally injected with 200 µg anti-mouse CD4, CD8, or GM1 antibodies 3 days before irradiation and once a week (n≥4 mice per group). All data are presented as mean±SEM *p<0.05, **p<0.01, ***p<0.001 with an unpaired two-tailed t-test. DMSO, dimethyl sulfoxide.
BR101801 enhances the infiltration of CD8α+ T cells into the tumor microenvironment after irradiation
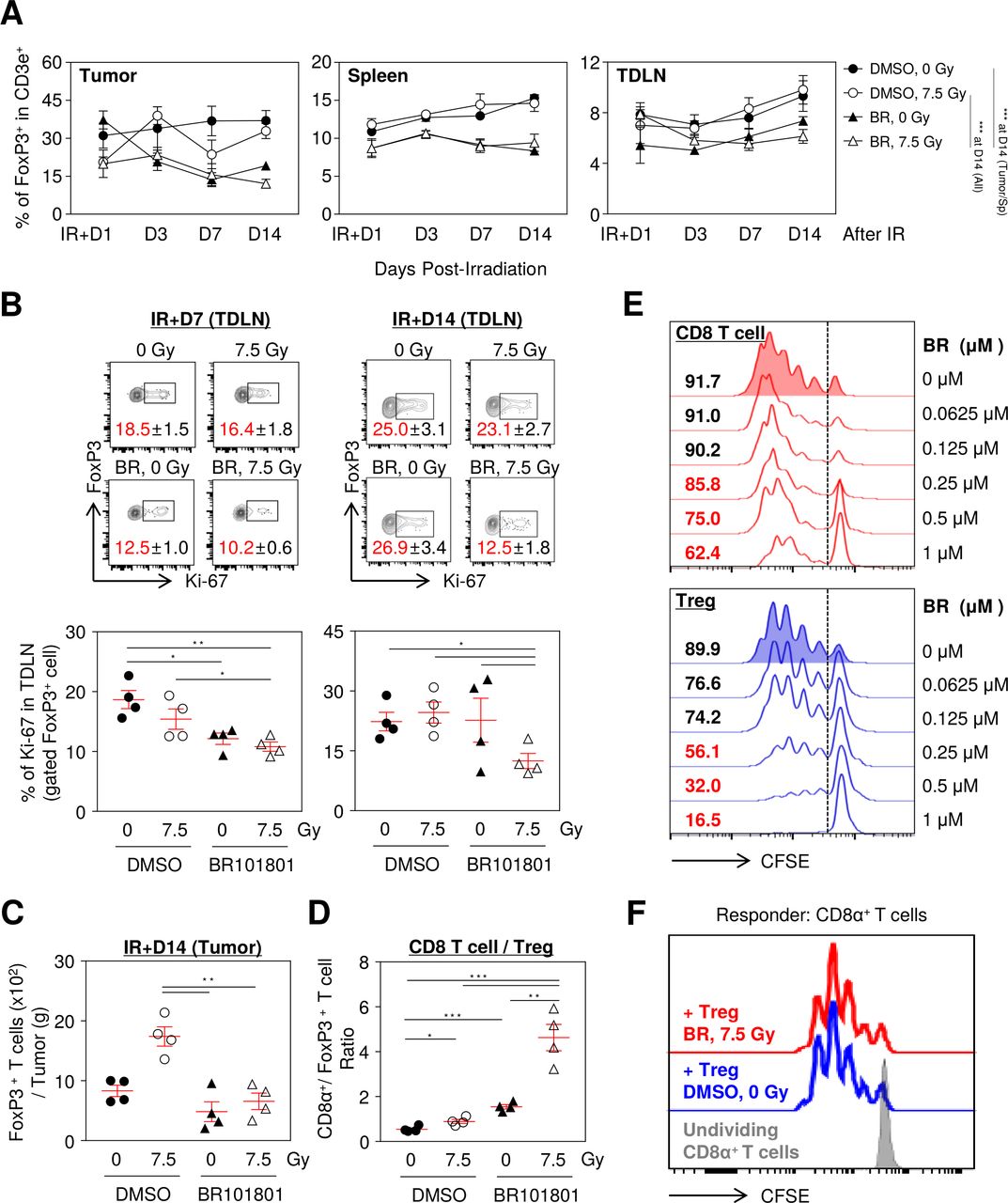
To assess changes in CD8α+ T cells in the tumor microenvironment after the combination treatment, we examined the intratumoral distribution of CD8α+ T cells on day 1, 3, 7, and 14 post-7.5 Gy irradiation. No statistical difference was observed from day 1 to 14 in the CD3e+ cell population in the tumor microenvironment and lymphoid organs (online supplemental figure 6A). Our data indicated that the CD8α+ T cell population of the combination treatment group was significantly increased by 54.5%±7% in CD3e+ cells at day 7 and was maintained by day 14 (figure 3A). In contrast, CD8α+ T cells in the 7.5 Gy irradiation alone group increased by 55.6%±9.9% at day 7 and then reverted to 29.1%±3.8% at day 14 (figure 3A). Indeed, the highest number of CD8α+ T cells calculated for tumors was from the combination group (figure 3B). In contrast, the overall CD4+ T cell population was decreased at day 7, except in the control group, and the combination group was the only one that sustained under 40% of the CD4+ T cell population in CD3e+ cells (online supplemental figure 6B). Further, proliferative CD8α+ T cells, as detected by Ki-67, were significantly increased in the tumor combination group (figure 3C), but there were no statistically significant differences in systemic CD8α+ T cells from the lymphoid organs (figure 3D,E). Therefore, we assumed that extrinsic determinants could affect the infiltration of CD8α+ T cells in the combination treatment.
