Article Text

Abstract
Background Several agents for oncolytic immunotherapy have been approved for clinical use, but monotherapy is modest for most oncolytic agents. The combination of several therapeutic strategies through recombinant and nanotechnology to engineer multifunctional oncolytic viruses for oncolytic immunotherapy is a promising strategy.
Methods An endothelium-targeting iRGD-liposome encapsulating a recombinant Newcastle disease virus (NDV), which expresses the dendritic cell (DC) chemokine MIP-3α (iNDV3α-LP), and three control liposomes were constructed. MIP-3α, HMGB1, IgG, and ATP were detected by western blotting or ELISA. The chemotaxis of DCs was examined by Transwell chambers. The phenotypes of the immune cells were analyzed by flow cytometry. The antitumor efficiency was investigated in B16 and 4T1 tumor-bearing mice. Immunofluorescence and immunohistochemistry were used to observe the localization of liposomes, molecular expression and angiogenesis. Synergistic index was calculated using the data of tumor volume, tumor angiogenesis and tumor-infiltrating lymphocytes.
Results Compared with NDV-LP, treatment with iNDV3α-LP and NDV3α-LP induced stronger virus replication and cell lysis in B16 and 4T1 tumor cells and human umbilical vein endothelial cells (HUVECs) with the best response observed following iNDV3α-LP treatment. B16 and 4T1 cells treated with iNDV3α-LP produced more damage-associated molecular pattern molecules, including secreted HMGB1, ATP, and calreticulin. Moreover, iNDV3α-LP specifically bound to αvβ3-expressing 4T1 cells and HUVECs and to tumor neovasculature. Tumor growth was significantly suppressed, and survival was longer in iNDV3α-LP-treated B16-bearing and 4T1-bearing mice. A mechanism study showed that iNDV3α-LP treatment initiated the strongest tumor-specific cellular and humoral immune response. Moreover, iNDV3α-LP treatment could significantly suppress tumor angiogenesis and reverse the tumor immune suppressive microenvironment in both B16-bearing and 4T1-bearing mice.
Conclusions In this study, iNDV3α-LP had several functions, such as tumor and vessel lysis, MIP-3α immunotherapy, and binding to αvβ3-expressing tumor and its neovasculature. iNDV3α-LP treatment significantly suppressed tumor angiogenesis and reversed the tumor immunosuppressive microenvironment. These findings offer a strong rationale for further clinical investigation into a combination strategy for oncolytic immunotherapy, such as the formulation iNDV3α-LP in this study.
- oncolytic virotherapy
- immunotherapy
- tumor microenvironment
- immunogenicity, vaccine
- combined modality therapy
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- oncolytic virotherapy
- immunotherapy
- tumor microenvironment
- immunogenicity, vaccine
- combined modality therapy
Introduction
Biotherapy with oncolytic viruses (OVs) is a promising method for treating cancer.1 2 OVs can infect and replicate in tumor cells and continue to infect others, leading to direct lysis of tumor cells.3–5 During this process, the infected tumor cells release many tumor-associated antigens, pathogen-associated molecular patterns, and damage-associated molecular patterns (DAMPS). These molecules can further stimulate adaptive immune responses against tumors, such as tumor immunogenic cell death (ICD), an immune process that can shift tumors from cold (immune desert) to hot (inflamed) tumors.6–8 However, OV monotherapy is unable to completely cure tumors, especially those that are deep and/or metastasized. Therefore, OVs require a combination of additional therapies to enhance their antitumor effects. This combination strategy has the added advantage of being able to deliver therapeutic transgenes to enhance antitumor activity through modulation of the host immune response. For example, oncolytic herpes virus (talimogene laherparepvec (T-VEC)) expressing granulocyte-macrophage colony-stimulating factor (GM-CSF) has been approved for the clinical treatment of non-resectable melanoma by the US Food and Drug Administration and the European Union.9–13 Intratumor administration of T-VEC led to a reduction in tumor size, but the clearance of the tumor remained incomplete. Case studies show that combining T-VEC with immune-checkpoint inhibitory monoclonal antibodies improves the antitumor effects.9–11 These clinical studies indicate that the combination of OVs with another immunotherapy strategy has additive or synergistic antitumor activities.
Angiogenesis, a process by which new blood vessels are formed or developed from existing blood vessels, plays an important role in tumor growth and metastasis.14 Antiangiogenesis therapies have been deeply investigated in various diseases, such as cancer and rheumatoid arthritis, for several decades, and they remain a research hotspot in this field.14–16 Various molecules, especially those that are overexpressed or specifically expressed on tumor neo-vasculature, have become the focus of therapeutic targets. Integrin alpha (v)beta (3) (αvβ3) receptor is an integrin family member that is overexpressed on both tumor neovasculature and some tumor cells.17 Several studies have investigated the use of an αvβ3 receptor conjugated to various nanoformulations, such as liposomes, for targeted therapy of cancers in murine models.18–20
iRGD is a cyclic nanopeptide with an amino acid sequence of CRGDKGPDC. iRGD has high tumor-targeting efficiency and tumor-penetrating abilities if it is chemically conjugated to nanoformulations or to other therapeutic agents.21–23 Additionally, iRGD has a specific capability of binding to the integrin αvβ3 receptor, which is highly expressed on tumor neovasculature but rarely on normal blood vessels.18–20 In a previous study, we engineered a recombinant Newcastle disease virus (NDV) that effectively expressed bioactive macrophage inflammatory protein-3α (MIP-3α), a specific dendritic cell (DC) chemokine, and promoted tumor-specific antitumor immunity and reversal of the suppressive tumor microenvironment; this NDV showed optimal suppression of tumor growth in several murine models.24 Thus, in the current study, we encapsulated recombinant MIP-3α NDV in an iRGD-liposome (iNDV3α-LP) to investigate its antitumor activities. Interestingly, we found that iNDV3α-LP treatment induced significant lysis of both tumor and endothelial cells and significantly promoted antitumor immunity. iNDV3α-LP treatment also significantly inhibited tumor neovasculature (the generation of new tumor vessels) and reversed the tumor suppressive microenvironment, resulting in a perfect inhibition of tumor growth in murine models.
