Article Text

Abstract
Background In lymphoid malignancies, the introduction of chimeric antigen receptor T (CAR-T) cells and bispecific antibodies (bsAbs) has achieved remarkable clinical success. However, such immunotherapeutic strategies are not yet established for acute myeloid leukemia (AML), the most common form of acute leukemia in adults. Common targets in AML such as CD33, CD123, and CLEC12A are highly expressed on both AML blasts and on normal myeloid cells and hematopoietic stem cells (HSCs), thereby raising toxicity concerns. In B-cell acute lymphoblastic leukemia (B-ALL), bsAbs and CAR-T therapy targeting CD19 and CD22 have demonstrated clinical success, but resistance via antigen loss is common, motivating the development of agents focused on alternative targets. An attractive emerging target is FLT3, a proto-oncogene expressed in both AML and B-ALL, with low and limited expression on myeloid dendritic cells and HSCs.
Methods We developed and characterized CLN-049, a T cell-activating bsAb targeting CD3 and FLT3, constructed as an IgG heavy chain/scFv fusion. CLN-049 binds the membrane proximal extracellular domain of the FLT3 protein tyrosine kinase, which facilitates the targeting of leukemic blasts regardless of FLT3 mutational status. CLN-049 was evaluated for preclinical safety and efficacy in vitro and in vivo.
Results CLN-049 induced target-restricted activation of CD4+ and CD8+ T cells. AML cell lines expressing a broad range of surface levels of FLT3 were efficiently lysed on treatment with subnanomolar concentrations of CLN-049, whereas FLT3-expressing hematopoietic progenitor cells and dendritic cells were not sensitive to CLN-049 killing. Treatment with CLN-049 also induced lysis of AML and B-ALL patient blasts by autologous T cells at the low effector-to-target ratios typically observed in patients with overt disease. Lysis of leukemic cells was not affected by supraphysiological levels of soluble FLT3 or FLT3 ligand. In mouse xenograft models, CLN-049 was highly active against human leukemic cell lines and patient-derived AML and B-ALL blasts.
Conclusions CLN-049 has a favorable efficacy and safety profile in preclinical models, warranting evaluation of its antileukemic activity in the clinic.
- immunity
- immunotherapy
- T-Lymphocytes
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Acute myeloid leukemia (AML) is the most frequent form of acute leukemia in adults, affecting 4–5 per 100 000 population.1 2 While several new treatments have recently been approved, including hypomethylating agents, the antibody drug conjugate gemtuzumab ozogamicin and various small molecule kinase inhibitors,3 induction and consolidation chemotherapy followed by allogeneic hematopoietic stem cell transplantation (HSCT) remains the most widely used curative treatment approach. However, more than 50% of patients are not eligible for the intensive pre-treatment chemotherapy regimens. In addition, remission-inducing chemotherapy and HSCT are associated with substantial treatment-associated morbidity and mortality, and many patients experience a subsequent relapse. Accordingly, prognosis of AML remains dismal with overall 5-year survival rates below 15%.4
Strategies to mobilize T cells against tumor cells via bispecific antibodies (bsAbs) or directly engineering T cells to express chimeric antigen receptors have achieved remarkable success in lymphoid malignancies, including acute lymphoid leukemia (B-ALL) and multiple myeloma.5–7 However, T cell engaging strategies for myeloid-derived neoplasias like AML are not yet clinically established.
A major challenge in AML is target selection. Antibody-based therapeutics currently in development for AML focus primarily on CD33, CD123 and CLEC12A as target antigens.8 However, beyond their well-documented expression on malignant cells, these targets are also found at similar levels on normal myeloid cells and hematopoietic stem cells (HSCs), raising concerns regarding a narrow therapeutic window.9 CD33 is expressed on approximately 30% of healthy bone marrow (BM) myeloid progenitors.10 11 CD123 is also expressed on many healthy cells such as myeloid progenitors, plasmacytoid dendritic cells (pDCs), monocytes, and basophils.12 Finally, CLEC12A is also expressed on normal myeloid cells including granulocytes, monocytes, macrophages, and DCs as well as granulocyte-macrophage progenitors.9 13 14
The receptor tyrosine kinase FLT3 is an attractive AML target with frequent expression on both blasts and leukemic stem cells, whereas expression on healthy myeloid cells is low and limited to myeloid DCs and HSCs.15–17 FLT3 is a proto-oncogene that plays a key role in promoting leukemic cell proliferation and survival, and expression is preserved in AML after relapse.18 Its essential role in disease progression has been validated by the clinical benefit of tyrosine kinase inhibitors that target mutant FLT3.19 20
Unlike kinase inhibitors that are specific to a particular FLT3 mutational context, antibody-based therapies that target the extracellular domain of FLT3 are independent of FLT3 mutations, allowing for treatment of a broader patient population. We recently developed an Fc-optimized monoclonal antibody (mAb) targeting FLT3, FLYSYN, which potently induces antibody-dependent cellular cytotoxicity. FLYSYN showed promising safety and preliminary efficacy in AML patients with minimal residual disease21 (clinicaltrials.gov, NCT02789254). However, for leukemia patients with higher disease burden, more potent treatment modalities are needed. To this end, we have constructed CLN-049, a FLT3xCD3 T cell-engaging bsAb, based on an Fc-silenced hIgG1 anti-FLT3 antibody with anti-CD3 single-chain antibodies (scFvs) fused to the C-termini of the heavy chains. This study provides a comprehensive preclinical characterization of CLN-049 as foundation for an upcoming clinical study in relapsed or refractory AML patients.
Methods
Biacore evaluation of CLN-049 binding
To determine monovalent affinity for FLT3, CLN-049 was captured onto a Protein A chip, and FLT3 (Stratech) was flowed over the biosensor. To determine avid affinity for FLT3 and FLT3-like orthologs, PDGFα, PDGFβ, VEGFR2, VEGFR3 (all from R&D Biosystems) or FLT3 was captured onto a CM5 sensor chip, and CLN-049 or positive control antibodies were flowed over the cell. A global fit algorithm (Biacore Insight Evaluation) was used to calculate KD, ka, and kd values.