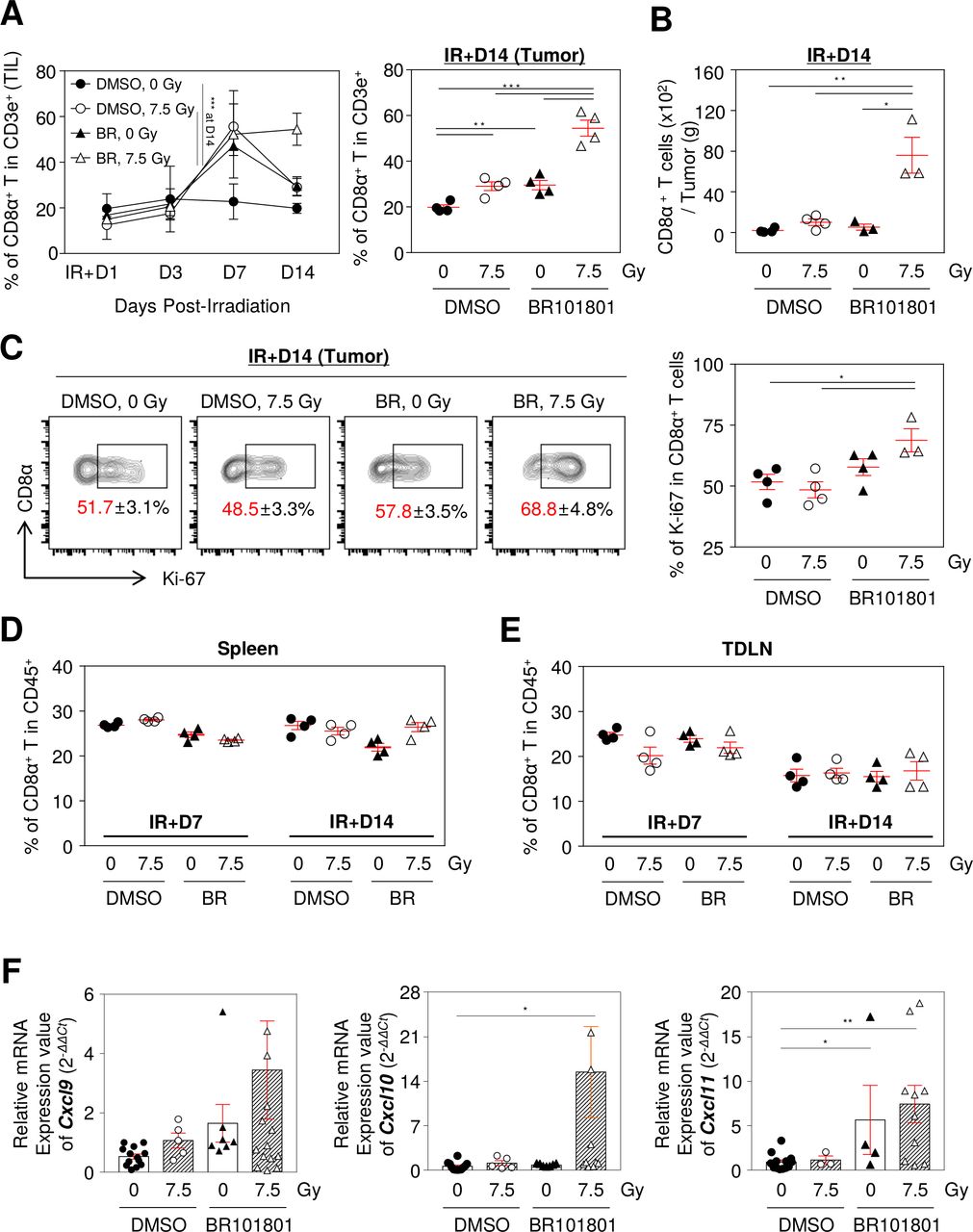
BR101801 enhances CD8α+ T cells infiltration in response to irradiation in tumor microenvironment. The flow cytometry analysis was performed on day 1, 3, 7, and 14 post-irradiation in the CT-26 tumor and lymphoid organs. (A) The percentage of tumor infiltrated CD8α+ T cells from day 1 to 14 after irradiation (left) and an individual graph of day 14 (right). (B) The absolute number of tumor infiltrated CD8α+ T cells per tumor (g) at day 14. (C) Representative contour plots depicting proliferating expression (Ki-67+) on tumor infiltrated CD8α+ T cells at day 14 (left) and quantitation of the percentages of Ki-67+ tumor infiltrated CD8α+ T cells (right). The percentages of CD8α+ T cells in (D) spleen and (E) TDLN. (F) mRNA expression of Cxcl9, 10, and 11 genes in tumor mass (n≥3 mice per group). All data are presented as mean±SEM *p<0.05, **p<0.01, ***p<0.001 with an unpaired two-tailed t-test. DMSO, dimethyl sulfoxide; TDLN, tumor-draining lymph nodes.
CXCR3 is expressed in activated CD8α+ T cells in tumors and attracts CXCL9, 10, and 11, secreted by various cancer cells.24 Since CXCL9, 10, and 11 are mainly induced by IFN-γ,24 we assumed that the excessive IFN-γ released from CD8α+ T cells could be associated with CXCL9, 10, and 11 expressions in the tumor microenvironment. We observed significantly increased gene expressions of Cxcl9, 10, and 11 in the tumor mass obtained from the combination group (figure 3F). Taken together, these data suggest that BR101801 in combination with irradiation affects the gene expression of Cxcl9/10/11 in the tumor milieu and consequently increases the infiltration of CD8α+ T cells.
BR101801 reduces regulatory T cell population following irradiation