Materials and methods
Virus and detection of its replication in vitro
The lentogenic NDV LaSota strain and its recombinant counterpart expressing MIP-3α (NDV3α) previously made by our group were used in this study.24 The NDV and NDV3α were cultured in embryonated chicken eggs. After serial dilution, the virus titers in A549 adenocarcinomic human alveolar basal epithelial cells were detected by immunofluorescence in a microplate system (ELX808IU, Bio-Tek, Winooski, Vermont, USA). Viral replication kinetics were detected in the following steps: 5×104 B16 murine melanoma, 4T1 murine breast cancer cells or human umbilical vein endothelial cells (HUVECs) were infected with the NDV virus (1×103 pfu) in six-well plates at 0.05, 0.5, 1.0, and 2.0 multiplicity of infection (MOI) in 100 µL. The culture medium was substituted with 1 mL of fresh medium, and the cells were continually cultured at 37°C in 1 mL Dulbecco’s modified Eagle’s medium or Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 250 ng/mL tosyl phenylalanyl chloromethyl ketone trypsin. Next, the virus replication at 24, 48, 72, and 96 hours was detected as previously reported.24 25
Preparation and characterization of iNDV3α-LP and the control liposomes
The iRGD-liposome loaded with NDV expressing MIP-3α (iNDV3α-LP), liposome loaded with NDV expressing MIP-3α (NDV3α-LP), liposome loaded with single NDV (NDV-LP), and empty liposome (LP) were prepared as previously reported.20 26 DSPE-PEG2000-iRGD was first prepared through a reaction between iRGD and DSPE-PEG2000-maleimide. In brief, iRGD and DSPE-PEG2000-maleimide were mixed at a 1.5:1 molar ratio in ultrapure water, followed by gentle stirring in a 4°C environment for 24 hours under nitrogen gas. To remove the unreacted impurities, the mixture was dialyzed in a dialysis bag (MWCO: 3500) for 48 hours in ultrapure water. The final solution in the dialysis bag was lyophilized for 48 hours and stored at −20°C until required. To prepare the iNDV3α-LP, DSPE-PEG2000-iRGD, DSPE-PEG2000, cholesterol, and dipalmitoylphosphatidylcholine were mixed in chloroformin at 1:4:40:55 molecular ratios and then volatilized to remove organic solvents under nitrogen flow until a lipid film was generated. The lipid film was further hydrated at 37°C with 1×108 pfu/mL NDV3α in 0.1 M phosphate buffered saline (PBS; pH 7.4) and vortexed for 5 min at room temperature. The resultant suspension was sequentially passed through a polycarbonate membrane of 200 nm pore size (Millipore, Bedford, Massachusetts, USA), and a Cellufine sulfate column was used to separate out the unconjugated NDVs. The control liposomes (LP, NDV-LP, and NDV3α-LP) were prepared using the same method without the addition of corresponding ingredients. The size, average grain diameter, and zeta potential of iNDV3α-LP and the control liposomes were tested using a laser-based analyzer (Zeta sizer Nano ZS90, Malvern, UK). The morphological characteristics of the liposomes were detected by transmission electron microscopy (TEM). The encapsulation efficiency (EE) of NDV or NDV3α was calculated using a previously reported formula.18 20
Cell lysis and cytotoxicity assays
The lactate dehydrogenase activity in the supernatant of B16, 4T1, and HUVECs was determined to evaluate the capability of the NDV or splenic monocytes to lyse tumor cells or HUVECs using a CytoTox 96 kit (Promega, Madison, Wisconsin, USA) as described previously.24 25 27 In brief, B16 cells, 4T1 cells, or HUVECs were infected with NDV virus (1×103 pfu) in six-well plates at 1 MOI in 100 µL. After infection, 50 µL aliquots were collected at 24, 48, 72, and 96 hours and transferred to another 96-well plate. After incubation for 60 min in the dark at room temperature, stop solution (50 µL) was added to each well, and the optical density (OD) values were detected at 492 nm by an ELX808IU microplate system (Bio-Tek). To detect the cytotoxic T lymphocytes (CTLs), tumor cells (target) in logarithmic growth were mixed with splenic lymphocytes (effectors) in a series of E (effectors):T (target) ratios (5–40:1) and then plated at a total volume of 100 µL in 96-well plates. The plates were subsequently incubated in a 5% CO2 atmosphere at 37°C for 8 hours. Following incubation, the 50 µL aliquots were transferred to another 96-well plate, incubated and detected for OD values. The target cell lysis was expressed as a percentage at each E:T ratio as described previously.27
Western blotting analysis
MIP-3α, high mobility group box 1 protein (HMGB1), integrin αvβ3 receptor, and sera IgG were analyzed by western blotting as described previously.24 The cells were infected with or without iNDV3α-LP, NDV3α-LP, NDV-LP, or LP at various MOIs for 24 hours, before collecting the cultured supernatants or cell lysates. Additionally, the sera from mice treated with different formulations were also collected. The cell lysates, sera, or supernatants were subjected to electrophoresis on 8%–10% SDS-PAGE gels. The proteins on the gels were transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Next, the membranes were blocked with 10% non-fat milk at 4°C for 1 hour, washed with PBS three times, and then incubated with monoclonal antibodies (Abcam) against MIP-3α, HMGB1, αvβ3 receptor, or IgG at a 1:200 dilution. Eventually, the PVDF membranes were stained with enhanced chemiluminescence (ECL) substrates for 30 min, and the resultant pictures were obtained using an ECL system (Amersham Biosciences, UK).
