Article Text

Abstract
Background Bladder cancer is a common disease worldwide with most patients presenting with the non-muscle-invasive form (NMIBC) at initial diagnosis. Postoperational intravesical instillation of BCG is carried out for patients with high-risk disease to reduce tumor recurrence and progression to muscle invasive disease. However, BCG can also have side effects or be ineffective in some patients because it cannot enter the cancer cells. Thus, to improve the efficacy of BCG immunotherapy is the long-term pursuit of the bladder cancer field.
Methods To increase the adhesion of BCG to the urothelium we overexpressed FimH, a mannose binding protein naturally used by uropathogenic Escherichia coli to adhere to human urothelium, onto the surface of BCG. The adhesion/internalization ability of rBCG-S.FimH was examined in mouse bladder by fluorescence microscopy. Preclinical evaluation of antitumor efficacy was carried out in orthotopic mouse models of bladder cancer and in human peripheral blood mononuclear cells. Mechanistic studies were carried out using toll-like receptor 4 (TLR4) knockout mice. Immune cells and cytokines in the serum, tumor and lymph nodes were analyzed by flow cytometry, PCR, ELISA and ELISPOT.
Results rBCG-S.FimH exhibited markedly improved adhesion and more rapid internalization into urothelial cells than wild-type BCG, resulting in more potent antitumor activity in orthotopic murine models of bladder cancer. To our surprise, rBCG-S.FimH elicited a much more prominent Th1-biased immune response known to be positively correlated with BCG efficacy. Mechanistic studies using TLR4 knockout mouse showed that rBCG-S.FimH could induce enhanced dendritic cell activation and tumor antigen-specific immune response in a TLR4-dependent manner. Furthermore, human peripheral blood mononuclear cells stimulated by rBCG-S.FimH also showed better tumoricidal effects than those using wild-type BCG.
Conclusion rBCG-S.FimH is a novel BCG strain with significantly improved efficacy against bladder cancer. Since intravesical BCG immunotherapy is the first-line treatment for NMIBC, which accounts for more than 70% of all bladder cancer cases, our results provide a compelling rationale for clinical development.
- Immunotherapy
- Urinary Bladder Neoplasms
- Translational Medical Research
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Bladder cancer is the most common malignancy of the urinary system, and non-muscle-invasive bladder cancer (NMIBC) accounts for 70%–80% of all clinical cases.1–3 Intravesical instillation of BCG after transurethral resection of bladder tumor is the gold-standard treatment for patients with high-risk NMIBC to prevent recurrence and progression to muscle-invasive disease.4 However, despite good success rates, about 30%~45% patients are refractory to BCG therapy and around 40% patients will eventually relapse after initial response. In addition, as many as 20% of patients develop BCG intolerance during treatment.5–7 Therefore, there is a long-lasting clinical need to augment the efficacy and reduce the side effects of intravesical BCG immunotherapy.
Although the antitumor mechanism of BCG is incompletely understood, it is generally accepted that BCG elicits both local and systematic immune responses.8 First, BCG adheres to the bladder urothelium through binding to fibronectin9–11; second, BCG is internalized by normal urothelial cells, bladder cancer cells and macrophages; thirdly, BCG induces an innate immune response, producing cytokines and chemokines to recruit infiltration of immune cells, including neutrophils, monocytes, macrophages, T cells, B cells, and natural killer cells12 13; lastly, repeated instillation of BCG leads to the induction of a strong T helper 1 (Th1) cell-biased adaptive immune response that may offer some protection against tumor recurrence and progression.14 Each of these steps has been explored to increase the efficacy of BCG. The most common strategy, however, is to boost the Th1 adaptive immune response either by direct coadministration of Th1 cytokines (interleukin (IL)-2, IL-12, IL-18, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, etc) or recombinantly overexpressing them in BCG.15–20 For example, two recently published works showed that recombinant BCG overexpressing the Th1 cytokine IL-15 or the S1 subunit of pertussis toxin prolonged the survival of mice with orthotopically implanted bladder tumor.21 22 However, to date no recombinant BCG strain has been proven to be superior to BCG in the clinic.23
Since BCG attachment to the bladder urothelium is the critical initial step for its antitumor effect,24 low attachment may account for treatment failure in some patients.11 25 26 Therefore, improving the ability of BCG to adhere to the urothelium may improve its antitumor effect. FimH is an adhesion protein found at the end of type 1 fimbria in Escherichia coli.27 Uropathogenic E.coli infection of the urinary tract relies on FimH binding to mannose residues on the surface of urothelial cells.28 The high binding affinity between FimH and mannose allows E.coli to adhere to the urothelium and could be intervened by analogs of D-mannose.29 Thus, we hypothesized that forced expression of FimH on the outer surface of BCG by genetic engineering will enhance BCG attachment to bladder urothelium and that this may induce a more pronounced adaptive immune response than previous methods, boosting the antitumor effects of BCG on bladder cancer.
In this study, we report the generation of a recombinant BCG strain that overexpresses the FimH protein on the outer surface. We found that rBCG-S.FimH exhibited enhanced attachment and internalization to bladder urothelium, and verified its antitumor effects in a murine orthotopic bladder cancer model. Mechanistically, rBCG-S.FimH induced stronger Th1 effect, dendritic cell activation, and tumor antigen-specific immunity than wild-type BCG. This novel recombinant BCG strain has the potential to significantly improve the efficacy of BCG immunotherapy in bladder cancer.
Methods
Animals
Female C57BL/6 and toll-like receptor 4 (TLR4)-KO transgenic mice, 6–8 weeks old, were obtained from SPF Biotechnology (Beijing, China), and kept under specific pathogen-free conditions. Mice were housed in a SPF facility and have free access to food and water. All animal experiments were approved by Nanjing University Institutional Animal Care and Use Committee.
Cell lines
The murine bladder cancer cell line MB49-luc is a gift from Dr Yi Luo (University of Iowa); human bladder cancer cell line 5637 (ATCC HTB-9) was purchased from ATCC. All cells were cultured in RPMI 1640 medium (Hyclone), supplemented with 10% fetal bovine serum (Hyclone), 100 units/mL penicillin, and 100 µg/mL streptomycin, at 37°C with 5% CO2.
BCG strains
A laboratory-maintained BCG Pasteur strain and its derivative 261BCG,30 which contains an empty pMV261 plasmid vector, are gifts from Dr Yi Luo (University of Iowa) and have been shown to be similar to commercial lyophilized BCG preparations in terms of immune stimulation.31 261BCG, rBCG-S.EGFP, rBCG-S.FimH-EGFP and rBCG-S.FimH all contain the kanamycin resistance gene and were cultured in Middlebrook 7H9 broth (Solarbio, LA7220) supplemented with 10% ADC (Solarbio, LA9560), 0.05% tween 80, and 30 µg/mL of kanamycin, at 37°C and shaken continuously. BCG concentration was quantified using absorbance at 600 nm, and converted to MOI by the equation: 1 OD600 unit=2.5×107 CFU/mL. The culture medium was centrifuged at 3000×g to collect the bacteria for passaging.
Construction and stability of recombinant BCG
The DNA fragments of FimH, 19ss-FimH, 19ss-EGFP and 19ss-FimH-EGFP were synthesized by GenScript (Nanjing, China), and inserted between BamHI and HandIII sites of the pMV261 plasmid. Enzyme digestion primers are: forward:5'CTGGTGCCGCGCGGCAGCCATATGATGAAACGAGTTATTACCCTGTTTGCTGT3'; reverse :5'AGTGGTGGTGGTGGTGGTGCTCGAGAAACTGGAAATCATCGCTGTTATAGTTGTT3'. After heat shock transformation of E. coli DH5α, the plasmids were amplified in the presence of kanamycin, harvested by a plasmid extraction kit (TSINGKE, PM0201-200), and verified by sanger sequencing. The plasmids were transformed into BCG by electroporation, and plated onto 7H10 solid medium supplemented with kanamycin (30 µg/mL). After 2–3 weeks, single clones were selected and transferred to 7H9 liquid medium containing kanamycin to continue the selection pressure. After four passages (1:100), the recombinant strains were aliquoted and stored at −80°. To test the stability of the plasmid, resuscitated rBCG was passaged eight times in medium with or without antibiotics. The same amount of samples were taken from each passage and plated onto 7H10 plate to assess the colony forming ability.