Next, alterations in the proliferation of immune-suppressive cells were assessed in the tumor microenvironment after day 1, 3, 7, and 14 post-7.5 Gy irradiation. Interestingly, BR101801 administration significantly reduced FoxP3+ Tregs in the spleen, TDLN, and tumors regardless of irradiation (figure 4A). Furthermore, BR101801 plus irradiation significantly reduced the proportion of Ki-67+ Tregs in TDLN on day 7 and 14 after irradiation (figure 4B), indicating that the proliferation of Tregs was impaired after combination treatment. BR101801 administration maintained the significant reduced number of intratumoral Tregs on day 14 irrespective of irradiation (figure 4C). Notably, the ratio of CD8α+ T cells to FoxP3+ Tregs, which are associated with a favorable prognosis in some cancers,25 exhibited a threefold increase in the combination group compared with other groups at day 14 (figure 4D). Inhibition of Tregs proliferation by direct exposure to BR101801 was more potent than CD8α+ T cells after CD3/CD28 stimulation (figure 4E). However, the suppressive function of splenic Tregs was not altered after the combination treatment, as shown by the significant inhibition of the proliferation of responder CD8α+ T cells compared with Tregs in the control group (figure 4F). Additionally, M1-like macrophages (CD206-/I-A/I-E+/F4/80+/CD11b+) and M2-like macrophage population (CD206+/I-A/I-E-/F4/80+/CD11b+) were significantly reduced. In contrast, the polymorphonuclear (PMN)-MDSC and monocytic (M)-MDSC populations were significantly increased in the combination group (online supplemental figure 7A,B). Dendritic cells were not affected by the combination treatment (online supplemental figure 7C), suggesting that myeloid cells may not be associated with the antitumor effect of the combination treatment. The data suggest that BR101801 in combination with irradiation reduces systemic and intratumoral FoxP3+ Treg subsets, which enhances the tumor-specific CD8α+ T cell function and tumor regression.