To evaluate binding to Fc gamma receptors (FcγRs), recombinant His6-tagged FcγRs were immobilized onto an anti-His6 antibody coated chip and CLN-049 was flowed over the biosensor at either pH 7.4 or 6.0. KD, ka, and kd values were determined by steady state analysis for each FcγR, except FcγR1, which was evaluated assuming 1:1 binding.
Sandwich ELISA
Recombinant FLT3 (Sino Biological) was coated to 96 well plates at 3 µg/mL. Plates were blocked and washed, then incubated with serially diluted CLN-049. After washing, 2 µg/mL biotinylated CD3ε/δ dimer (Acro Biosystems) was added and detected with streptavidin-HRP. Dose response curves were fit using a 4-parameter logistic regression model.
Cell binding of CLN-049
Target cell lines expressing FLT3 (NALM-16) or T cells expressing CD3 (Jurkat) were incubated with CLN-049 for 30 min at 4°C. After washing, cells were incubated with PE-labeled goat anti-human Fc or goat anti-human F(ab)2, then assessed via flow cytometry. To compare binding to FLT3 and FLT3 N399D, ExpiCHO-S cells were transfected with wild-type FLT3 or FLT3 N399D. After 4 days, binding analysis was performed as described above.
To evaluate binding to primary cells, cryopreserved samples, either peripheral blood mononuclear cells (PBMC) or isolated T cells from human donors were thawed prior to addition of live/dead viability dye, Fc block, and staining solution. Samples were bound to CLN-049-allophycocyanin (APC), isotype control-APC, or incubated with anti-CD3-APC (clone UCHT1) or anti-FLT3-PE (clone 4G8) to confirm target positivity. In PBMC samples, DC-enriched cells were identified by flow cytometry as CD45+CD14-CD20-CD4-CD8-HLA-DR+.
FLT3 expression on cell lines and primary cells
AML cell lines or Jurkat cells were cultured for approximately 1 week. For normal FLT3+ primary cells, healthy donor PBMC were isolated from buffy coats, and cryopreserved BM samples were thawed at 37°C overnight. Cells were stained with PE-labeled anti-FLT3 antibody (clone 4G8 or BV10), evaluated by flow cytometry, and quantified by calibrating PE MFI with Quantibrite beads (BD Biosciences). pDCs were defined as CD45+HLA-DR+CD11 c-CD123+ in PBMC, and CD34+ cells were defined as CD45+CD3-CD34+ cells in BM samples, after exclusion of lineage markers.
Evaluation of FLT3 expression on AML patient samples: 20 BM samples from adult AML patients were stained with anti-FLT3-PE (clone 4G8) and MFI values converted to FLT3 molecules per cell.
Evaluation of FLT3 expression on primary B-ALL patient samples: cells were labeled with anti-CD45 (clone HI30), anti-FLT3 (clone 4G8), anti-CD19 (clone J3-129), anti-CD20 (clone 2H7), anti-CD22 (clone HIB22), and live/dead viability dye. B-ALL cells were defined by the CD45+CD19+ double-positive population.
In vitro pharmacology
To evaluate activity against AML cell lines, leukemic cell lines were labeled with cell proliferation dye then cocultured with PBMC at a 1:1 effector:target (E:T) ratio in RPMI-1640 + 10% fetal bovine serum (FBS) in the presence of CLN-049 for 72 hours, unless otherwise indicated. If included, sFLT3 or sFLT3L were pre-cultured with MOLM-13 target cells for 2 hours prior to addition of CLN-049 and PBMC. Via flow cytometry, 7-AAD uptake in CD14-CD33+ proliferation dye+ cells and CD69 expression on T cells were used to determine target cell viability and T cell activation, respectively. Cytokine concentrations in supernatants were assayed by Luminex (Bio-Rad). EC50 values were determined using 4-parameter logistic regression model.
To evaluate killing of primary AML and B-ALL samples, cells were cultured with CLN-049 in RPMI-1640 + 10% FBS for 72 hours. The number of viable cells was determined by excluding 7-AAD+ cells from the CD33+ cell population (AML) or CD19+ cell population (B-ALL), and E:T ratios were determined by relative ratio of cell types in control group.
In vitro toxicology
Cytotoxicity of normal FLT3+ cells: pDC depletion was evaluated using PBMC from nine healthy donors isolated from buffy coats. CD34+ depletion was evaluated using cryopreserved BM samples from five human donors that were recovered at 37°C overnight. Cells were treated with CLN-049 for 72 hours, either in RPMI-1640 + 10% FBS (pDC) or X-vivo media (CD34). pDC viability was assessed using Live/Dead eFluor780 uptake in CD45+HLA-DR+CD11c-CD123+cells, and CD34+ cell viability was assessed using 7-AAD uptake in CD45+CD3-CD34+ cells, in both cases after exclusion of lineage markers.
To determine T cell activation in the absence of target cells, T cells were isolated from three healthy donors and incubated for 72 hours with CLN-049, or anti-CD3 antibodies OKT3 or parental UCHT1. CD8+ and CD4+ T cell activation after 72 hours was measured by expression of CD69 by flow cytometry. Alternatively, PBMC were used instead of isolated T cells, and CD69 was evaluated both by percent positivity and MFI.
In vivo efficacy studies
MOLM-13 study: 1×105 MOLM-13 cells were inoculated intravenously by tail vein injection into NCG mice. One day after tumor cell inoculation (Day 1), mice were randomly sorted and received 2×107 PBMC intraperitoneally and subsequently administered CLN-049 IV, which continued weekly until end of study. Animals were regularly monitored and euthanized if moribund. On Day 15, 0.1 mL blood was sampled and assayed for CD45+CD33+ MOLM-13 tumor cells via flow cytometry. For PK analysis, 0.08 mL blood was collected and processed for serum from 3 mice/timepoint/dose group by mandibular bleeds. Serum was collected at 0.5, 2, 8, 24, 72, and 168 hours post-dose and concentrations measured via FLT3/CD3 sandwich immunoassay.
Primary AML study: Immunodeficient NSG mice were intravenously engrafted with 8×106 PBMC from an AML patient. On the same day, 8×106 PBMC from a healthy donor were transferred IV, followed by 20 µg/mouse CLN-049 or control antibody (MOPCxCD3) injected intravenously 4 hours later. CLN-049 treatment (10 µg/mouse IV) was repeated on days 3 and 10. At day 24, leukemic burden (ratio hCD45+hCD33+/mCD45+ cells) in BM was determined by flow cytometry.