Western blotting
BCG and rBCG-S.FimH were cultured in the 7H9 medium. The bacterial culture was centrifuged at 3000×g for 15 min, washed with phosphate buffered saline (PBS) thrice, and dissolved at room temperature with 20KU/mL lysozyme (Beyotime, #ST206) for 30 min, followed by addition of medium-strength lysis solution (Beyotime, # P0013C) and sonication for 5 min at 15 s intervals; the lysates were then placed on ice for 2 hours, centrifuged to collect the supernatant. In order to extract membrane proteins, samples were divided into two identical parts, 3% T-114 (MDBio, #2190701) was added to the membrane protein sample and incubated on ice for 2 hours. The sample was then heated in a 30°C water bath for 5 min, after liquid phase separation, the bottom layer is the membrane protein. Total proteins were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes, and subjected to western blotting using anti-FimH antibodies.
Horseradish peroxidase incubation experiment
BCG, rBCG-S.EGFP and rBCG-S.FimH at a concentration of 2.5×105 CFU/mL were first blocked for 1 hour with 5% Fetal Bovine Serum (FBS) in PBS, followed by incubation with horseradish peroxidase (1:1000) at room temperature for 2 hours. The wells were then washed thrice with blocking buffer, enhanced chemiluminescence (ECL) reagent (Vazyme, #E412-02-AA/B) were then added and chemiluminescence signal detected on a gel imaging system (Tanon 5100, Tanon, Shanghai, China).
Adhesion and internalization of BCG
MB49 and 5637 cells were plated in 6-well plates and cultured for 24 hours. BCG, rBCG-S.EGFP and rBCG-S.FimH-EGFP (5×105 CFU/mL) were then incubated with the cells and collected at different time points. D-mannose (100 µM) was added at the beginning as a competitive inhibitor of FimH. Excess BCGs were washed off, cells were then fixed with 4% paraformaldehyde for 15 min. The adhesion ability of BCG is determined by the residual amount of green fluorescence on the cell surface. To assess BCG internalization, cells were incubated with PBS, rBCG-S.EGFP and rBCG-S.FimH-EGFP for 12 hours and treated with amikacin (200 µg/mL) for 2 hours to kill residual BCG on the surface. Cells were then treated with 0.1% Ttiton-X for 10 min and sonicated for 15 s. The amount of internalized BCG in cells were then quantified using colony formation on 7H10 plates.
Mouse orthotopic models of bladder cancer
Female C57BL/6 or TLR4-KO transgenic mice (8 weeks old) were used for the establishment of orthotopic models of bladder cancer according to published literature.32 MB49-luc mouse bladder cancer cells stably expressing luciferase were prepared and concentrations adjusted to 2×107 cells/mL in 50% mouse serum. The mice were anesthetized with 5% sodium pentobarbital and residual urine in the bladder were removed by gentle pressing. An indwelling needle (0.7×19 mm, BD 8234041) was gently inserted into mouse bladder through the urethra for injection of fluids and cells. Silver nitrate (AgNO3, 5 µL, 0.3 M) was gently injected intravesically to erode the mouse bladder epithelium for 15 s, followed by a quick wash with PBS. A 50 µL cell suspension containing 1×106 MB49-luc cells was instilled into the bladder and retained for 1 hour. The next day, mice were randomized into different treatment groups and intravesically instilled with BCG or proper controls twice a week. In vivo imaging system (IVIS) imaging was performed every 7 days to monitor tumor growth. At the end of experiment, the whole bladder, the inguinal lymph nodes, the tumor-draining lymph nodes, the spleen and serum were harvested from each mouse and immediately processed for subsequent experiments.
Evaluation of BCG-induced cystitis and BCG infection in mice
The frequency of urination in mice was measured using the void spot assay according to literature.33 Briefly, mice were gently put into cages covered with Whatman filter paper (Fisher Scientific #0 57 163W), one mouse per cage. The whole experiment lasted for 4 hours during which time mice were fed with food but without water. After the experiment, filter paper stained with urine was imaged using ultraviolet light and void spots were analyzed using Image J Software (V.1.46r). Pelvic pain in mice was measured using the von Frey test as previously described.34 Briefly, mice were placed gently in a plexiglass box with a metal mesh base in a behavioral test room. Mice were stimulated with Von Frey filaments at the left hindlimb plantar and the lower abdominal area in the general vicinity of the bladder for 3~4 s each time. The test was repeated with an interval of 5 min. Three types of reactions were counted as positive reaction: (1) the abdomen retracts sharply; (2) mice lick or scratch the itch immediately; (3) jumping. The histology of detrusor muscle and suburothelial layer (including submucosa and lamina propria but excluding urothelium) was evaluated according to literature.35 To evaluate BCG infection in mouse organs, acid fast staining was performed in the liver, lung, spleen and kidney; PCR experiments were also carried out to detect the presence of BCG in these organs.
Antibodies
Polyclonal antibody against FimH was generated in rabbit using the full length FimH protein. The following antibodies were purchased from BioLegend (New Jersey, USA): isotype control (IgG1, IgG2a or IgG2b), CD11c-PE-Cy7 (N418, #117317, 1:200), CD8α-APC (53–6.7, #100711, 1:200), CD40-PE (FGK45, #157505, 1:200), CD80-PE (16–10 A1, #104707, 1:200), CD86-PE (GL-1, #105007, 1:200), anti-MHC class I-AlexaFluor647 (AF6-88.5, #116511, 1:40), anti-MHC class II-pacific blue (M5/114.15.2, #107620, 1:40), CD11b-PE-Cy7 (M1/70, #101216, 1:200), CD3-APC (17A2, #100236, 1:200), anti-IFN-γ (XMG1.2, #505801, 1:200), anti-TNF-α (MP6-XT22, #506323, 1:40). The following antibodies were purchased from Abcam (Cambridge, UK): anti-p38(#ab32142, 1:1000), anti-p38 (phospho T180 +Y182) (#ab195049, 1:1000), anti-JNK (#ab179461, 1:1000), anti-JNK (phospho T183 +T183+T221) (#ab124956, 1:1000), anti-ERK (#ab184699, 1:1000), anti-ERK (phospho T202 +T185) (#ab201015, 1:1000), anti-Iκβ (#ab32518, 1:1000). Antibodies CD45-APC-Cy7 (30-F11, #557659, 1:200) and CD4-BV421 (GK1.5, #562891, 1:200) were obtained from Becton Dickinson (New Jersey, USA). Anti-tubulin antibodies were purchased from Beyotime (#AT819, 1:500).
Flow cytometry
Cells were incubated with unlabeled isotype control antibody and Fc blocking antibody for 15 min (BioLegend, New Jersey, USA). After that, fluorescently coupled antibodies were added, and the cells were incubated on ice for 30 min. After washing with PBS, cells were analyzed on FACS Fortessa (Becton Dickinson, Franklin Lakes, New Jersey, USA) using FlowJo V.10.1 software (Tree Star, California, USA). Cell debris and dead cells were excluded by forward and side scatter gating and 7-amino-actinomycin D (BioLegend) staining.
Analysis of mouse lymphatic dendritic cells
The inguinal lymph nodes (iLNs) of mice were digested with collagenase for 30 min at room temperature. Cells were washed with PBS and passed through a 70 µm single cell screen. The cells were then centrifuged at 1700×g for 10 min, resuspended, and stained with fluorescently labeled monoclonal antibodies for 1 hour. The antibodies used were: anti-B220-FITC (RA3-6B2, #103205, 1:200), anti-CD3-FITC (17A2, #100203, 1:200), anti-CD49b-FITC (DX5, #108905, 1:200), anti-Gr1-FITC (RB68C5, #108405, 1:200), anti-Thy1.1-FITC (OX-7, #202503, 1:200) and anti-TER-119-FITC (TER-119, #116205, 1:200). These were added as pedigree markers. Lymphatic dendritic cells (DCs) are defined as lineage−CD11c+ cells and further separated into CD8α+ and CD8α− subpopulations.
Intracellular cytokine staining
Cells were fixed and permeabilized with 1% paraformaldehyde and 2% T-100 (Sigma, #9002-93-1) at room temperature for 30 min, and then stained with anti-cytokine antibodies in PBS containing 2% T-100 for 1 hour at room temperature. After washing off excess antibodies, cells were stained with Cy3-labeled Goat anti-Rat IgG (Beyotime, #A0507) for 30 min at room temperature in the dark. Isotype control IgG staining was performed in all experiments.
ELISA
The concentrations of IL-2, IFN-γ, IL-12p40 and TNF-α in mouse serum were measured using respective ELISA kits (Multi Science, Shanghai, China) according to the manufacturer’s instructions.