Isolation of single cells
Murine bone marrow derived dendritic cells (BMDCs), spleen lymphocytes and tumor-infiltrating lymphocytes (TILs) were isolated for analysis. BMDCs were isolated from 8 to 10 week-old female BALB/c or C57BL/6 mice as previously described.28 Following depletion of the erythrocytes, the isolated bone marrow cells (1×105/well) were cultured in RPMI-1640 (HyClone, Thermo Scientific, Waltham, Massachusetts, USA) supplemented with GM-CSF (100 U/mL) and IL-4 (20 ng/mL). The non-adherent cells were depleted on days 2 and 4, while the adherent cells were transferred to another six-well plate on day 6. The cells were incubated for a further 6 days, and the resultant non-adherent cells with typical morphological characteristics of BMDCs were collected for subsequent experiments. To isolate the spleen lymphocytes, spleens were minced into small pieces with a scissor and then pressed through 100 µm cell strainers (BD Biosciences, Franklin Lakes, New Jersey, USA). The erythrocytes in the cell suspensions were further lysed with ACK buffer and then filtered through a cell strainer (100 µm). The resultant cell suspensions were used for subsequent experiments. To isolate single tumor cells, B16 and 4T1 tumor tissues were minced with a TissueLyser II instrument (Qiagen, Germany) and digested in culture medium with 0.8 mg/mL Dispase II, 0.2 mg/mL collagenase P, and 0.1 mg/mL DNase I (all from BD Biosciences, San Jose, California, USA) for 30 min at 37°C. The isolated cells from B16 and 4T1 tumors were filtered through a cell strainer (70 µm) and purified on a Ficoll gradient as previously described.24
In vitro DC chemotaxis assay
The in vitro DC chemotaxis assay was performed in a 24-well Transwell chamber kit (Corning Costar, New York, USA) as previously reported.24 27 In brief, 5×104 DCs dissolved in 200 µL bovine serum albumin medium (5%) were plated in the upper chamber, while indicated percentages of supernatants from iNDV3α-LP-treated, NDV3α-LP-treated, NDV-LP-treated, or LP-treated tumor or HUVECs were added to a volume of 750 µL in the bottom chamber. The Transwell chambers were placed in an incubator supplemented with 5% CO2 at 37°C for 3 hours. Subsequently, the filter between the upper and bottom chamber was removed and washed with Hanks’ Balanced Salt Solution, fixed, and stained on a slide according to the Transwell chamber kit protocol. The number of chemotactic DCs on the bottom chamber was counted using a microscope (80i, NIKON, Japan) at 200× magnification.
Flow cytometry (FCM) analysis
FCM analysis was performed to analyze the expression of marker molecules, including CD11c, DEC205, CD4, CD8, CD45, CD25, Foxp3, and Gr-1, on the cell membranes, as well as intracellular cytokines, including IFN-γ, TNF-α, and IL-2.24 Cells stained with fluorescent dyes, such as CFSE and DiI, were also analyzed by FCM. The cells isolated from various tissues were incubated with the corresponding fluorescent dyes or monoclonal antibodies conjugated to FITC, PE, PerCP-Cy5.5, or APC for 30 min at 4°C in 1:100–150 dilutions. The following monoclonal antibodies were purchased from eBioscience (San Diego, California, USA) or BD Biosciences: anti-DEC205, anti-CD11c, anti-CD45, anticalreticulin (anti-CRT), anti-CD8, anti-IFN-γ, anti-TNF-α, anti-CD4, anti-CD25, anti-FOXP3, anti-Gr-1, anti-CD206, and anti-F4/80. Before intracellular cytokine staining, 2×106 splenocytes were stimulated with tumor lysates (5 µg/mL) in culture medium with 10% fetal calf serum and 2 µg/mL brefeldin A (BD Bioscience) for 6 hours at 37°C. The intracellular cytokines in splenocytes or TILs were stained using the Cytofix/Cytoperm kit (BD Bioscience) as per the manufacturer’s protocol. The stained cells were detected by FCM (CyFlow Cube 6; Sysmex, Japan), and the resultant data were analyzed to capture the FCM images with FlowJo software version 10 (Tree Star, Ashland, Oregon, USA).
Fluorescent observation of cell morphology
Fluorescent staining with dyes or monoclonal antibodies was performed to observe whether CRT was present on the cell surface and whether the fluorescence-labeled liposome iNDV3α-LP targets the αvβ3 receptor in vitro and the blood vessels in vivo, as previously reported.15 16 28 For in vitro observation, B16, 4T1, or HUVECs (1×105/well) were incubated in six-well plates and infected with NDV-encapsulated or NDV3α-encapsulated liposome formulations with equivalent 1 MOI NDV or treated with empty LP. The cells were first incubated at 37°C in a 5% CO2 atmosphere for 10 min and then washed thrice with PBS. After being fixed in 4% paraformaldehyde at room temperature for 30 min, the cells were stained with anti-CRT or anti-αvβ3 antibodies (BD Bioscience) in 1:250 dilutions at 4°C overnight and then washed three times with cold PBS. Following washing, the cells were stained with a FITC-labeled secondary antibody at 1:300 dilutions for 60 min at room temperature. Additionally, the cell membrane and nuclei were stained with DiI (MedChemExpress, USA) and DAPI (Beyotime Biotechnology, China) for 15 min. Images were captured by a confocal microscope (FV1000, Olympus, Japan). To observe whether iNDV3α-LP targets blood vessels in vivo, B16- or 4T1-bearing mice were randomly grouped and intravenously injected with DiI-conjugated iNDV3α-LP, NDV3α-LP, NDV-LP, or LP. Thirty minutes after injection, 10 mg/mL fluorescein isothiocyanate conjugate (FITC)-Dextran (Sigma, USA) was intravenously injected into the mice by tail vein. Ninety minutes after FITC-dextran injection, the mice were sacrificed, and the tumors were removed into cryo-embedding compound OCT (Tissue-Tek, Sacura) for frozen sectioning, and the images were captured by a confocal fluorescence microscope (FV1000, Olympus, Japan).