Primary B-ALL study: Immunodeficient NSG mice were intravenously engrafted with 4×106 PBMC from a B-ALL patient. On day 7, 10×106 PBMC from a healthy donor were transferred IV, followed by 30 µg/mouse CLN-049 or control antibody (MOPCxCD3) injected intravenously 4 hours later. CLN-049 treatment was repeated on day 11, using 30 µg/mouse intravenous. At day 17, leukemic burden (ratio hCD45+hCD19+/mCD45+cells) in BM was determined by flow cytometry.
Results
Generation and biochemical characterization of CLN-049
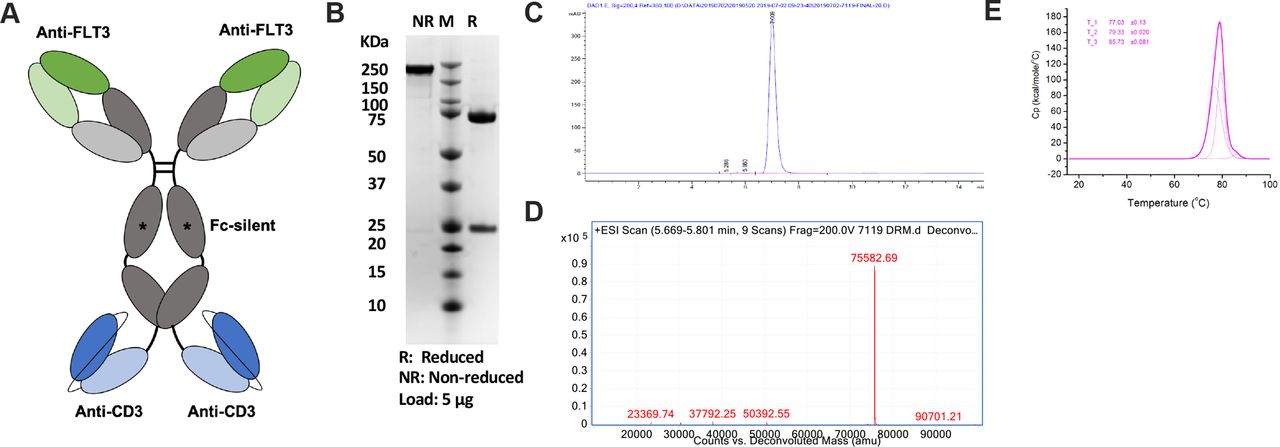
CLN-049 has specificity for FLT3 in its two Fab arms and specificity for CD3 in two single-chain variable (scFv) fragments fused to the heavy chain C-termini (figure 1A). The FLT3-binding Fab arms were derived from humanization of murine clone 4G8, which binds to the membrane-proximal domain 4 of FLT3.22 Membrane proximity of epitopes may critically enable the antitumor activity of bsAbs.23–25 The CD3-binding scFvs were derived from humanized mAb UCHT1, as described previously.22 Effector functions in the Fcγ1 domain of CLN-049 were silenced by mutations, as previously reported,26 in order to prevent FcγR-mediated activation of T cells. CLN-049 was expressed at a titer of 3 g/L in CHO cells and purified by Protein A to high purity as assessed by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (figure 1B) and size exclusion chromatography (figure 1C). Mass spectrometry confirmed the anticipated molecular weight of the heavy chain/scFv fusion (figure 1D). CLN-049 showed a high overall melting temperature of 77°C as determined by differentiating scanning calorimetry.
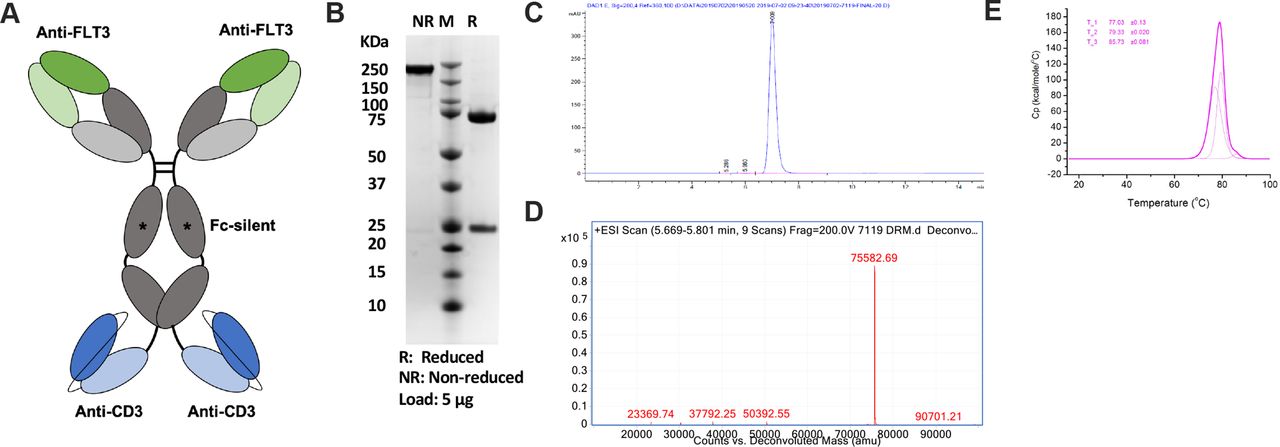
Structure and characterization of CLN-049 (A) Design of CLN-049. (B) SDS-PAGE analysis of CLN-049 under reducing and non-reducing conditions. (C) SEC trace of CLN-049 following Protein A-based affinity purification. (D) Mass spectrum of CLN-049. (E) Heat flux curve from DSC evaluation of CLN-049 determining three melting temperatures for CLN-049. DSC, differentiating scanning calorimetry; SDS-PAGE, sodium dodecyl-sulfate polyacrylamide gel electrophoresis; SEC, size exclusion chromatography.