Reverse transcription PCR and real-time PCR
Total RNA was extracted from the cells and reverse transcribed into cDNA by Oligo (dT) and reverse transcriptase (Vazyme, #R101-01, Nanjing, China). On the LightCycler 480 real-time PCR system (Roche, Basel, Switzerland), cDNA was subjected to 40 cycles of real-time PCR at an annealing and extension temperature of 62°C. For RT-PCR, a one-step RT-PCR kit (Vazyme, #P612-01) was used. After 20 cycles, semiquantitation of the cytokines was done by agarose gel electrophoresis. The primer sequences used are: mouse
GAPDH forward, 5'-CGGGAAGCCCATCACCATCT-3', reverse, 5'-TCACGCCACAGCTTTCCAGA-3';
IL-2 forward, 5’-AAGCACAGCAGCAGCAGCAG-3’, reverse, 5ʹ-GCCGCAGAGGTCCAAGTTCATC-3ʹ;
IL-4 forward, 5’-TGTCATCCTGCTCTTCTTTCTC-3’, reverse, 5ʹ-CTTGGACTCATTCATGGTGC −3ʹ;
IL-5 forward, 5’-AGACGGAGGACGAGGCAGTT −3’, reverse, 5ʹ-TGCAACGAAGAGGATGAGGG-3ʹ;
IL-10 forward, 5’-CTGCTATGCTGCCTGCTCTTACTG-3’, reverse, 5ʹ-AGCCGCATCCTGAGGGTCTTC-3ʹ;
IL-12p40 forward, 5’-CACTCACATCTGCTGCTCCACAAG-3’, reverse, 5ʹ- TATTCTGCTGCCGTGCTTCCAAC-3ʹ;
IFN-γ forward, 5′-TGGCTGTTTCTGGCTGTTACTGC-3′, reverse, 5′-GACGCTTATGTTGTTGCTGATGGC-3′;
TNF-α forward, 5′-GCGACGTGGAACTGGCAGAAG-3′, reverse, 5′-GTGGTTTGTGAGTGTGAGGGTCTG-3′;
T-bet forward, 5ʹ-CAACAACCCCTTTGCCAAAG-3ʹ, reverse, 5ʹ-TCCCCCAAGCATTGACAGT-3ʹ;
Tumor specific antigen peptides
S12 (KLVVVGASGVGKS), G12 (KLVVVGAGGVGKS) and Smcy (KCSRNRQYL) were purchased from Sangon Biotech (Shanghai, China).
Splenic T cell proliferation assay
Mouse spleen cells were grounded and counted to 107 cells/mL, suspended in serum-free medium (Invitrogen, San Diego, California, USA) containing 5 µM carboxyfluorescein succinimidyl ester (CFSE) and incubated for 10 min, and then incubated with 50% cold FBS for 5 min to stop the reaction. CFSE-labeled cells were stimulated by antigen peptides for 4 days, flow cytometry (Attune NxT, Thermo Fisher, Waltham, Massachusetts) was then used to determine the proliferation of splenic T cells (CD3 positive) by analyzing CD4, CD8 and CFSE fluorescence intensity. Flow data were analyzed using the FlowJo V.10.1 software (Tree Star, San Diego, California).
Splenic T cell cytotoxicity assay
The stimulated or unstimulated spleen cells were sorted by flow, CD3+ T cells were incubated with preseeded MB49-luc cells in 96-well plates for 8 hours. The effector to target cell ratio were set at 1:100, 1:50, 1:25, 1:12.5 and 1:6.5. Alma blue (10 µl) was added to each well and incubated for 4 hours, fluorescence signals were then measured using a microplate reader (excitation wavelength =550 nm, emission wavelength =590 nm).
Isolation and culture of bone marrow-derived DCs
Femurs from wild type C57BL/6 mice were smashed and bone marrow cells were collected by filtering through 40 µm filter membranes. After lysis of red blood cells, the rest of the cells were cultured in the presence of mouse GM-CSF (20 ng/mL) and IL-4 (10 ng/mL). After 3 days, cells were collected and plated with fresh GM-CSF and IL-4. On day 7, CD11c+ cells were isolated as bone marrow-derived DC (BMDCs) by FACS and used in following experiments.
ELISPOT assay
Mouse IFN-γ levels in the mononuclear cells of tumor-draining lymph nodes was measured using an ELISPOT kit according to the manufacturer’s protocol (Mabtech, Nacka Strand, Sweden). In short, tumor-draining lymph nodes were harvested from mice, and mononuclear cells separated by density gradient centrifugation. The cells were seeded on the pre-coated plate at a density of 2.5×104 cells per well and stimulated with 10 µg/mL antigen peptide or negative control peptide at 37°C for 48 hours. The AID ELISPOT Reader System (Germany) was used to automatically count the spots in the ELISPOT plate, the number of spots observed with the control peptide were subtracted from that with the specific antigen peptide in each sample.
Immunohistochemistry and immunofluorescence
For immunohistochemistry, whole mouse bladders were fixed in 4% paraformaldehyde solution for 72 hours. The fixed bladders were then embedded in paraffin and cut into a thickness of 5 µm. The sections were placed on a glass slide, deparaffinized and hydrated, and subjected to standard H&E staining. Alternatively, the slides were permeabilized with 2% T-100 for 1 hour and incubated with anti-IFN-γ/anti-TNF-α, and developed by DAB (Servicebio, Wuhan, China) for 1–15 min, and finally observed under a microscope. For immunofluorescence, the sections were also permeabilized with 2% T-100 for 1 hour and stained with cy3-labeled anti-tubulin and DAPI (Beyotime, #P0131).
Human peripheral blood mononuclear cell assays
Human whole blood samples were donated by healthy volunteers and processed at the Department of Urology, the First Affiliated Hospital of Nanjing Medical University. Ficoll Histopaque was used to separate human peripheral blood mononuclear cells (PBMC) from whole blood by density gradient centrifugation, then the PBMCs were incubated with BCG for 48 hours, and the supernatant was collected to measure IL-2 expression. Stimulated PBMCs were collected by centrifugation, incubated with KU-7 cells for 8 hours, and 10 µl of Alma blue was added to each well, after 4 hours of incubation, the percentage of cell killing was calculated by fluorescence spectrophotometry. The supernatant was also collected to measure IL-2 and IFN-γ levels by ELISA.
Statistical analysis
All statistical tests were performed using the open-source statistics package R or using GraphPad Prism software V.8 (San Diego, California). Data are presented as the mean±SD or mean±SEM. Differences were considered statistically significant at p<0.05. Normality and equal variances between group samples were assessed using the Shapiro-Wilk test and Brown-Forsythe tests, respectively. When normality and equal variance were achieved between sample groups, one-way analysis of variance (ANOVA) (followed by Bonferroni’s multiple comparisons test), two-way ANOVA (followed by Bonferroni’s multiple comparisons test) or t-tests were used. Where normality or equal variance of samples failed, Kruskal-Wallis one-way ANOVA (followed by Dunn’s correction) or Mann-Whitney U tests were performed.
Data availability
The data generated in this study are available within the article and its online supplemental data files.