BR101801 reduces regulatory T cell population following irradiation. The flow cytometry analysis was performed on day 1, 3, 7, and 14 postirradiation in the CT-26 tumor and lymphoid organs. (A) The percentage of Tregs from day 1 to 14 after irradiation in tumor (left), spleen (middle), and TDLN (right). (B) Representative contour plots depicting Ki-67+ expression on Tregs at day 7 (top-left) and 14 (top-right) and the percentages of Ki-67+ Tregs (bottom). (C) The absolute number of tumor infiltrated Foxp3+ T cells per tumor (G) at day 14. (D) CD8α+ / Treg ratio at day 14 after irradiation. (E) The proliferative capacity of isolated CD8α+ T cells (top) and CD4+/CD25+ Tregs (bottom) after 3 days in absence or presence of gradient concentration of BR101801 (μM). (F) Representative CFSE dilution flow cytometric histograms showing the suppressive capacity of purified CD3+/CD4+/CD25+/CD127- splenic Tregs against Responder CD8 T cells for 4 days in the presence of immobilized anti-CD3 and soluble anti-CD28. The splenic Tregs were isolated from PI3Kδ/γ inhibition and irradiation treated and DMSO treated mice, and Responder CD8 T cells were isolated from naïve (non-treated) mice (n≥3 mice per group). All data are presented as mean±SEM *p<0.05, **p<0.01, ***p<0.001 with an unpaired two-tailed t-test. DMSO, dimethyl sulfoxide; TDLN, tumor-draining lymph nodes.
BR101801 promotes the immune-stimulatory tumor microenvironment in response to irradiation
To investigate the transcriptional changes in CD45+ intra-tumoral leukocytes, transcriptomic assessments were performed using the NanoString nCounter platform. Irradiation triggers immune-stimulatory effects due to the accumulation of cytoplasmic double-stranded DNA, similar to viral infections.26 Indeed, genes related to the viral defense systems, such as innate immune response, inflammatory response, and T cell activation, were highly elevated in the combination group, and 91 genes were selected as differentially expressed between the control and the combination group with a p<0.01 cut-off (figure 5A). Overall, the gene expression in the combination therapy group was highly upregulated compared with that in the other groups (figure 5B) and NanoString-based multiple categories of genes were enhanced by the combination therapy (figure 5C). Owing to the combination therapy, a variety of genes in the immunological gene set changed when comparing each group (figure 5D). Notably, immune-stimulatory pathways, such as viral infection and autoimmune disease, were highly enhanced in the combination therapy group compared with that in the irradiation alone group (figure 5E and online supplemental figure 8). Collectively, our data suggest that BR101801 promotes the immune-stimulatory tumor microenvironment after irradiation in CT-26 tumors.

Combining BR101801 and irradiation promotes the immune-stimulatory antitumor microenvironment. The gene copy number of CD45+ cells from CT-26 tumor was analyzed using NanoString nCounter immunology panel 7 days after irradiation (n=3 mice per group). (A) Heatmap of mean fold-change in gene expressions of other treatment groups vs DMSO along group for genes that are differently expressed (p<0.01). (B)Volcano plot representation of differential expression analysis of transcripts between two groups as indicating on each panel. (C) Bar graphs indicating undirected global significance scores measured the overall differential expression of the selected genes set relative to DMSO single treatment. (D) Venn diagram of differently expressed genes of other treatment groups vs DMSO alone group. (E) enrichment terms between BR101801, 7.5 Gy group vs DMSO, 7.5 Gy group. Circle size and color indicates the number of genes and statistic significances, respectively. DMSO, dimethyl sulfoxide.