To demonstrate two-armed FLT3 binding by CLN-049, which can confer slower dissociation rates and increase functional affinity, we measured binding rate constants for recombinant FLT3 by surface plasmon resonance (SPR). Under avid conditions, the apparent KD was >10 fold lower than for monovalent conditions (15.7 nM vs 182 nM), which was largely driven by a slower dissociation rate constant (1.53×10−3 1/s vs 3.80×10−2 1/s) (figure 2A). Binding of CLN-049 was highly specific with no detectable binding to other members of the receptor family, including PDGFRα, PDGFRβ, VEGFR2, and VEGFR3 (figure 2A). A critical element of the binding interface between CLN-049 and FLT3 was revealed by the lack of binding to murine and cynomolgus monkey FLT3 by SPR (online supplemental figure 1A). Both species contain the divergent mutation D399N in FLT3 domain 4, which completely abrogated binding by CLN-049 (figure 2B). Figure 2C shows D399 in the presumed CLN-049 binding interface within the structure of human FLT3. Furthermore, we confirmed by SPR that mutations introduced into the Fcγ1 domain of CLN-049 completely abrogated binding to FcγRs but preserved binding to FcRn (online supplemental file 1B).
Supplemental material

Binding characteristics of CLN-049. (A) Summary of Biacore binding data of CLN-049 using FLT3 and FLT3-related proteins. (B) Binding of CLN-049 to CHO cells expressing WT FLT3 or FLT3 N399D. (C) Human FLT3 ligand-receptor complex in dimeric form, based on PDB 3QS9. Dimeric ligands are colored in raspberry. Receptor ectodomains are colored as follows: domain one in yellow, domain two in green, domain three in cyan, domain four in blue, and domain five in gray. N399 in domain four is colored in red. (D) Binding of CLN-049 to FLT3+ NALM-16 cells and CD3+Jurkat cells by flow cytometry. (E) Flow cytometry plots depicting HLA-DR+ cells from PBMC of a representative donor stained with PE-labeled FLT3 mAb clone BV10 (Untreated), an APC-labeled IgG1 isotype control antibody, or APC-labeled CLN-049. The panel below depicts quantification across three donors, with the percentage of CLN-049 calculated as %CLN-049+ - %IC+. (F) Histograms showing purified T cells from a representative donor stained with APC-labeled IgG1 isotype control antibody (red) or APC-labeled CLN-049 (blue). The panel below shows quantification across three donors, where the percentage of CLN-049 is calculated as %CLN-049+ - %IC+. (G) Sandwich ELISA with FLT3 capture, incubation with titrations of CLN-049, and CD3-biotin based detection. APC, allophycocyanin; DC, dendritic cell; PBMC, peripheral blood mononuclear cells.
Binding of CLN-049 to FLT3-expressing NALM-16 cells and to CD3-expressing Jurkat cells revealed half maximal binding at 3.2 nM and 8.4 nM, respectively (figure 2D). Approximately 5%–10% of HLA-DR+ DCs in peripheral blood (PB) were found to express FLT3 and were bound by CLN-049 (figure 2C). CD4+and CD8+ primary T cells were also bound by CLN-049 (figure 2D).
Simultaneous binding of CLN-049 to both human FLT3 and human CD3 was investigated by a sandwich ELISA which revealed half maximum dual binding at 88.5 ng/mL, or 0.45 nM (figure 2E).
CLN-049 induces TDCC against AML cell lines expressing a broad range of FLT3 levels
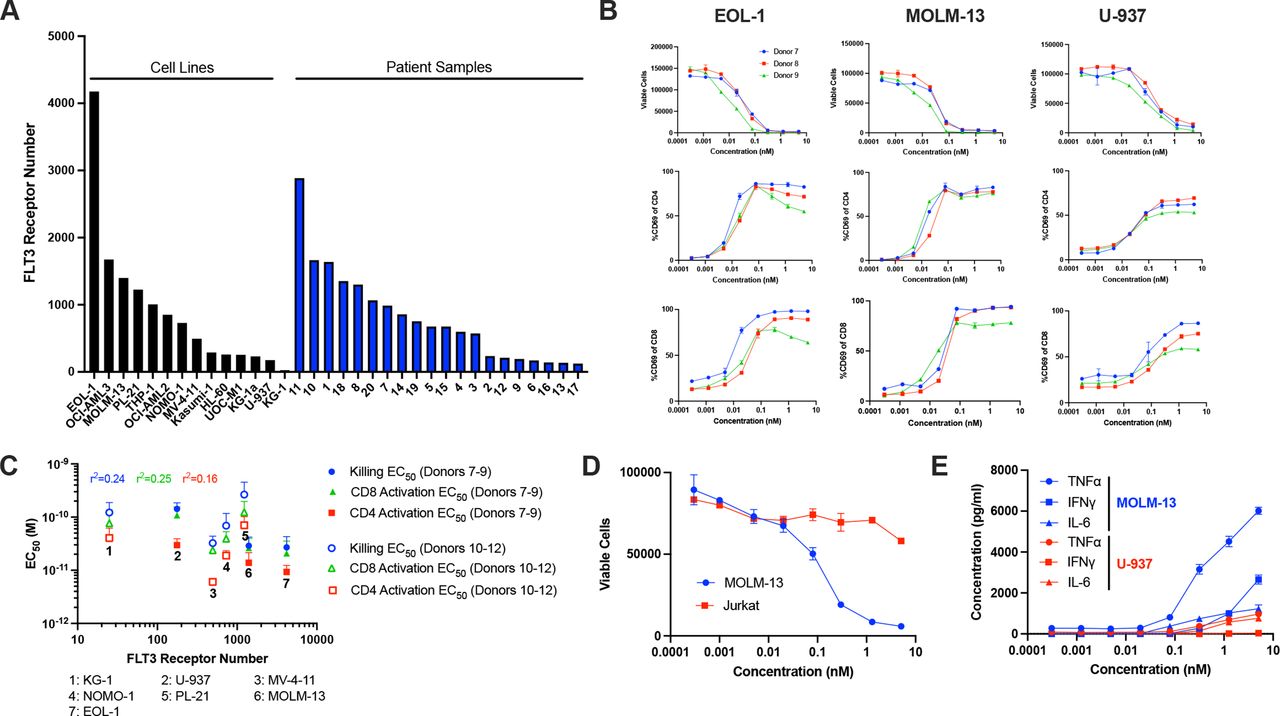
FLT3 expression was determined for 14 human AML cell lines by quantitative flow cytometry, which revealed a broad range of surface receptor levels, ranging from fewer than 100 FLT3 copies per cell in KG-1 cells to 4000 copies per cell in EOL-1 cells (median of 600 molecules). Similar results were obtained on analysis of primary BM-derived blasts from 20 AML patients, where expression ranged from 100 to 3000 FLT3 molecules (median of 700 molecules) (figure 3A).