Mice tumor models and liposome treatment
The syngeneic C57BL/6 or BALB/c mice (6–8 weeks old) were chosen for the establishment of B16 or 4T1 tumor models. In brief, B16 or 4T1 cells (5×105 in 100 µL PBS) were subcutaneously implanted in the right flank. The mice were randomly divided into four groups (n=10 per group) based on receiving tail vein injection of iNDV3α-LP, NDV3α-LP, NDV-LP ((NDV or NDV3α 2×107 pfu), or LP in 100 µL PBS. The injections were performed on day 3 after tumor cells were implanted as previously reported.25 The treatment was repeated once a week for a total of four treatments. The tumor volumes and images were taken in 3-day intervals by a handheld tumor-detecting instrument (TM900, Peira Scientific Instruments, Belgium). At the same time, the mouse survival was recorded.
Calculation of synergistic indexes (SIs)
In this study, we calculated an SI using the tumor volume, tumor angiogenesis, and TILs to evaluate whether the iNDV3α-LP treatment induced synergistic antitumor activities through synergistic inhibition of neovasculature formation and reversal of the tumor suppressive microenvironment as described previously.15 The mean values of these parameters were used to calculate the SI for each group. In brief, an observed relative ratio (ORR) of the NDV-LP, NDV3α-LP, and iNDV3α-LP groups was obtained by dividing the mean of each group by that of the LP group. The expected relative ratio (ERR) of the iNDV3α-LP group was obtained by multiplying the ORR of the NDV-LP group and that of the NDV3α-LP group. The SI of iNDV3α-LP treatment was calculated using the formula: SI=ERR/ORR (ORR <1) or ORR/ERR (ORR >1), where SI>1 indicates a synergistic effect.
ELISA and enzyme-linked immunospot (ELISPOT) assay
The concentrations of HMGB1 and ATP in the supernatants of tumor cells treated with iNDV3α-LP, control, and IgG antibody and its subtypes in pooled sera from the liposome-treated mice were quantified by commercial ELISA kits (Wuhan Boster Biological Technology, China) according to the manufacturer’s protocol. Additionally, ELISPOT was performed to enumerate the number of splenocytes that produced IgG antibodies specific to B16 or 4T1 tumors as previously reported.27 In brief, tumor lysate (30 µg/mL) was first added to PVDF-bottomed 96-well plates (Millipore, Billerica, Massachusetts, USA). Next, the various splenocytes isolated from the spleen of the mice treated with iNDV3α-LP, NDV3α-LP, NDV-LP, or LP were incubated in the plates at 37°C in a 5% CO2 atmosphere for 6 hours. The plate was then washed thrice with PBS and stained with ECL substrates using the protocol outlined for western blotting. The number of spots on the PVDF membrane was considered to indicate the number of IgG-secreted cells.
Immunohistochemistry
Immunohistochemistry was performed to detect the microvessels in B16 and 4T1 tumor sections as described previously.24 In brief, frozen sections (5 µm) from tumors treated with iNDV3α-LP, NDV3α-LP, NDV-LP, or LP were placed on slides, fixed with acetone for 20 min, and air-dried for 60 min. Next, the slides were washed three times with PBS and then incubated in PBS containing 5% normal goat serum for 90 min at room temperature. The slides were further incubated overnight at 4°C in PBS with rat antimouse antibodies against CD41 (BD Bioscience) at a 1:50 dilution. Subsequently, the sections were further stained with streptavidin biotin-labeled reagents (Dako LSAB kit, peroxidase; Dako), and the images were obtained under a microscope (80i, NIKON, Japan).
Alginate encapsulation assay
An alginate encapsulated tumor cell assay to quantify tumor microvessels was performed as described previously.15 16 In brief, B16 or 4T1 cells (1×106) in a solution of 1.5% sodium alginate (Sigma, St. Louis) were added dropwise to a swirling calcium chloride solution (250 mM). During this process, alginate beads containing approximately 1×104 tumor cells in each bead were formed. Four beads for each mouse were implanted subcutaneously into an incision in the dorsal side. On day 14, the mice were injected with 10 mg/kg FITC–dextran in 100 µL PBS via the tail vein (Sigma). The alginate beads were surgically removed and squashed in a TissueLyser II (Qiagen). The fluorescence intensity of the beads was quantified against a standard curve of FITC-dextran by a microplate reader (ELX808IU, Bio-Tek).
Statistical analysis
The data are presented as the mean±SD, and all data were analyzed by GraphPad-Prism Software (San Diego, California USA). One-way or two-way analysis of variance (ANOVA), followed by a Tukey’s multiple comparisons test, was performed to compare the differences among the experimental groups. Animal survival was graphed as Kaplan-Meier survival curves and analyzed with the log-rank (Mantel-Cox) test.
Results
Characterization of engineered liposomes
The procedure for the preparation of iRGD-liposome loaded with NDV expressing MIP-3α (iNDV3α-LP) is illustrated in figure 1A. Other liposomes used for controls, such as empty LP, NDV-LP or NDV3α-LP, were also prepared by following a similar procedure but without corresponding ingredients. Successful preparation of the four liposomes was confirmed by their typical morphology visualized by TEM (figure 1B). The diameter of iNDV3α-LP was slightly larger than that of LP and NDV-LP but almost equal to that of NDV3α-LP (figure 1C,D). The diameters, zeta potential, and polydispersity are shown in figure 1D, showing no significant difference among the four liposomes. Additionally, the EE of NDV and NDV3α was similar (approximately 70%) in NDV-LP, NDV3α-LP, and iNDV3α-LP (figure 1D). These data indicate that LP, NDV-LP, NDV3α-LP, and iNDV3α-LP are suitable for use in the study.