Supplemental material
Results
Recombinant rBCG-S.FimH showed enhanced attachment and internalization to bladder urothelium
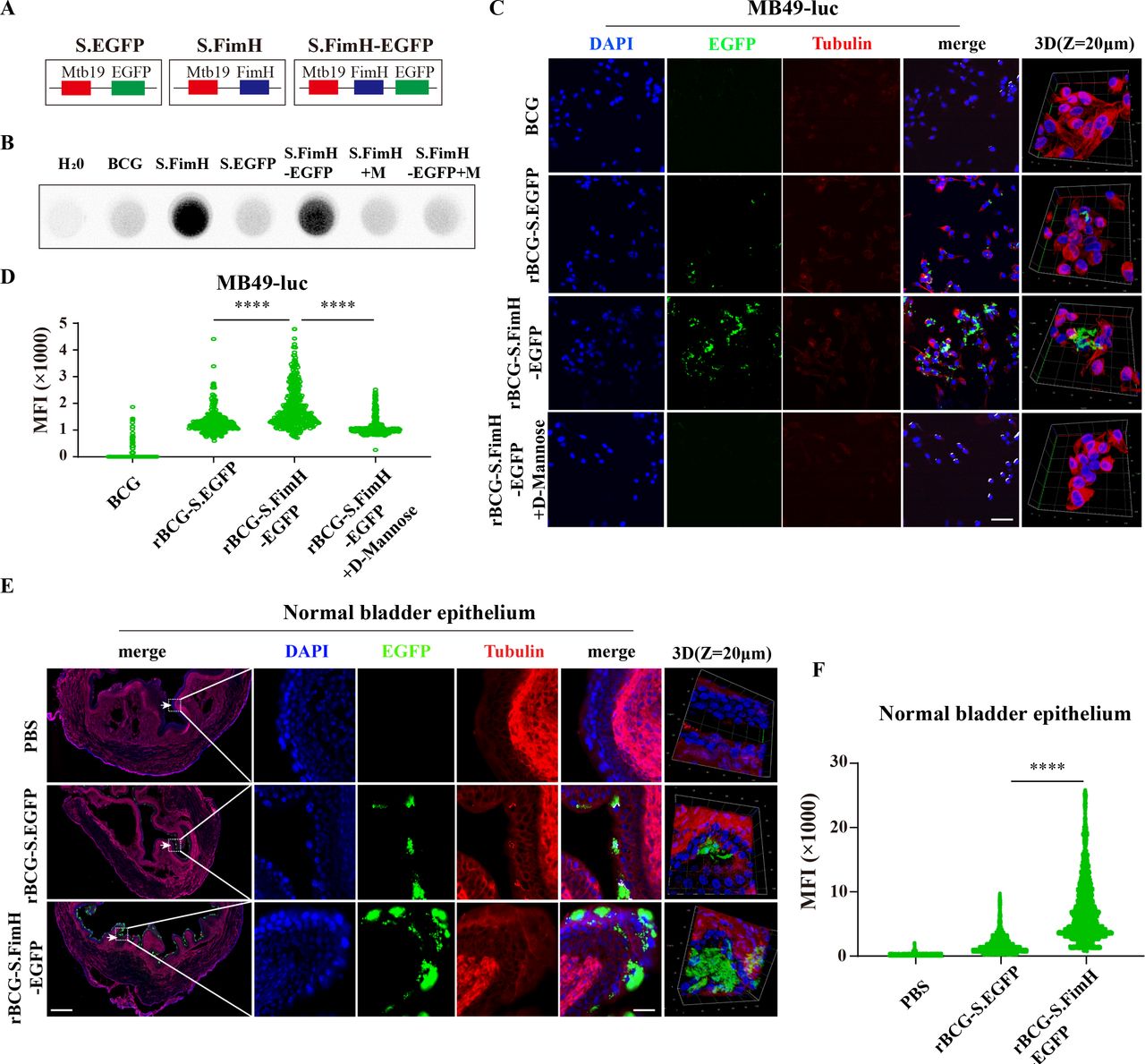
To develop recombinant BCG strains that can overexpress FimH on the outer surface, we used the fusion protein strategy and attached FimH to the C-terminus of Mycobacterium tuberculosis 19 kDa lipoprotein (Mtb19), a signal peptide known to assist the expression of heterologous proteins on the surface of BCG.36 37 The Mtb19-FimH construct was generated using the classic pMV261 plasmid backbone, transformed into BCG via electroporation, followed by clonal expansion and screening for recombinant BCG strains that express high level of FimH on the outer surface (referred to as rBCG-S.FimH) (figure 1A and online supplemental figure 1A,B). In order to facilitate experimental visualization, we also constructed fluorescently labeled strains rBCG-S.FimH-EGFP and rBCG-S.EGFP following the same procedures (figure 1A and online supplemental figure 1A,B), both of which gave strong autofluorescence signals (online supplemental figure 1C). The successful expression of FimH was confirmed in both rBCG-S.FimH and rBCG-S.FimH-EGFP by RT-PCR (online supplemental figure 1D) and immunoblotting with a polyclonal antibody against FimH made in the laboratory (online supplemental figure 1E,F). Notably, FimH was not expressed in wild-type BCG (261BCG, a commonly used BCG strain with empty pMV261vector, was used as control and referred to as wild-type BCG below) and was mostly expressed on the membrane fraction of rBCG-S.FimH (online supplemental figure 1F).

rBCG-S.FimH exhibited stronger bladder adhesion and internalization ability in vitro and in vivo. (A) Schematic representation for the construction of plasmids expressing FimH, EGFP or FimH-EGFP fused to the membrane protein Mbt19. (B) Only recombinant BCGs expressing FimH can bind to mannose residues in the HRP protein (BCG: wild-type BCG; S.FimH: rBCG-S.FimH; S.EGFP: rBCG-S.EGFP; S.FimH-EGFP: rBCG-S.FimH-EGFP; M: 100 µM D-mannose). (C–D) Mouse bladder cancer cell line MB49-luc was incubated with rBCG-S.EGFP and rBCG-S.FimH-EGFP for 4 hours, fluorescence images were taken by confocal (C) and fluorescence intensity quantitated using image J (D). Scale bar: 100 µm. (E–F) rBCG-S.FimH-EGFP showed higher adhesion to the urothelium in normal mouse bladder. C57BL/6 mice were deprived of water for 8 hours, followed by intravesical injection with PBS, rBCG-S.EGFP or rBCG-S.FimH-EGFP (5×105 CFU/mL, 50 µL/mice), the bladders were collected 4 hours later and frozen sections were subjected to immunofluorescence staining and confocal microscopy (E), fluorescence intensity were quantitated using image J (F). Scale bars: 250 µm and 40 µm. ****P<0.0001. HRP, horseradish peroxidase; PBS, phosphate buffered saline.
Next, we tested whether the overexpressed FimH on the outer surface of rBCG retained the ability to bind to mannose. Because HRP, an enzyme commonly used in secondary antibodies for immunoblotting, contains a lot of mannose residues, we used HRP as the binding target and the ECL reagent as the reporter to characterize the binding ability of FimH to mannose. As shown in figure 1B, both rBCG-S.FimH and rBCG-S.FimH-EGFP yield strong positive signals, which were inhibited in the presence of 100 µM free D-mannose, suggesting that FimH displayed on the surface of BCG readily binds to mannose residues in proteins. Thus, we have successfully constructed recombinant BCG with functional FimH proteins expressed on the surface.
We then compared the cell adhesion and internalization capabilities between wild type and recombinant BCG. When the MB49 mouse bladder cancer cell line was incubated with rBCG-S.FimH-EGFP or the rBCG-S.EGFP control for 4 hours, we observed a significantly higher fluorescence signal in cells incubated with rBCG-S.FimH-EGFP, which was abolished by the addition of free D-mannose (figure 1C,D). Similar results were obtained by using the human bladder cancer cell line 5637 (online supplemental figure 2A,B). We next measured the kinetics of recombinant BCG adhesion/internalization in the MB49 and 5637 cell lines. We found that in both cell lines the rBCG-S.FimH-EGFP group reached the peak fluorescence signal much earlier than the rBCG-S.EGFP group, and its peak signal was also higher (online supplemental figure 2C-F). To determine whether such effect could be attributed to BCG internalization rather than adhesion, we collected the bladder cancer cells after BCG incubation, washed off the attached BCGs on the cell surface with amikacin (an antibiotic to kill mycobacteria), and analyzed the amount of internalized BCG by a colony formation assay. Compared with rBCG-S.EGFP, rBCG-S.FimH-EGFP showed a higher level of cell internalization (online supplemental figure 2G). Next, we evaluated the adhesion/internalization ability of rBCG-S.FimH-EGFP in vivo in the bladders of normal C57BL/6 mice and tumor-bearing bladders from an orthotopic bladder cancer model (established by intravesical inoculation of MB49 cells) after intravesical instillation. Confocal imaging of the frozen bladder sections showed that rBCG-S.FimH-EGFP induced higher fluorescence signals than rBCG-S.EGFP in the urothelium of both normal and tumor-bearing bladders (figure 1E,F and online supplemental figure 3A,B). Finally, we measured the retention time of BCG and rBCG-S.FimH in the mouse bladder at 4, 8, 12 and 24 hours post instillation and found that rBCG-S.FimH could dwell much longer than BCG (online supplemental figure 3C). Taken together, these data demonstrated that the FimH-expressing recombinant BCG had stronger adhesion/internalization ability than wild-type BCG both in vitro and in vivo.
rBCG-S.FimH exhibited superior anticancer effect in vivo
To evaluate the antitumor effect of recombinant BCG in vivo, we used the classical mouse orthotopic bladder cancer model generated by intravesical inoculation of the mouse MB49 bladder cancer cells onto bladder urothelium.32 One day after tumor inoculation, mice were subjected to intravesical instillation with PBS, 2.5 mg/kg FimH, 2.5×105 CFU/mL BCG or rBCG-S.FimH two times a week for a total of 4 weeks, and the bladders were harvested on day 28 (figure 2A). We found that rBCG-S.FimH inhibited tumor growth significantly better than wild-type BCG, as evidenced by both IVIS imaging (figure 2B,C) and bladder weight measurement (figure 2D,E). Notably, while seven of the eight bladders in the BCG group contained tumors at the end of treatment, four of the eight rBCG-S.FimH bladders were tumor-free as analyzed by H&E staining (figure 2F). Therefore, rBCG-S.FimH exhibited stronger antitumor effect than wild-type BCG in orthotopic models of bladder cancer.