CLN-049 potently induces T cell activation and TDCC. (A) FLT3 receptor number on 14 AML cell lines and 20 patient BM blast samples. (B) AML cell lines were labeled with cell proliferation dye, cocultured with PBMC from three different healthy donors at an E:T ratio of 2:1, in the presence or absence of the indicated concentrations of CLN-049 for 72 hours. Shown are lysis curves, as flow cytometrically assessed by 7-AAD uptake in AML cells and CD69 expression profiles as marker for activation of CD4+ and CD8+ T cells. (C) Plot of FLT3 receptor number relative to CLN-049 mediated T cell activation killing and target cell lysis as measured by flow cytometry. Numbers identify the different AML cell lines used as targets. Correlation constants were calculated using log-normalized values for both FLT3 receptor number and EC50 values. (D) MOLM-13 and Jurkat cell lysis was determined as described in (B). (E) Supernatants from cocultures of MOLM-13 cells or U-937 cells with PBMC as in (B) were analyzed for the indicated cytokines by Luminex. AML, acute myeloid leukemia; BM, bone marrow; PBMC, peripheral blood mononuclear cells; TDCC, T cell dependent cellular cytotoxicity; 7-AAD, 7-aminoactinomycin D.
TDCC of CLN-049 was investigated with the human AML cell lines EOL-1, MOLM-13, and U-937, which displayed high, medium, and low FLT3 expression levels, respectively. PBMC from healthy donors were co-cultured with AML cells in the presence or absence of CLN-049 followed by analysis of target cell lysis via 7-AAD uptake. In all cases, potent target cell lysis was detected at sub-nM concentrations of CLN-049, with EC50 values ranging from 16 to 150 pM and with >85% lysis observed in all three cell lines with all donor PBMC (figure 3B). The analysis was expanded with additional donor PBMC to include KG-1, MV-4–11, NOMO-1, and PL-21 AML cell lines, confirming CLN-049 activity at similar EC50 values. No obvious correlation was found between FLT3 expression levels on target cells and CLN-049 potency (r2=0.24), indicating that CLN-049 was capable of redirecting T cells even against AML cells with low level FLT3 expression (figure 3C). Of note, the potency of CLN-049 was unaffected by the mutational status of FLT3 in the evaluated AML cell lines (online supplemental figures 2A,B). In spite of exquisite sensitivity against low FLT3-expressing cell lines, lysis was confirmed to be FLT3-dependent by the sparing of Jurkat cells (figure 3D), which express no FLT3 by flow cytometry (online supplemental figure 3).
Soluble FLT3 ligand (sFLT3L) and shed FLT3 extracellular domain (sFLT3) have been reported in AML patient sera at concentrations ranging from 0.001 to 0.1 ng/mL and 1–100 ng/mL, respectively.27 28 We investigated supraphysiological levels of recombinant sFLT3L (10 ng/mL) or recombinant sFLT3 (150 ng/mL) for a possible interference with CLN-049-induced TDCC using MOLM-13 as target cells. No impact on the efficacy of CLN-049 was observed under either condition (online supplemental figure 4).

In vitro safety of CLN-049 (A) FLT3 receptor number of normal cells shown in red, compared with FLT3 receptor number of AML cell lines in black. Numbers show the frequency of live events of the indicated cell type. Each point represents data from an individual donor. (B) PBMC from healthy donors were incubated with increasing concentrations of CLN-049 for 72 hours followed by assessment of pDCs lysis by flow cytometry. (C) BM cells from healthy donors were incubated with increasing concentrations of CLN-049 for 72 hours followed by assessment of CD34+ cells lysis by flow cytometry. (D) Purified T cells from the indicated donors were incubated with increasing concentrations of the indicated CD3-specific antibodies for 72 hours followed by assessment of CD4+ and CD8+ T cell activation via CD69 analysis by flow cytometry. (E) PBMC from healthy donors were incubated with increasing concentrations of the indicated CD3-specific antibodies for 72 hours in the presence or absence of MOLM-13 cells. Subsequently, CD4+ and CD8+ T cell activation was assessed via analysis of CD69 expression by flow cytometry. AML, acute myeloid leukemia; BM, bone marrow; pDCs, plasmacytoid dendritic cells; PBMC, peripheral blood mononuclear cells.
Concomitant with TDCC, both CD4+ and CD8+ T cells were robustly activated by CLN-049 in the co-culture assays as measured by CD69 expression, with CD4+ T cells appearing to be more potently activated than CD8+ T cells. EC50 values for inducing T cell activation were consistently lower than for inducing target cell lysis (figure 3C). CLN-049 also dose-dependently stimulated release of TNFα, IFNγ, and IL-6 in co-cultures of AML cell lines with PBMC (figure 3E). Unlike TDCC activity, where no significant correlation between FLT3 expression and cytotoxicity was observed, cytokine release was significantly lower in the presence of U-937 cells (~200 FLT3 molecules/cell) as compared with MOLM-13 cells (~1400 FLT3 molecules/cell).
Normal FLT3-expressing cells are less sensitive to CLN-049-induced TDCC than AML cells
We determined FLT3 expression levels on DCs from PB (online supplemental figure 5A,B) and CD34+ cells from BM of healthy donors (online supplemental figure 5C). cDC1 cells (HLA-DR+CD11c+CD141+CD1c-) expressed on average 600 FLT3 molecules per cell and their frequency in PBMC was only 0.05%. cDC2 cells (HLA-DR+CD11c+CD141-CD1c+) and pDCs (HLA-DR+CD11c-CD123+) were of higher frequency (0.65% and 0.25%, respectively), but expressed lower levels of FLT3: 300 and 70 molecules per cell, respectively. CD34+ cells from BM expressed approximately 100 molecules per cell (figure 4A).