Construction and characteristic evaluation of the liposome formulation NDV-MIP3α. (A) Schematic presentation of the construction of iNDV3α-LP and three control liposomes NDV3α-LP, NDV-LP and Lp. (B) TEM images of liposomes LP, NDV-LP, NDV3α-LP, and iNDV3α-LP. (C) Size distribution of LP, NDV-LP, NDV3α-LP, and iNDV3α-LP. (D) Basic characteristics of LP, NDV-LP, NDV3α-LP, and iNDV3α-LP. LP, empty liposome; MIP-3α, macrophage inflammatory protein-3α; NDV; Newcastle disease virus; NDV-LP, liposome encapsulated with wild-type LP NDV; NDV3α-LP, liposome encapsulated with the recombinant MIP-3α NDV; iNDV3α-LP, iRGD-directed liposome encapsulated with the recombinant MIP-3α NDV.
Recombinant MIP-3α NDVs in iRGD-liposomes increase the oncolytic activities of ανβ3-expressing cells and express similar bioactivities of MIP-3α
Previous studies have shown that 4T1 breast cancer cells and vascular endothelial cells express ανβ320 and that NDV proliferation is the most efficient in B16 tumor cells.24 25 Thus, we first observed whether the NDVs encapsulated in iNDV3α-LP, NDV3α-LP, and NDV-LP retained their oncolytic ability and replicative capacity in 4T1 and B16 tumor cells and HUVECs. The cells treated with equivalent titers of NDV or NDV3α in iNDV3α-LP, NDV3α-LP, and NDV-LP showed similar levels of viral replication (figure 2A) and cell lysis (figure 2B) as the wild-type NDV in B16 cells. However, the level of viral replication and cell lysis in both 4T1 and HUVECs was significantly increased, especially when the MOI was low (figure 2C,D and online supplemental figure S1A,B). These data suggest that recombinant NDV3α encapsulated in iNDV3α-LP or NDV3α-LP had similar bioactivities in B16 cells but induced more cell lysis in CT41 cells and HUVEC compared with wild-type NDV encapsulated in NDV-LP. In agreement with our previous study,24 1 MOI was a moderate titer of NDV or NDV3α for tumor cell lysis (figure 2A–D and online supplemental figure S1A,B). Considering that robust cell lysis induced by NDVs was not necessary for the induction of antitumor immunity, we chose 1 MOI of NDV3α and its wild-type counterpart (NDV) to perform the subsequent experiments in order to simplify our study and save resources.
Supplemental material

Evaluation on replication and tumor-lysis capabilities of the NDVs and MIP-3α activity in the liposomes. (A and C) vVrus replication of 1 MOI NDV or NDV3α at the indicated time points in B16 (A) and 4T1 (B) cells. (B and D) Lysis of B16 (B) or 4T1 (D) cells by indicated formulations at the indicated MOI and time points. (E) Expression of MIP-3α in B16 and 4T1 cells treated with indicated formulations. (F) In vitro chemotaxis of DCs induced by the supernatants from B16 cells infected with 1 MOI NDV or NDV3α in the indicated formulations. (G) In vivo chemotaxis for DCs in mouse abdomen injected with supernatants from B16 or 4 T1 cells treated with the indicated formulations. Data are plotted as mean±SD; two-way ANOVA with Tukey multiple comparisons: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; DCs dendritic cells; MOI, multiplicity of infection; NDV3α, recombinant NDV encoding MIP-3α.
Western blot analysis verified the expression of MIP-3α in B16, 4T1, and HUVECs infected with iNDV3α-LP and NDV3α-LP but not in the cells infected with NDV-LP (figure 2E). The bioactive MIP-3α in the culture supernatants was confirmed by a DC chemotaxis assay in Transwell chambers and directly in an in vivo mouse model.24 27 28 As predicted, the supernatants of B16, 4T1, and HUVECs infected with iNDV3α-LP and NDV3α-LP showed significant chemotactic effects for DCs compared with the NDV-LP-infected cells (figure 2F and online supplemental figure S1C,D). In the in vivo mouse model, the supernatants from B16, 4T1, and HUVECs infected with iNDV3α-LP, NDV3α-LP, or NDV-LP were injected into the abdominal cavities. On day 3 after supernatant injection, cells in the abdominal cavities were collected for FCM analysis. Compared with the NDV-LP group, more CD11c and DEC205 double-positive DCs were found in the iNDV3α-LP and NDV3α-LP groups (figure 2G and online supplemental figure S1E). Taken together, these data indicate that the MIP-3α expressed in iNDV3α-LP and NDV3α-LP is functionally bioactive.