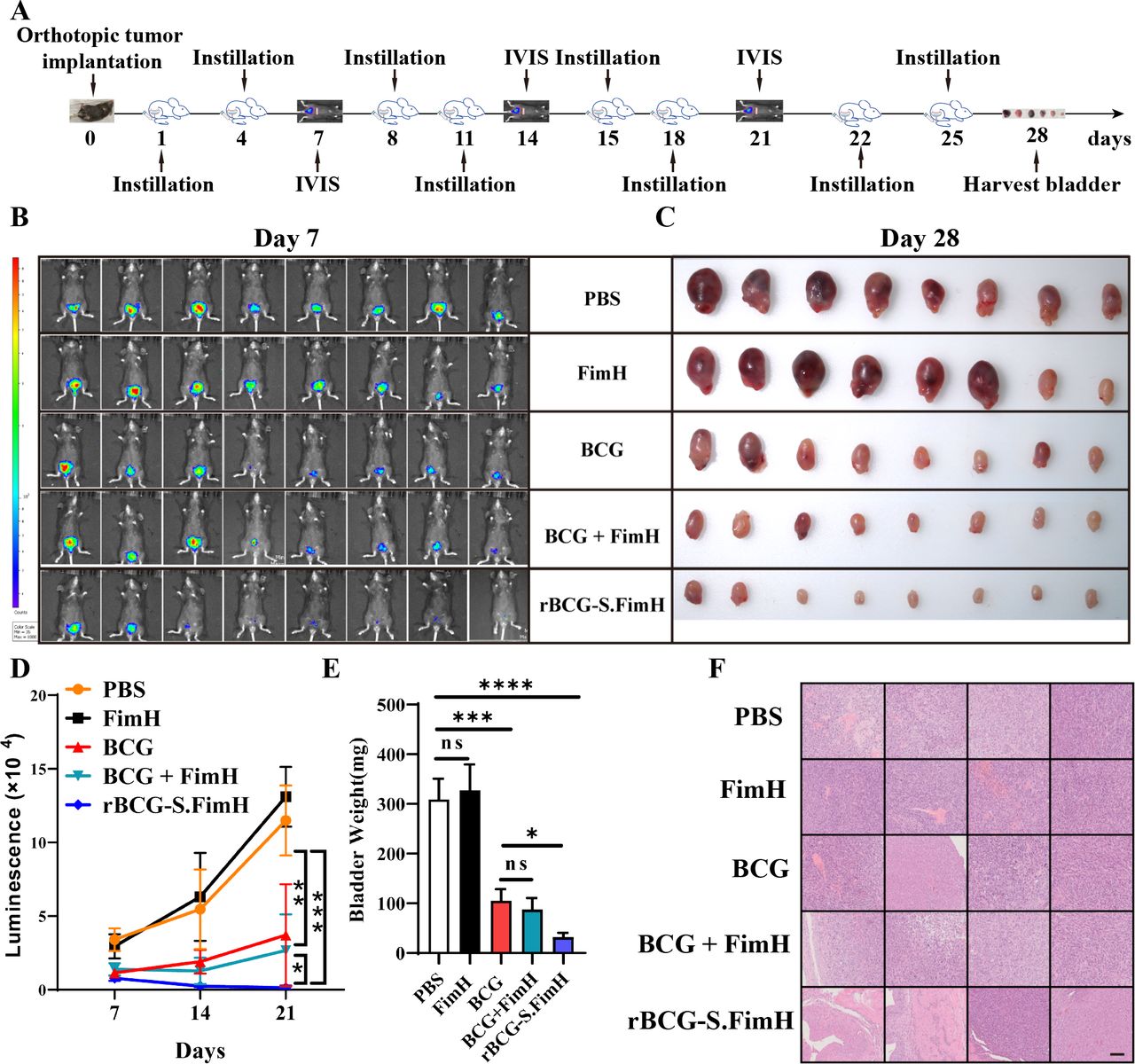
rBCG-S.FimH exhibited enhanced antitumor effect than wild-type BCG in vivo. (A) Schematic representation of the experimental design. C57BL/6 mice were intravesically inoculated with 1×106 MB49-luc cells, and treated with PBS, 2.5 mg/kg FimH, 2.5×105 CFU/mL BCG, 2.5×105 CFU/mL BCG with 2.5 mg/kg FimH or 2.5×105 CFU/mL rBCG-S.FimH two times a week for 4 weeks. Bladders were harvested 28 days after treatment. (B) IVIS images of mice from different treatment groups at day 7. (C) Image of the bladders at the end of the experiment. (D) The growth curve of tumors as monitored by IVIS from day 7 to day 21 (n=8 mice, one-way analysis of variance (ANOVA), mean ± SD). (E) Weight of the bladders at the end of the experiment (n=8 mice, one-way ANOVA, mean ± SEM). (F) Representative H&E staining of the bladders at the end of the experiment showing reduced tumor burden in the rBCG-S.FimH group. scale bar: 100 µm. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. PBS, phosphate buffered saline. IVIS, in vivo imaging system.
Because BCG instillation therapy often causes local side effects including BCG-induced cystitis and bacterial infection (BCGitis), we also compared the safety profile between rBCG-S.FimH and wild-type BCG by evaluating cystitis symptoms and BCG infection in mice after a full course of treatment. For cystitis symptoms, we measured the frequency of urination using the void spot assay (online supplemental figure 4A) and pelvic pain using the von Frey test (online supplemental figure 4B). We also evaluated the histology of detrusor muscle and suburothelial layer (including submucosa and lamina propria but excluding urothelium) (online supplemental figure 4C). For BCGitis, we performed RT-PCR and acid-fast stain experiments on the liver, lung, spleen and kidney to evaluate BCG infection (online supplemental figure 4D–F). In all these experiments, rBCG-S.FimH exhibited similar side effect profile compared with wild-type BCG, demonstrating the safety of this new BCG strain. rBCG-S.