BR101801 enhances the abscopal effect following irradiation
Since our data showed BR101801 plus irradiation induced systemic antitumor immunity via activation of effector CD8α+ T cells and inhibition of Tregs, we investigated whether BR101801 plus irradiation induces the abscopal effect after irradiation. Five days after the first CT-26 tumor implantation, the second tumor was implanted on the opposite flank of mice. Only the first implanted tumor was locally irradiated following the schedule (figure 6A). While the combination group showed significantly delayed tumor growth at both sites, irradiation or BR101801 treatment alone had no impact on the tumor growth at the non-irradiated site (figure 6B). Seven of 13 mice exhibited tumors less than 50 mm3 in size at the non-irradiated site (figure 6C). Therefore, our data suggest that BR101801 augments the abscopal effect of local irradiation in metastatic CT-26 tumors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
BR101801 enhances the abscopal effect of irradiation by modulating the immunogenic tumor microenvironment. BALB/c mice-bearing CT-26 colon cancer were orally administrated (q.d.) with 50 mg/kg BR101801 for 23 days. (A) Schematic representation of the schedules for abscopal effect model. (B) Mean tumor volume of subcutaneous (CT-26) implants in BR101801 administration and irradiation treated mice at the irradiated site (first tumor, top) and non-irradiated site (second tumor, bottom). (C) Individual CT-26 tumor growth curves of the non-irradiated site (second tumor, left) with the number of mice and pie charts showing under 50 mm3 sized tumor at the endpoint and individual CT-26 tumor growth curves of the irradiated site (first tumor, right) (n=13 mice per group) (the black line indicates the mean tumor volume of each group, the gray line indicates individual tumor growth). DMSO, dimethyl sulfoxide.
Discussion
Radiotherapy enhances the antitumor immunity; however, the infiltration of immunosuppressive cells into the tumor microenvironment is a major hurdle in successful radiation therapy against cancer.2–6 Thus, targeting radiation-induced immunosuppression is essential to enhance the antitumor immunity after irradiation. It is well established that the combination of immune checkpoint blockade and radiotherapy induces potent antitumor immunity and an abscopal effect in solid tumors3; however, the application of these combination therapies as a treatment strategy remains limited.27 Here, we found that PI3Kδ/γ inhibition with irradiation promotes systemic antitumor immunity and results in an abscopal effect in the CT-26 tumor model. Our data showed that the PI3Kδ/γ inhibitor BR101801 combined with radiotherapy enhanced the activity of intratumoral effector CD8 T cells and inhibition the proliferation of Tregs, thereby promoting an immunologically responsive tumor microenvironment in the CT-26 syngeneic mouse model. Therefore, this is the first report describing the role of PI3Kδ/γ in radiation-induced antitumor immunity in solid tumors.
Several PI3Kδ/γ inhibitors such as duvelisib and idelalisib have been used for the treatment of leukemia and lymphoma patients.28–30 The use of BR101801 as a new PI3Kδ/γ inhibitor was recently reported in phase Ⅰ clinical trial of lymphoma treatment (NCT04018248).21 22 Between four different PI3Kδ or γ inhibitors including duvelisib (PI3Kδ/γ), idelalisib (PI3Kδ), IPI-549 (PI3Kγ), and BR101801 (PI3Kδ/γ), BR101801 was selected as the PI3Kδ/γ inhibitor, as it maintained the highest number of intratumoral CD8α+ T cells and showed the most effective Treg inhibition capacity in the CT-26 tumor model (online supplemental figure 9). BR101801 also inhibited DNA-dependent protein kinase (DNA-PK) (online supplemental figure 1). However, we observed that BR101801 marginally modulated the radiosensitivity of CT-26 cells in vitro (online supplemental figure 4), suggesting that rather than the inhibition of DNA-PK, inhibition of PI3Kδ/γ by BR101801 is predominant in antitumor immunity in the combination treatment regimen. Thus, in our experiment, BR101801 was used to investigate the role of PI3Kδ/γ in radiation-induced antitumor immunity.
Our data provide novel evidence for the enhanced antitumor immunity in solid tumors due to PI3Kδ/γ inhibition in combination with radiotherapy. According to a recent study, PI3Kδ/γ inhibition alone has only a moderate antitumor effect in solid tumors.31 Similarly, we also observed that BR101801 alone had marginal tumor regression in the CT-26 tumor model (figure 1B–D). Notably, our study demonstrated that BR101801 enhances the antitumor effect of radiotherapy through CD8α+ T cell-mediated antitumor immunity in solid tumors. Therefore, our data provide translational evidence for the potential use of a PI3Kδ/γ inhibitor as a radiosensitizer for radiotherapy to boost the antitumor immunity in solid tumors.