TDCC of CLN-049 against cDC1 and cDC2 cells could not be evaluated because cells did not remain viable in cell culture for the 72 hours required to assess TDCC (online supplemental figure 3B). For ~50% of donors, however, a subset of pDCs were evaluable for analysis, which revealed no significant lysis in four of five donors (figure 4B). CLN-049 did not induce significant lysis of BM-derived CD34+ cells from any of the five evaluated donors (figure 4C).
T cell activation by CLN-049 is dependent on FLT3 binding
Although CLN-049 has two CD3-binding scFvs, they seem to bind the CD3ε subunits in the TCR in a mutually exclusive fashion, as recently shown for a PSMA/CD3-bispecific sister molecule which shares the Fcγ1 domain and anti-CD3 scFvs with CLN-049.29 To confirm this for CLN-049, we evaluated the activation of purified T cells via CD69 expression in the absence of target cells. Positive control antibodies were the agonistic anti-CD3 murine mAbs OKT3 as well as UCHT1, the latter being the parental murine anti-CD3 mAb used for construction of CLN-049. Whereas OKT3 induced T cell activation in 3/3 donors and UCHT1 in 2/3 donors, CLN-049 induced no significant T cell activation over background in any of the donor T cells tested (figure 4D).
Healthy PBMC contain a low frequency of FLT3-expressing cells (figure 4A). When donor PBMC were incubated with CLN-049, both CD4+ and CD8+ T cells in the PBMC samples showed a slight dose-dependent increase of CD69 expression (figure 4E). When MOLM-13 AML cells were added, treatment resulted in robust T cell activation. The lack of activation with purified T cells, the low-level activation of T cells in PBMC, and the profound activation of T cells in the presence of AML cells demonstrate that CLN-049 stimulates T cells in a strictly FLT3-dependent manner.
Anti-leukemic activity of CLN-049 in a xenograft model of human AML
To evaluate the PK and efficacy of CLN-049 in vivo, NCG mice were engrafted with human PBMC and MOLM-13 AML cells, and then treated with CLN-049 as outlined in figure 5A,B. In the PK cohort, a 0.01 mg/kg dose yielded a terminal half-life of 5.6 days, consistent with a typical IgG molecule (figure 5C). In the efficacy cohort (figure 5B), mice were treated with CLN-049 weekly. On day 15, PB was sampled, and CLN-049 at 0.001 mg/kg reduced the number of detectable MOLM-13 cells by 87%, while CLN-049 at 0.003 mg/kg and 0.01 mg/kg did so by >95% (figure 5D). CLN-049 significantly increased median survival compared with IgG control by 27% at 0.001 mg/kg, 49% at 0.003 mg/kg, and 76% at 0.01 mg/kg (figure 5E).
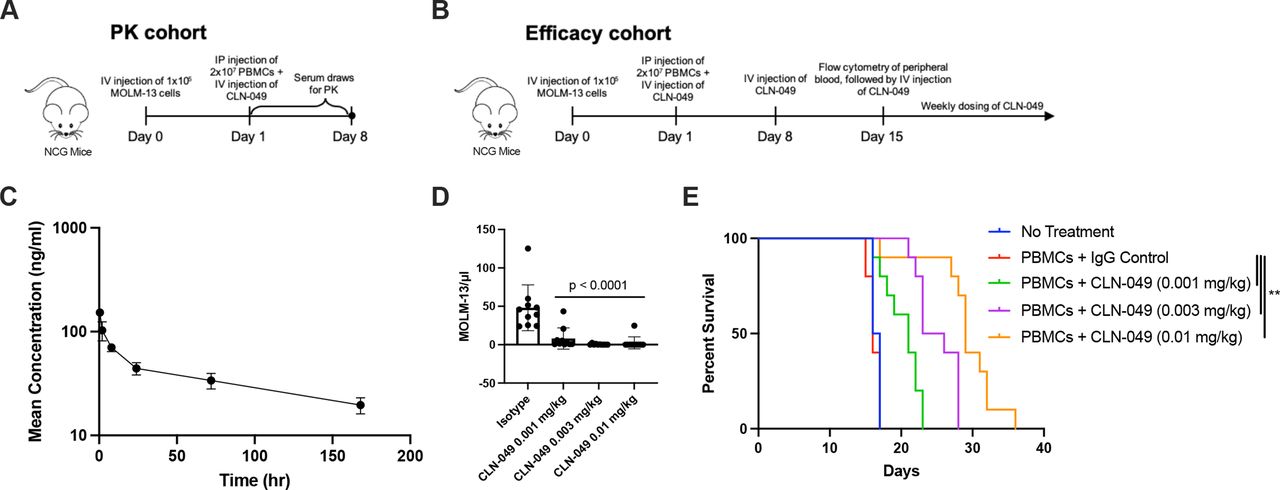
In vivo activity of CLN-049 (A, B) Study designs of CLN-049 administered to NCG mice engrafted with 1×105 MOLM-13 AML cells and 2×107 human PBMC, consisting of (A) a PK cohort and (B) an efficacy cohort. (C) PK curve of CLN-049 injected intravenously at 0.01 mg/kg. (D) MOLM-13 counts in peripheral blood at day 15. (E) Kaplan-Meier survival curves. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple comparisons test (D) or Mantel-Cox test (E). AML, acute myeloid leukemia; ANOVA, analysis of variance; PBMC, peripheral blood mononuclear cells; PK, pharmacokinetics.
Activity of CLN-049 against primary human AML cells
We next assessed whether CLN-049 could mediate lysis of primary AML blasts at physiological E:T ratios. Out of three ex-vivo samples from AML patients examined, two exhibited E:T ratios below 1:1 (figure 6A). In all cases, CLN-049 induced lysis of AML blasts by autologous T cells to near-completion at antibody concentrations below 10 nM (figure 6A).