FimH enhanced BCG-induced Th1 but not Th2 immune response
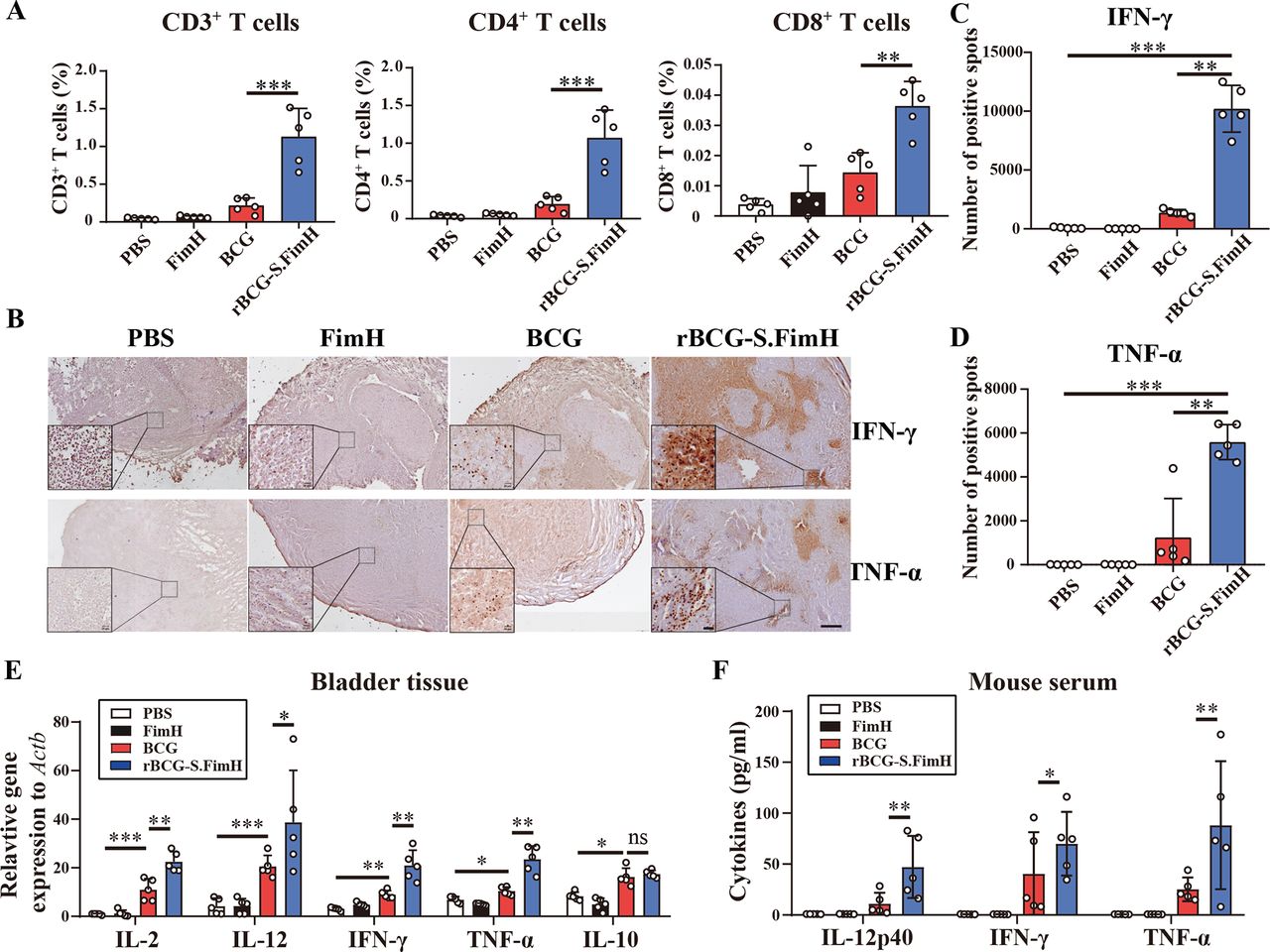
Activation of both CD4+ T helper cells and CD8+ cytotoxic T cells are required for an effective immune response to BCG.38 We sorted out T cells (CD3+) from the above mouse bladder tumors by flow cytometry, and further analyzed CD4 and CD8 expression (online supplemental figure 5A,B). Compared with wild-type BCG, the rBCG-S.FimH group showed more T-cell infiltration in the bladder, and a significant increase in both CD4+ T helper cells and CD8+ cytotoxic T cells (figure 3A), indicating enhanced activation of both Th and Tc response. Although both Th1 and Th2 CD4+ T helper cells are involved in the immune response elicited by BCG instillation, it is well established that the efficacy of BCG in bladder cancer patients is positively correlated with the induction of Th1 response, which is characterized by the production of Th1 cytokines such as IL-2, IL-12 and IFN-γ.39–41 Immunohistochemical analysis of the mouse bladder showed significantly higher levels of IFN-γ (Th1 cytokine) and TNF-α (Tc1 cytokine) in the rBCG-S.FimH group than the BCG group (figure 3B–D). Compared with BCG, treatment with rBCG-S.FimH also caused a significant upregulation in the messenger RNA levels of IL-2, IL-12, IFN-γ and TNF-α in the bladder tissues, but no significant change was observed for Th2 immune response genes IL-10, IL-4 and IL-5 (figure 3E and online supplemental figure 5C). Moreover, rBCG-S.FimH greatly increased the production of IL-12, IFN-γ, and TNF-α in mouse serum (figure 3F). These data indicated that the expression of FimH markedly enhanced BCG-induced Th1 and Tc1 immune response, a favorable biomarker for positive therapeutic outcome.

rBCG-S.FimH enhanced Th1 and Tc1 immune response in mice. C57BL/6 mice were intravesically inoculated with 1×106 MB49-luc cells, and treated with PBS, 2.5 mg/kg of FimH, 2.5×105 CFU/mL BCG or rBCG-S.FimH instillation two times a week for 4 weeks. Mouse bladder were harvested 28 days after treatment. (A) Flow cytometry analysis of total CD3+ T cells and the CD4+ and CD8+ subpopulation in the bladder tissue. (B–D) Immunohistochemical analysis of IFN-γ and TNF-α in bladder tissue. (E) messenger RNA levels of the Th1 and Th2 cytokines in mouse bladder tissue were measured by RT-PCR. (F) Serum levels of the Th1/Tc1 cytokines IL-12p40, IFN-γ and TNF-α in mice treated with PBS, FimH, BCG or rBCG-S.FimH were measured by ELISA. Scale bars: 200 µm, 20 µm for inset. Data are mean± SD *p<0.05, **p<0.01, ***p<0.001. IFN, interferon; IL, interleukin; PBS, phosphate buffered saline; TNF, tumor necrosis factor.
rBCG-S.FimH enhanced activation of DCs via TLR4 in mice
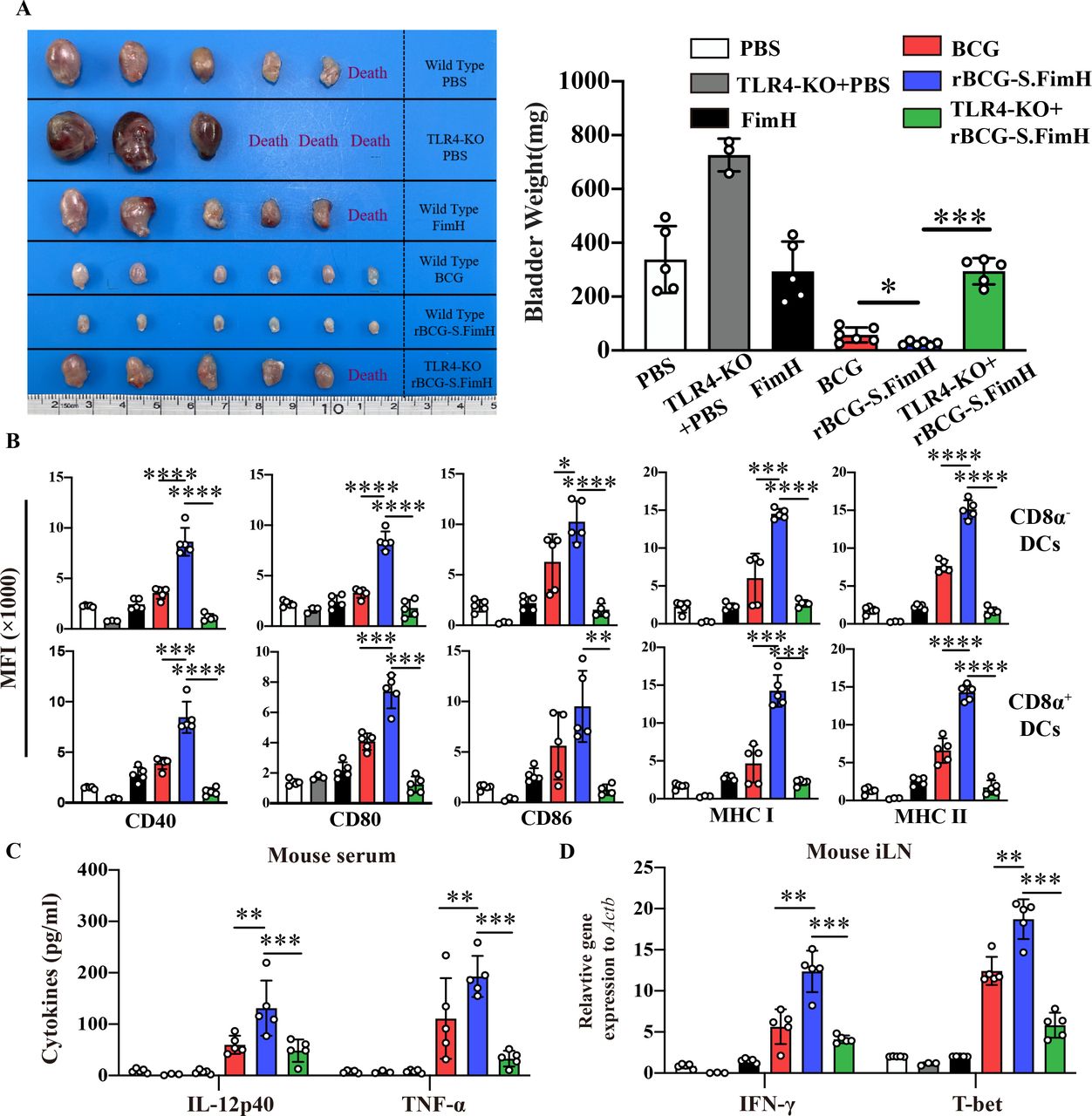
Antigen-presenting DCs plays a central role in the induction of adaptive immune response to BCG.42 43 To test whether rBCG-S.FimH can better promote the activation of DCs, we repeated the BCG instillation experiment in the MB49 orthotopic bladder cancer model in both wild type and TLR4 knockout mice (FimH protein was shown to be an agonist of the TLR4 pathway44). The enhanced antitumor effect of rBCG-S.FimH was significantly inhibited in TLR4 knockout mice (figure 4A and online supplemental figure 6A,B). At the end of the experiment, both the mouse bladders and iLN were harvested, and DCs in the iLNs (CD11c+) were sorted by flow cytometry and further divided into CD8α+ and CD8α− subpopulations (online supplemental figure 6C). Analysis of the surface markers showed that rBCG-S.FimH significantly upregulated the expression levels of CD40/CD80/CD86 costimulatory molecules and MHC class I and II in both CD8α+ and CD8α− DCs (figure 4B). Notably, TLR4 knockout brought down the expression of these costimulatory molecules in the rBCG-S.FimH group to the level of wild-type BCG (figure 4B). In agreement with previous experiments, treatment with rBCG-S.FimH greatly increased the production of Th1 cytokines (IL-12, IFN-γ) and Tc1 cytokines (T-bet, TNF-α) but not Th2 cytokines (IL-10) in both the serum and iLN of mice compared with wild-type BCG (figure 4C,D and online supplemental figure 6D). TLR4 knockout also markedly blocked the induction of these cytokines by rBCG-S.FimH (figure 4C,D and online supplemental figure 6D). To further dissect the molecular mechanisms underlying TLR4-mediated DC activation by rBCG-S.FimH, we investigated the activation status of the signaling pathways downstream of TLR4 in rBCG-S.FimH treated mouse BMDC cells. Compared with wild-type BCG, rBCG-S.FimH induced a more rapid increase in JNK phosphorylation and Iκβ degradation as well as IL-1β, IL-12 and TNF-α expression, which was blocked by the inhibition of MyD88, an effector protein directly downstream of TLR4 (online supplemental figure 7A,B). MyD88 inhibition also significantly blocked rBCG-S.FimH induced upregulation of CD40/CD80/CD86 and MHCI/II expression in BMDC cells (online supplemental figure 7C). Together, these data indicated that rBCG-S.FimH enhanced BCG-induced DC activation via the TLR4 pathway.