Our data clearly demonstrated that the antitumor effect of BR101801 combined with irradiation was mediated by effector CD8α+ T cells. Although PI3Kδ or γ inhibition directly impairs the activation and proliferation of T cell subsets,17 31 32 a recent study showed that a blockade of PI3Kδ preferentially suppressed Tregs more significantly than effector CD8 T cells.32 Similarly, we observed that PI3Kδ/γ inhibition suppressed TCR-mediated proliferation of CD8α+ T cells and Tregs; however, the proliferation of Tregs was much more susceptible to PI3Kδ/γ inhibition in vitro compared with the CD8α+ T cells (figure 4E). Tregs are highly sensitive to IL-2 compared with conventional T cells because of their constitutive expression of the IL2Rα chain (CD25) and they also express higher levels of PTEN that regulate the PI3K/AKT pathway.33 34 However, recent reports have shown that the inhibition of PI3K decreases PTEN expression and the depletion of PTEN in Tregs leads to loss of CD25 and FoxP3, which is critical for the maintenance of Treg stability.35 36 Therefore, it is possible that BR101801 strongly attenuated Tregs before irradiation, and the effector CD8α+ T cells were potentiated by irradiation, which converted to an immunogenic tumor microenvironment. Subsequently, tumor-specific effector CD8α+ T cells could be unleashed by the low number of Tregs, leading to an enhanced systemic antitumor immunity and tumor regression.
Preclinical studies have revealed that the PI3K subunit inhibitors alter the immune-suppressive capacities of Tregs, MDSCs, and M2-like macrophages in solid tumor models.18–20 In particular, PI3Kδ depletion shows outstanding effectiveness in diminishing functional ability and decreasing the number of Tregs in draining lymph nodes and decreasing MDSCs in the spleen, thereby leading to tumor regression.18 32 Blocking PI3Kγ also reduces immune-suppressive myeloid cell phenotype, leading to higher antitumor T cell activity and tumor regression.19 20 Our data show that a constant reduction in Tregs and CD206+ M2-like macrophages was observed after treatment with BR101801 and irradiation, but not in MDSCs (figure 4A and online supplemental figure 7A,B). Irradiation promotes TAMs to have more diverse and broad-spectrum roles in suppressing and supporting antitumor immunity after irradiation.37 This suppression of antitumor immunity is probably due to M2-like macrophages, which are polarized by Th2 cytokines, such as IL-4 and IL-13, whereas Th1 cytokines differentiate precursor cells into M1-like macrophages with antitumor immunity.38 Contrary to our expectation, proportions of M1 macrophages and M2 macrophages show a significant reduction in tumors in combination therapy at day 14 postirradiation (online supplemental figure 7A). Populations of PMN-MDSCs and M-MDSCs also showed a statistically significant increase; however, we could not prove whether inhibition of PI3Kδ/γ with BR101801 impacts the populations of MDSCs (online supplemental figure 7B), and there was no significant change in the dendritic cell population (online supplemental figure 7C). The activation of PI3Kγ suppresses NF-κB and activates C/EBPβ, contributing immunosuppressive property of the myeloid population.19 Our data showed that type 1 IFN signaling was significantly increased by the combined treatment of BR101801 and irradiation (figure 5A). Type 1 IFN signaling is crucial for maintaining CD8 T cell priming by dendritic cells as well as for suppressing Treg function to maintain antitumor immunity.39 40 In contrast, type 1 IFN signaling acts as a mechanism that limits the acquisition of suppressive function by MDSCs.41 Based on previous reports, it could be presumed that the increase in MDSCs in the combined therapy with BR101801 plus irradiation is due to the increase in type1 IFN signaling at the tumor site. Therefore, we concluded that the immune-suppressive activities of myeloid cells might have a weak correlation with the antitumor effect induced by BR101801 in combination with radiotherapy.
The density and diversity of tumor-infiltrating immune cells are closely related to the prognosis and prediction of radiotherapy efficacy.42 Radiotherapy is known to increase antitumor immunity through CD8 T cell priming43; however, radiation-induced tumor-infiltrating CD8 T cells are limited by an immune-suppressive tumor environment, the so-called cold tumor.44 Clinical studies of chemo-radiation therapy showed that the high CD8 T cell infiltration group had significantly longer disease-free survival and overall survival compared with the low CD8 T cell infiltration group.45 Thus, an increase in intratumoral CD8 T cells is important for successful radiotherapy. Chemokines are one of the main factors that influence T cell trafficking, and radiotherapy contributes to the enhanced chemokine expression.46 An elevated chemokine (C-X-C motif) ligand (CXCL) 9, 10, and 11 expressions are produced by the monocytes, cancer cells, and stromal cells.24 These chemokines are correlated with T cell infiltration in colorectal cancer and are related to the prognosis of patients.47 Moreover, CXCR3 ligands, including CXCL9, 10, and 11, are induced by IFN-γ stimulation.24 Similarly, our data showed that IFN-γ production strongly correlated with the gene expression of Cxcl9, 10, and 11 (figures 2A–D and 3). Besides, our transcriptome data showed that CXCR3, a counterpart of CXCL9, 10, and 11, was significantly increased in the BR101801 and radiotherapy combination therapy group (online supplemental figure 10). CXCR3 is mainly expressed on T cells, NK cells, and NKT cells, and it is also induced by IFN-γ stimulation.47 CXCR3 levels are strongly correlated with metastasis and prognosis of cancer patients.48 Therefore, it is possible that BR101801 in combination with irradiation increases the infiltration of CD8α+ T cells in the tumor mass by inducing CXCR3 ligands.