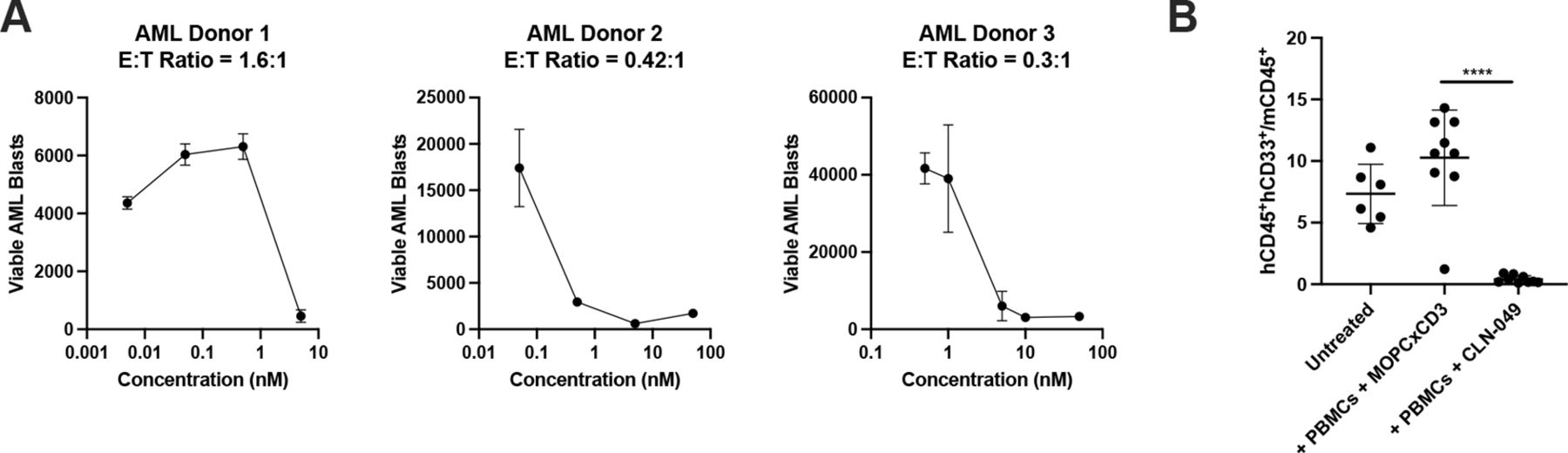
Cytotoxicity against primary AML cells (A) PBMC from three AML patients were incubated with increasing doses of CLN-049 for 72 hours followed by assessment of target cell lysis by flow cytometry. (B) NSG mice were coengrafted with 8×106 PBMC from an AML patient and 8×106 PBMC from a healthy donor, and then treated with CLN-049 or a control antibody (MOPCxCD3) on days 0, 3, and 10 (1 mg/kg on day 0, and 0.5 mg/kg on days 3 and 10). Leukemic burden was evaluated by flow cytometry of BM on day 24 and quantified by the ratio of human AML cells (hCD45+hCD33+) to murine hematopoietic cells (mCD45+). ****p<0.0001. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple comparisons test (B). ANOVA, analysis of variance; AML, acute myeloid leukemia; BM, bone marrow.
To evaluate the activity of CLN-049 against primary leukemic cells in vivo, we engrafted NSG mice intravenously with AML patient cells and with PBMC from a healthy donor as effector cells, followed by treatment with CLN-049 or a control bsAb (MOPCxCD3). On day 24, compared with animals receiving the control antibody, treatment with CLN-049 resulted in a 96% reduction in tumor burden in the BM (figure 6B).
Activity of CLN-049 against primary human B-ALL cells
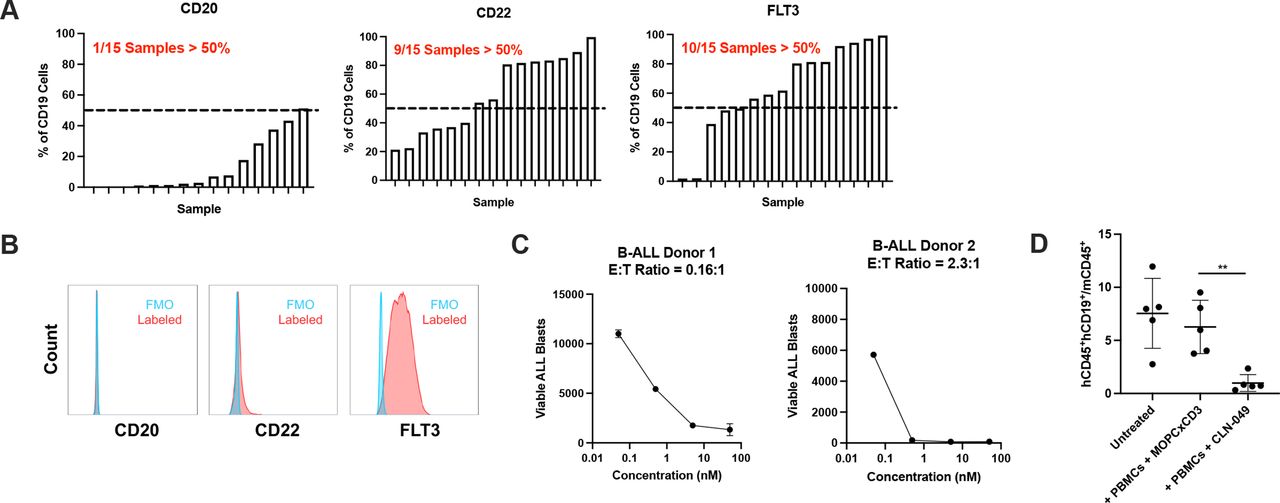
As FLT3 expression has also been reported for B-ALL,30 31 we investigated how expression compares to other B cell leukemia targets. Cell surface expression of FLT3 as well as that of CD20 and CD22 was evaluated in samples from 15 B-ALL patients by flow cytometry. FLT3 was detectably expressed in 10/15 samples, which was a greater fraction than observed for CD20 (1/15) or CD22 expression (9/15) (figure 7A). In a subset of samples, including the example shown (figure 7B), CD20 and CD22 were poorly expressed compared to FLT3. As with AML, CLN-049 could mediate lysis of primary B-ALL blasts to near-completion, including E:T ratios below 1:1 (figure 7C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Efficacy of CLN-049 in B-ALL (A) Fifteen samples of B-ALL patients were evaluated for CD20, CD22, and FLT3 expression by flow cytometry. B-ALL blasts were identified as CD19+. Shown in each graph is a rank-ordered distribution of expression patterns for each target. (B) Representative donor with low expression of CD20 and CD22 and high expression of FLT3. (C) PBMC from two B-ALL patients were incubated with increasing doses of CLN-049 for 72 hours followed by assessment of target cell lysis by flow cytometry. (D) NSG mice were coengrafted with 4×106 PBMC from a B-ALL patient and 10×106 PBMC from a healthy donor, then treated with CLN-049 or a control antibody (MOPCxCD3) on days 7 and 11 (1.5 mg/kg). Leukemic burden was evaluated by flow cytometry of the BM on day 17 by the ratio of human B-ALL cells (hCD45+hCD33+) to murine hematopoietic cells (mCD45+). **p<0.01. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple comparisons test (D). ANOVA, analysis of variance; B-ALL, B-cell acute lymphoblastic leukemia; BM, bone marrow.