rBCG-S.FimH enhanced tumor antigen-specific T cell activation
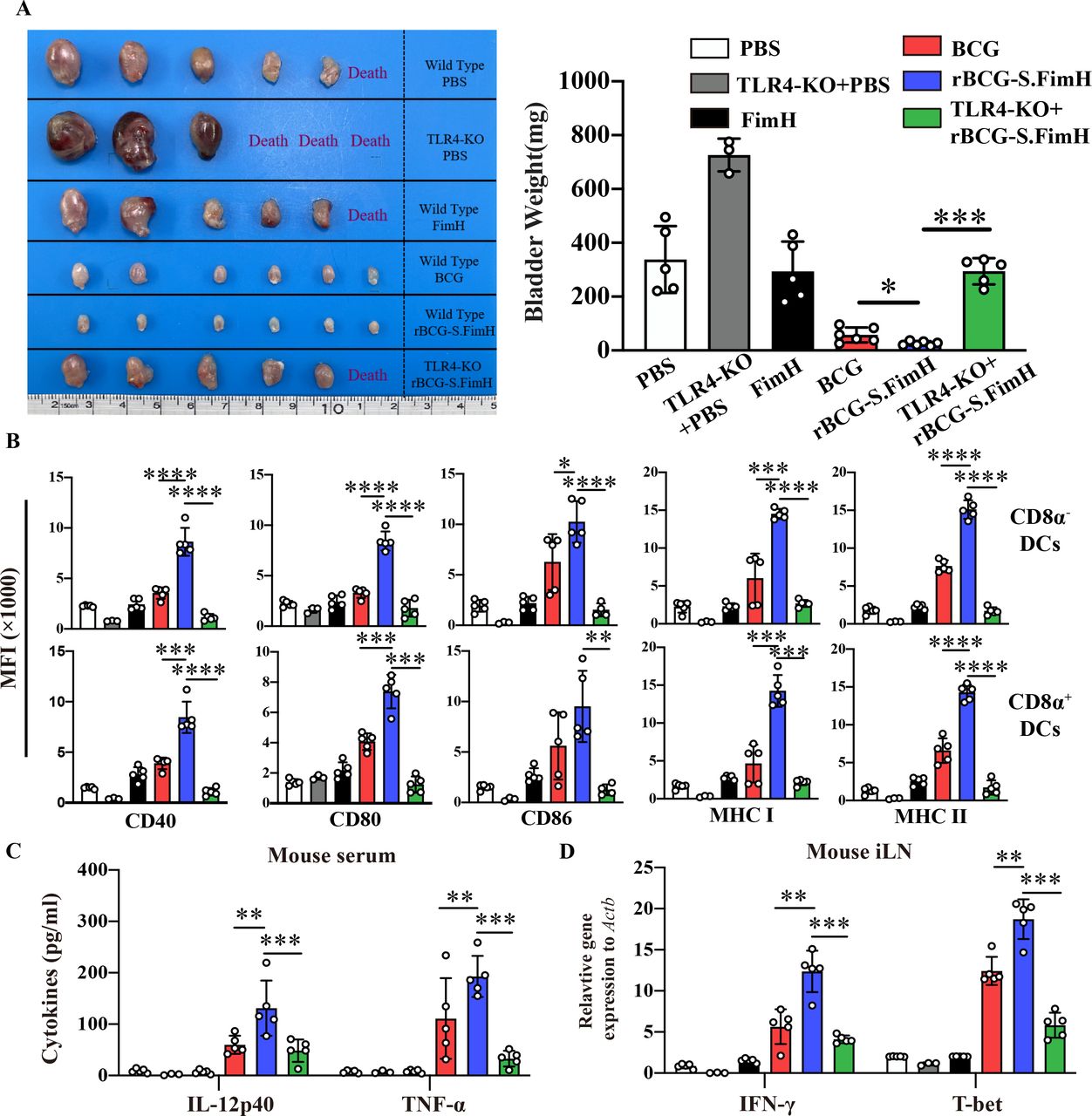
rBCG-S.FimH enhanced the activation of dendritic cells in inguinal lymph nodes in tumor-bearing mice. C57BL/6 mice (n=6 per group) were intravesically inoculated with 1×106 MB49-luc cells, and treated with PBS, 2.5 mg/kg of FimH, 2.5×105 CFU/mL BCG or rBCG-S.FimH instillation two times a week for 4 weeks. Mouse bladder and inguinal lymph nodes (iLN) were harvested 28 days after treatment. (A) Image and weight of mouse bladder at the end of the experiment (mean ± SEM). (B) The expression levels of CD40, CD80, CD86, MHC I and II on CD8α+ and CD8α– dendritic cells in the iLN of mice were determined by flow cytometry (mean ± SD). (C) Serum concentrations of IL-12p40 and TNF-α in mice serum were determined by ELISA (mean±SD). (D) Messenger RNA expression levels of IFN-γ and T-bet in the iLN were determined by RT-PCR (mean±SD). *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IFN, interferon; IL, interleukin; PBS, phosphate buffered saline; TLR4, toll-like receptor 4; TNF, tumor necrosis factor.
Since rBCG-S.FimH could enhance the activation of DC and T cells in mice, we next evaluated whether it could promote tumor antigen-specific immune activation in tumor bearing mice. To do this, we harvested the spleen cells and tumor-draining lymph nodes from the mice of the above experiment. The spleen cells were first labeled with CFSE, and then incubated with the S12 peptide (a specific antigen generated from the MB49 cell line) or the control G12 peptide.45 Four days later, the proliferation and activation of spleen T cells (CD3+) were analyzed by flow cytometry. A significantly higher proliferation of CD4+ and CD8+ T cells were observed in the rBCG-S.FimH group compared with wild-type BCG (figure 5A–C). rBCG-S.FimH also significantly increased the ability of T cells to produce IFN-γ and TNF-α (figure 5D–F). When S12-stimulated spleen T cells were co-cultured with MB49-luc at different effector to target cell ratios, the rBCG-S.FimH group showed higher specific cytotoxicity (figure 5G). Furthermore, we extracted the mononuclear cells from the tumor-draining lymph nodes and stimulated them with tumor-specific peptides S12 or Smcy46 for 48 hours and measured the production of cytokines by the mononuclear cells using the ELISPOT method. The rBCG-S.FimH group also produced much higher levels of IFN-γ in this assay (figure 5H–I). These data indicated that rBCG-S.FimH was more effective in promoting the proliferation and activation of tumor antigen-specific T cells in tumor-bearing mice.