Our data suggest two feasible usages for an effective antitumor strategy using a combination of PI3Kδ/γ inhibitor and radiotherapy. The first one is the potential antitumor effect as an in situ vaccination. Local injury of cancer cells leads to the release of multivalent tumor antigens and subsequently results in the stimulation of the immune system similar to in situ vaccination. Radiotherapy is often referred to as a powerful tool for in situ vaccination. However, radiotherapy alone induces immunosuppressive cells related to tumor development; therefore, adjuvants are required to exhibit a proper vaccination effect.49 Our data showed that BR101801 in combination with irradiation promoted an immunostimulatory tumor milieu by reducing the Treg population and increasing the number of tumoricidal CD8 T cells, and these effects were maintained for a long time. Concurrently, tumor-specific memory T cells were generated, rejecting tumor reimplantation. Thus, the combination of PI3Kδ/γ inhibitors and radiotherapy would be beneficial to prevent tumor relapse.
The second usage is for the abscopal effect of the combination of PI3Kδ/γ inhibitors and radiotherapy. Many oncologists are eager to establish regimens in combination with radiotherapy because the abscopal effect can be a promising solution for metastatic cancer patients and prevent tumor relapse.3 Before applying combinatory radiotherapy, elaborate regimens should be prescribed depending on the patient cohorts by considering immunotherapeutic agents, radiotherapy dose, and timing, which would be linked to favorable prognostic.50 Retrospective studies show that the elaborate immunological mechanism behind the abscopal effect is unclear, but CD8 T cells are identified as a key part of the abscopal effect.3 Our data showed that combining PI3Kδ/γ inhibitor and radiotherapy enhanced tumor-specific effector CD8 T cells and inhibited immune-suppressive Tregs, leading to an abscopal effect. Therefore, our data showed the enhancement of the abscopal effect using pretreatment with a PI3Kδ/γ inhibitor to repress immunosuppressive cells before radiotherapy, which could provide a promising regimen.
In summary, our data showed that BR101801 in combination with radiotherapy significantly decreased systemic accumulation of Tregs, resulting in increased tumor antigen-specific CD8 T cell responses leading to tumor regression. Subsequently, the tumor-specific CD8 T cell response induced by the combination therapy promoted the immunological memory and abscopal effects. For the first time, our study provides evidence that the blockade of PI3Kδ/γ following radiotherapy improves local and systemic antitumor immunity against solid tumors. Therefore, combining PI3Kδ/γ inhibitors and radiotherapy could be a new promising strategy for preventing metastasis and cancer treatment in solid tumors.
Data availability statement
Data are available in a public, open access repository.
Ethics statements
Patient consent for publication
Ethics approval
Mice were maintained in specific pathogen-free facilities, and all studies were performed in accordance with relevant ethical regulations for animal testing and research and the protocols approved by the IACUC at the Korea Institute of Radiological and Medical Science Animal Center (kirams 2020-0023, kirams 2021-0042).
Acknowledgments
We thank Boryong Co. for providing the BR101801 compound and supporting the grant.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YNY and EL contributed equally.
Contributors Conceived/designed experiments: YNY, EL, T-JK and J-SK; performed the experiments: YNY, EL, and Y-JK; analyzed the data: YNY, EL, J-AG, T-JK and J-SK; wrote the paper: YNY, T-JK and J-SK. All authors reviewed the manuscript.
Funding This study was supported by a grant from the Korea Institute of Radiological and Medical Sciences (KIRAMS), funded by the Ministry of Science and ICT (MSIT), The Republic of Korea (No. 50531-2021), and the National Research Foundation of Korea (NRF-2020M2D9A2094153, NRF-2020R1A2C1004775).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.