To evaluate the in vivo efficacy of CLN-049 against primary human B-ALL blasts, we engrafted NSG mice with B-ALL patient cells and PBMC from a healthy donor as effector cells, followed by treatment with CLN-049 or the control bsAb MOPCxCD3. On day 17, compared with animals receiving the control antibody, treatment with CLN-049 resulted in a 85% reduction in tumor burden in the BM (figure 7D).
Discussion
While T cell-redirecting therapies have significantly improved the treatment of certain B cell hematologic malignancies,6 7 there remains a dearth of approved agents of this class in AML. One shortcoming in the field is the quality of target antigens. While several highly specific B cell markers have been successfully targeted, including CD19, CD20, CD22 and BCMA, without significant toxicities,32 the situation for AML is different. The most frequently exploited targets for T cell therapies in AML are CD33, CD123, and CLEC12A,8 which are all broadly expressed in the large and complex myeloid cell compartment. As a result, T cell-engaging bsAbs against these targets are expected to have a narrow therapeutic index both due to ablation of normal myeloid cells and as a consequence of broad systemic T cell activation which can potentially lead to cytokine release syndrome.
Here, we selected FLT3 as target antigen for several reasons: (1) FLT3 is expressed in ~80% of AML patients16; (2) FLT3 is expressed in B-ALL patients, in some cases to a greater extent than CD20 or CD22; (3) FLT3 expression on normal cells is limited to subsets of myeloid DCs in PB, and expression levels on progenitor cells in the BM are low; (4) FLT3 is a well-characterized oncogenic driver of disease, reducing the risk of tumor escape by target loss.
CLN-049 was designed for two-armed FLT3 target binding and functionally single-armed CD3 binding by employing a symmetric IgG fusion protein format with a silent Fc domain. As shown here, CLN-049 is able to bind cells expressing low levels of FLT3 by an avidity effect but cannot activate T cells in the absence of FLT3. Alternative engineering strategies to achieve monovalent CD3 binding include asymmetric IgG-based fusions or tandem single-chain Fv constructs, so called BiTE antibodies. Such formats typically do not exploit an avidity effect for target binding. Due to its symmetric, IgG-based design, CLN-049 can be produced and purified at the high yields and with the stability typical of conventional mAbs, allowing for a conventional antibody manufacturing processes.
In cocultures of human PBMC with various AML cell lines, CLN-049 consistently drove complete target cell lysis at sub-nM EC50 values, suggesting that responses may be achieved in patients at very low doses. As observed with other T cell-engaging antibodies,33 34 neither EC50 values for TDCC nor for T cell activation significantly correlated with levels of FLT3 expression on target cells. This is in accordance with typical cytotoxic T cell function, where single digit numbers of pMHC complexes are sufficient to trigger formation of cytolytic immune synapses and target cell lysis.35 The high potency of CLN-049 may be facilitated by avid binding to FLT3 and specifically via a membrane-proximal epitope. This feature of CLN-049 should be confirmed in animal models bearing low FLT3-expresing tumors. If survival is significantly improved in this setting, as would be predicted from the in vitro co-culture data, then this would suggest that CLN-049 could address a broad patient population due to a low FLT3 expression threshold required for clinical benefit. As previously described,36 we found target expression level to affect cytokine release more so than cytotoxicity. Although in vitro cytokine release is not always predictive of cytokine release syndrome in the clinic, and these studies were not reproduced in an in vivo model, the trends described herein suggest that patients with low FLT3 expressing blasts may have reduced cytokine-related toxicities yet maintain benefit from CLN-049 treatment.
Despite the high potency of CLN-049 against AML cell lines, we did not observe significant depletion of normal FLT3-expressing cells, including DCs from PB and CD34+ cells in BM samples. In contrast, autologous T cells in PBMC from AML and B-ALL patients mediated complete lysis of AML blasts on treatment with CLN-049, even at very low E:T ratios. This suggests that normal FLT3-expressing cells are less susceptible to TDCC by CLN-049 than AML cells expressing similarly low levels of FLT3, the mechanistic basis for which warrants further investigation.
Given the low threshold for FLT3 expression levels, CLN-049 may have applications beyond AML. For example, CLN-049 may impart clinical benefit in premalignant myeloid disorders, including myeloproliferative neoplasms and myelodysplastic syndrome, especially patients that present as refractory anemia with excess blasts that have a high likelihood of progressing to AML.37 While canonically considered a marker and driver of myeloid disease, we also detected robust FLT3 expression on B-ALL patient samples, in line with published data.30 31 In a subset of samples, expression of FLT3 was higher than expression of CD22, an emerging target for immunotherapy of B-ALL. In accordance with these findings, CLN-049 was able to drive the depletion of patient-derived B-ALL both in vitro and in vivo. In conclusion, we expect CLN-049 to have robust antitumor activity in the clinic in both AML and related myeloid disorders as well as in FLT3-expressing B-ALL patient populations.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
NKM and MP contributed equally.
Contributors NKM and MP wrote the manuscript, with support by KM, BL, PAB, JSM, GJ and HRS. Experiments were planned by NKM, KM, JSM and PAB. MP, IS, FV, MM, JSH, JK, LO, LZ, H-JB, SM and SH provided samples, reagents, and shared experimental protocols. Funding was secured by GJ and HRS. JSM was guarantor for the manuscript.
Funding Part of this work was conducted with funding by Helmholtz Validation Fund.
Competing interests NKM, KM, BL, PAB, JSM, GJ and HRS have ownership in Cullinan Florentine, which is seeking to commercialize CLN-049.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.