iNDV3α-LP increased the production of DAMPs in tumor cells and HUVECs
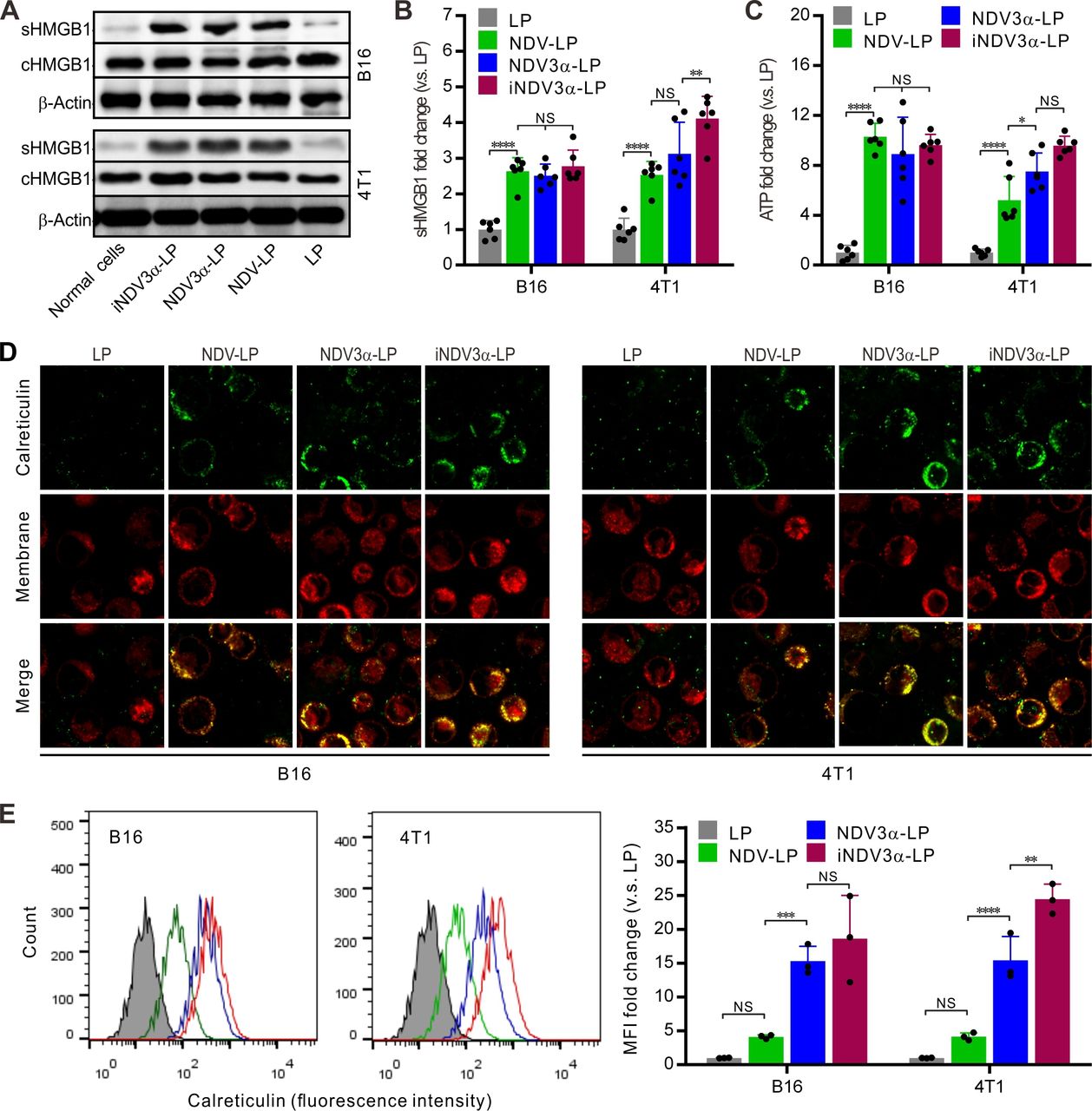
The production of DAMPs induced by OVs is considered a potential strategy for tumor immunotherapy.29 30 Thus, DAMP markers were detected in B16 and 4T1 tumor cells infected with iNDV3α-LP, NDV3α-LP, or NDV-LP at 1 MOI. Compared with cells infected with NDV-LP, the expression (c) and secretion (s) of HMGB1 were at least equivalent or even significantly higher in both B16 and 4T1 cells infected with iNDV3α-LP or NDV3α-LP (figure 3A). Additionally, sHMGB1 in the supernatant was detected by ELISA and demonstrated similar results as western blot analysis in B16 and 4T1 cells (figure 3B). Consistent with the HMGB1 expression, the release of ATP in the supernatants from the cells infected with iNDV3α-LP or NDV3α-LP was the same, or even increased, compared with that from the cells infected with NDV-LP (figure 3C). Additionally, CRT on the outer leaflet of the cell membrane (ecto-CRT) was directly observed by immunofluorescence staining and quantitatively analyzed by FCM at 24 hours after the B16 and 4T1 cells were infected with iNDV3α-LP, NDV3α-LP, NDV-LP, or LP. Compared with the LP-treated cells, the results showed significant translocation of CRT onto the cell surface in both B16 and 4T1 cells treated with iNDV3α-LP, NDV3α-LP, NDV-LP (figure 3D). Interestingly, the FCM results showed that MIP3α-expressing NDV liposomes promoted an increase in CRT translocation, which was more significant in 4T1 cells (figure 3E). Taken together, these data demonstrate that both iNDV3α-LP and NDV3α-LP were superior in their ability to induce DAMP release in B16 and 4T1 cells, and the phenomenon was more significant in iNDV3α-LP-infected cells.
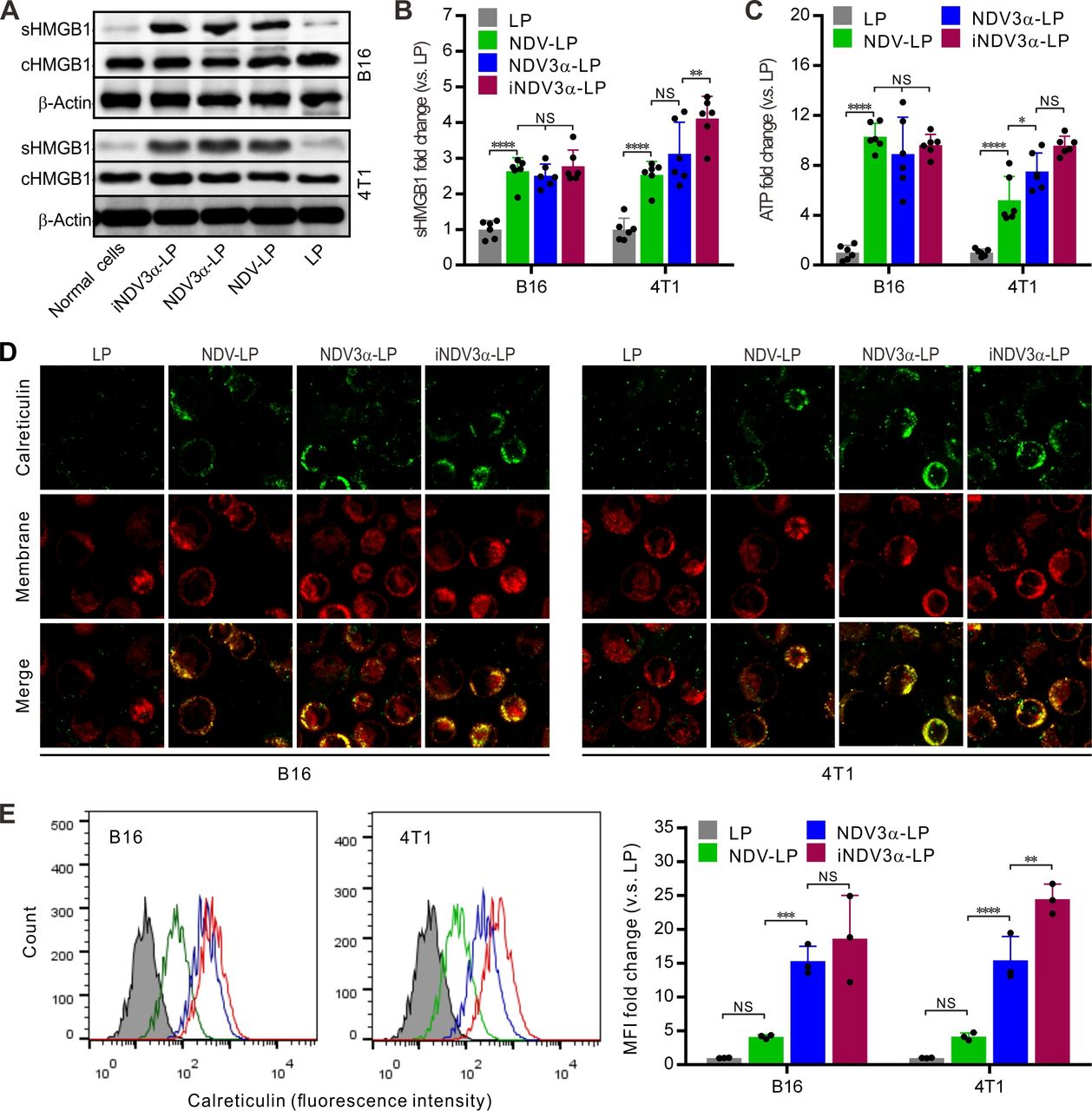
iNDV3α-LP treatment induces the immunogenic tumor cell death. (A) HMGB1 in B16 and 4T1 cell supernatants (sHMGB1) and whole-cells (cHMGB1) treated with the indicated formulations by western blot. (B) The titers of sHMGB1 in B16 and 4T1 cell supernatants by ELISA. (C) ATP released in B16 or 4T1 cell supernatants by ELISA. (D) CRT translocation to B16 or 4T1 cell surfaces by two counterstains with an FITG-conjugate anti-CRT antibody (green) and DiI (red). (E) FCM analysis of the CRT intensity on the B16 and 4T1 cell surface, showing the representative images and MFI fold change. CRT, calreticulin; FCM, flow cytometry; HMGB1, high mobility group box 1; iNDV3α-LP, iRGD-liposome loaded with NDV expressing MIP-3α; sHMGB1, secreted HMGB1.
iNDV3α-LP targets iRGD-specific receptors on tumor cells and HUVECs
Our previously mentioned results (figures 2 and 3) show that iNDV3α-LP induced more significant intensities of cell lysis and DAMP production in 4T1 cells than in B16 cells. We hypothesized that these phenomena are related to iRGD-specific receptors on the cells. Therefore, we detected whether αvβ3 was expressed on B16, 4T1, and HUVECs, by western blotting. As predicted, αvβ3 expression was found in 4T1 and HUVECs but not in B16 cells (figure 4A). To identify whether iNDV3α-LP specifically adhered to αvβ3-expressing cells, iNDV3α-LP, NDV3α-LP, NDV-LP, and LP were labeled with FITC (green), the cell membrane was labeled with DiI (red), and the nucleus was stained with DAPI. We then observed the cells using confocal microscopy, before FCM analysis. Consistent with the western blotting results, colocalization of green and red signals (yellow) was found on 4T1 and HUVECs treated with iNDV3α-LP but not on B16 cells (figure 4B). As predicted, no colocalization of green and red signals was found in the three cell lines treated with NDV3α-LP, NDV-LP, or LP (online supplemental figure S1F). These findings were further confirmed by FCM analysis (figure 4E).

iNDV3α-LP targets to αvβ3-expressing tumor cells and HUVECs and to tumor vasculature. (A) Immunoblotting detection of integrin αvβ3 expression in B16, 4T1, and HUVECs. (B–D) confocal fluorescent images showing that FITC-conjugated iNDV3α-LP (green, see also online supplemental figure S1F) binds to 4T1 (C) and HUVEC (D) cell membranes (DiI strained, red) but not to B16 (B); scale bar=5 µm. (E) FCM analysis of the green fluorescence intensity on the cell surface, showing the representative images and MFI fold change of the B16 cells, 4T1 cells, and HUVECs treated with the indicated formulations. (F) Uptake analysis of 4T1 cells by flow cytometry. (F) Confocal fluorescent images of B16 and 4T1 tumor sections, showing that only DiI-labeled iNDV3α-LP (red) specifically binds to FITC-dextran-marked blood vessels (green), which are merged as yellow (arrow). HUVECs, human umbilical vein endothelial cells; iNDV3α-LP, iRGD-liposome loaded with NDV expressing MIP-3α.
To detect whether iNDV3α-LP could directly target tumor blood vessels, B16 and 4T1 tumor models were established in syngeneic mice. After the tumor masses were palpable, FITC-conjugated dextran and DiI-labeled liposomes were injected into tumor-bearing mice, and 20 min later, the tumor masses were removed for frozen sectioning and then observed by fluorescence microscopy. The results showed that colocalization of green and red signals (yellow) was only observed in the sections of 4T1 tumors injected with DiI-labeled iNDV3α-LP but not in the sections of 4T1 tumors injected with NDV3α-LP, NDV-LP, or LP (figure 4F). As predicted, no colocalization of green and red signals was found in any sections of B16 tumors (figure 4F). Taken together, these data strongly suggest that iNDV3α-LP targets iRGD-specific receptors on tumor cells and HUVECs.
iNDV3α-LP induces antitumor effects in mouse tumor models
The antitumor activities of the engineered liposomes iNDV3α-LP, NDV3α-LP, NDV-LP, and LP were evaluated in B16 and 4T1 tumor models. These liposomes were injected into the tail vein of the model mice when the tumor masses were palpable on day 5 after inoculation of tumor cells. Figure 5A shows the tumor masses in 4T1-bearing mice on day 25 after inoculation of tumor cells; the results show effective antitumor activities of NDV-LP, NDV3α-LP, and iNDV3α-LP, but the most effective activities were observed in mice treated with iNDV3α-LP. Treatment with NDV-LP, NDV3α-LP, or iNDV3α-LP suppressed tumor growth and increased survival rates in both B16- and 4T1-bearing mice, but treatment with iNDV3α-LP was superior to other formulations (figure 5B,C).