rBCG-S.FimH promotes tumor antigen specific T cell activation. C57BL/6 mice were intravesically inoculated with 1×106 MB49-luc cells, and treated with PBS, 2.5 mg/kg of FimH, 2.5×105 CFU/mL BCG or rBCG-S.FimH instillation two times a week for 4 weeks. Mouse spleens and tumor-draining lymph nodes were harvested 28 days after treatment. (A–C) The proliferation of CD4+ and CD8+ T cells in mouse spleens after stimulation with MB49 tumor specific antigen S12. The division index was calculated using the FlowJo software (A) and the average percentage of proliferating cells were shown in (B and C). (D–F) Flow cytometry analysis of IFN-γ and TNF-α levels in T cells from mice spleen. Percentage of T cells in the spleen producing IFN-γ and TNF-α are shown in (E and F), respectively. (G) S12 or G12-stimulated spleen T cells were co-cultured with MB49-luc at different effector to target cell ratios, cytotoxicity was measured by the Alamar blue assay. (H–I) IFN-γ production on stimulation by the S12 or Smcy tumor-specific peptide in tumor-draining lymph node mononuclear cells were determined by ELISPOT analysis. Shown are representative ELISPOT images (H) and the quantitation of average number of spots (I). Data are mean±SD *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. CFSE, carboxyfluorescein succinimidyl ester; IFN, interferon; PBS, phosphate buffered saline; TLR4, toll-like receptor 4; TNF, tumor necrosis factor.
rBCG-S.FimH enhanced the activation of PBMC
To gain insight to the potential clinical translation of our findings, we tested whether rBCG-S.FimH could enhance the activation of PBMC because PBMCs are the primary source of innate immune response cells in patients with NMIBC receiving BCG instillation. We sorted human PBMCs from four healthy donors by gradient density centrifugation and incubated them with wild-type BCG or rBCG-S.FimH for 48 hours. Compared with wild-type BCG, rBCG-S.FimH significantly promoted the secretion of IL-2 and IFN-γ in human PBMC (figure 6A). Moreover, when these activated PBMCs were co-cultured with the human bladder tumor cell line KU-7, the rBCG-S.FimH treated group also showed a stronger secretion of IL-2 and IFN-γ (figure 6B) and a more potent cell killing effect towards KU-7 bladder cancer cells (figure 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
rBCG-S.FimH enhanced the activation of human peripheral blood mononuclear cells (PBMC). Human PBMC from four healthy volunteers were treated with PBS, 2.5×105 CFU/mL BCG or rBCG-S.FimH for 48 hours. (A) The IL-2 and IFN-γ concentration in the culture medium was measured by ELISA. (B–C) The stimulated PBMC and human bladder cancer cell KU-7-luc were co-cultured for 72 hours, IL-2 and IFN-γ concentration in the culture medium was measured by ELISA (B); cell killing effect on KU-7 was determined by cell counting under a fluorescence microscope (C). Data are mean±SD *p<0.05, **p<0.01, ***p<0.001. IFN, interferon; IL, interleukin; PBS, phosphate buffered saline; PBMC, peripheral blood mononuclear cells.
Discussion
Postoperative intravesical instillation with BCG is the first-line treatment for patients with high-risk NMIBC to prevent tumor recurrence and progression. Patients who are resistant to BCG instillation or patients who relapse after initial response often have to receive radical cystectomy or systematic chemotherapy resulting in poor quality of life. Therefore, many studies have attempted to modify BCG to enhance its antitumor efficacy in the past 40 years, yet few showed better therapeutic effect than wild-type BCG in the clinic. Our strategy is based on the theory that the adhesion ability of BCG to the bladder urothelium is critical for its overall antitumor effect, which is supported by the clinical observation that low attachment accounts for treatment failure in many patients.11 25 26 FimH is a naturally existing adhesion protein that is used by uropathogenic E.coli to attach to the urinary tract.27 FimH specifically binds to mannose residues on the surface of urothelial cells, a feature that has recently been exploited to treat uropathogenic Escherichia coli (UPEC) infections with FimH antagonists.14 Inspired by this feature of FimH, we successfully overexpressed this E.coli protein on the surface of BCG and markedly increased the adhesion ability of BCG to the urothelium. Intravesical instillation of rBCG-S.FimH rendered faster, stronger adhesion and internalization, and longer retention time in the bladder urothelium, all of which contribute to the overall enhanced therapeutic effect in vivo.
It is well established that the induction of Th1 cell-biased adaptive immune response is associated with better BCG efficacy in patients.14 Therefore, it is not surprizing that the most common strategy of modifying BCG is to boost the Th1 response either by direct combination with Th1 cytokines or recombinantly overexpressing them in BCG.15–20 However, none of these approaches worked in the clinic, suggesting that only boosting one cytokine might not be sufficient. rBCG-S.FimH, on the other hand, was able to elicit a much more prominent Th1 response as evidenced by the significant induction of a wider spectrum of Th1 cytokines. Moreover, compared with rBCG-SIPT which induced an increase in both Th1 (IL-2 and IFN-γ) and Th2 (IL-10) responses,21 22 rBCG-S.FimH only induced increased Th1 cytokines but not Th2 cytokines in the serum, bladder or lymph nodes. Therefore, rBCG-S.FimH is an efficient and powerful way to induce local and systematic Th1 effect.
Recently and during the preparation of this manuscript, Zhang et al reported that FimH protein alone could function as an adjuvant for tumor immune therapy in a TLR4-dependent manner.44 Systematic administration of FimH protein could efficiently promote DC activation by upregulating the expression of co-stimulatory molecules and MHC-II, and provoke the production of IL-12 via TLR4-dependent signaling pathway.44 47 48 Lung mucosal exposure to FimH was also able to accelerate Th1-type immunity by enhancing DC antigen presentation and Th1-cell priming.49 BCG itself is a weak TLR4 agonist.50 51 TLR4 is highly expressed in DCs, macrophages, lymphocytes, but lowly expressed in epithelial and endothelial cells as well as cancer cells.48 Activation of DCs by TLR4 agonist leads to specific activation of Th1 cytokines.52 53 Consistent with these researches, rBCG-S.FimH improved the antigen presenting ability of DCs via the TLR4-MyD88-pJNK-NF-κb axis. The adjuvant effect of rBCG-S.FimH was further confirmed by the tumor antigen-specific immune activation both in the MB49 bladder tumor orthotopic mouse model and in the KU7 human bladder cancer cell line treated with human PBMC. Therefore, we reasoned that BCG itself could be considered an immune adjuvant while recombinant FimH further boosted its adjuvant function.
One caveat of BCG therapy is that it cannot distinguish between bladder epithelial cells and tumor cells, although rBCG-S.FimH provides no advantage in selectivity, our results demonstrated that rBCG-S.FimH elicited a much stronger antitumor effect than wild-type BCG but a similar safety profile in mouse orthotopic models of bladder cancer. rBCG-S.FimH did this by both enhancing adhesion to urothelial cells and boosting Th1 immune response. This unique approach provides compelling preclinical data that merits further translational development.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
The collection of human blood samples was approved by the Internal Review and Ethics Boards at the First Affiliated Hospital of Nanjing Medical University (2021-SR-083). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Dr Yi Luo from the University of Iowa for providing BCG strains, MB49-luc cells and technical suggestions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YZ, FH and QC contributed equally.
Contributors Conceptualization: QL, PL, CY; Methodology: YZ, FH, QC, RJ, QH, ZAW; Investigation: YZ, FH, QC, RJ, QH ; Visualization: YZ, QC, RJ; Funding acquisition: DT, QL, PL, CY; Project administration: PL, CY; Supervision: QL, PL, CY; Writing—original draft: YZ, FH; Writing—review and editing: ZAW, DT, QL, PL, CY. Guarantor: CY.
Funding American Cancer Society grant ACS 134386-RSG-20-038-01-DDC (ZAW); National Institutes of Health grant R01CA075115 (DT); Hairun Biotechnology, Xuzhou, China (CY).
Competing interests CY receives a research grant from Hairun Biotechnology, Xuzhou, Jiangsu Province, China.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.