iNDV3α-LP treatment induces specific antitumor effects. (A) Representative images of 4T1 tumor masses treated with the indicated formulations on day 28 after tumor cell injection. (B and C) Data of the tumor volumes and survival rates of the tumor-bearing B16 and 4T1 mice treated with the indicated formulations at the indicated time points. (D) The synergistic indexes (SIs) calculated with the mean tumor volumes at the indicated time points in (B) and (C). ORR=mean tumor volume in the NDV-LP, NDV3α-LP, or iNDV3α-LP group/the mean tumor volume in the LP group; ERR=ORR of the NDV-LP group × ORR of NDV3α-LP; SI=ERR/ORR (ORR <1) or ORR/ERR (ORR >1), where SI>1 indicates a synergistic effect. ERR, expected relative ratio; iNDV3α-LP, iRGD-liposome loaded with NDV expressing MIP-3α; NDV3α-LP, liposome loaded with NDV expressing MIP-3α; NDV-LP, liposome loaded with single NDV; ORR, observed relative ratio.
The previous data (figures 2–4) indicate that iNDV3α-LP, but not NDV3α-LP, can directly lyse and bind to HUVECs in vitro and target blood vessels in vivo. The engineered liposome iNDV3α-LP used in this study contains several strategies, such as iRGD-directed HUVEC lysis, tumor-vessel targeting, and immune enforcing by MIP-3α. Thus, treatment with the iNDV3α-LP liposome could be considered as a combination therapy, NDV3α-LP as NDV combined with MIP-3α immunotherapy, NDV-LP as only NDV monotherapy, and LP as not treatment control. To investigate whether the combination of these additional strategies by iNDV3α-LP could enhance antitumor immunity, the mean tumor volume of both B16 and 4T1 cells at different time points was used to calculate the SI. The resultant SIs at most of the time points were >1 (figure 5D), suggesting that the combination of the above-mentioned strategies of iNDV3α-LP induces stronger antitumor activities in both B16 and 4T1 models.
iNDV3α-LP induces tumor-specific cellular immunity and humoral immunity
Both cellular and humoral immunity are major arsenals against tumors. In the current study, tumor-specific CTLs were detected using splenocytes as effector cells and 4T1 or B16 cells as target cells. Target cell lysis was significantly evidenced in the iNDV3α-LP, NDV3α-LP, and NDV-LP groups, but the greatest lysis was observed in the iNDV3α-LP group (figure 6A,B). In this study, an in vivo method was used to detect tumor-specific CTLs by injecting CFSE-conjugated 4T1 or B16 cells into the abdominal cavity of syngeneic mice to analyze the cell proliferation by FCM. The results show that the CD45-negative CFSE-conjugated 4T1 or B16 cells proliferated for four generations, with an increased cell number observed in the LP group compared with that in the iNDV3α-LP, NDV3α-LP, and NDV-LP groups. However, cell proliferation was significantly inhibited, and the tumor cell number was markedly decreased in the iNDV3α-LP, NDV3α-LP, and NDV-LP groups, with the most significant decrease in the iNDV3α-LP group (figure 6C,D and online supplemental figure S1G). Additionally, the CD45+ immune cells from both 4T1 and B16 tumor tissues were gated and analyzed by FCM. CD8+ cells from iNDV3α-LP-treated tumors expressed more active cytokines, such as IFN-γ (figure 6E and online supplemental figure S2A,B), TNF-α (figure 6F and online supplemental figure S2C,D), and IL-2 (figure 6G and online supplemental figure S2E,F). We also used the mean per cent of these immune cell subsets to calculate their SIs, most of which were >1 (figure 6H). These results also suggest that combination therapy induced by iNDV3α-LP provokes more tumor-specific cellular immunity in both 4T1 and B16 tumor-bearing mice.
Supplemental material

iNDV3α-LP treatment enhances antitumor immunity. (A and B) CTL response using B16 (A) and 4T1 (B) as target cells and the splenocytes from the mice treated with the indicated formulations as effector cells. (C and D) The in vivo CTL response was examined by injecting CFSE-labeled B16 (D) or 4T1 cells into the mouse abdomen treated with the indicated formulations to analyze the proliferation of CD45− tumor cells by FCM and calculate the percentage of cell lysis (C, see also online supplemental figure S1). (E–G) The percentage of CD8+ splenocytes secreting IFN-γ (E), TNF-α (F), and IL-2 (G) from B16- or 4T1-bearing mice treated with the indicated formulations. (H) The synergistic indexes (SIs) calculated with the mean percentage of CD8+ cell subtypes in (E–G). (I) Western blot detection of the tumor-specific IgG in the serum from mice treated with the indicated formulations. (J and K) ELISPOT detection of the splenocytes secreting IgG specific to B16 or 4T1 cells (J) and the average number of IgG-secreting cells in 105 splenocytes (K). (L and M) The IgG titer in the serum from B16-bearing or 4T1-bearing mice treated with the indicated formulations (L) and the IgG subtypes (M). ELISPOT, enzyme-linked immunospot; iNDV3α-LP, iRGD-liposome loaded with NDV expressing MIP-3α.
Moreover, we found that iNDV3α-LP treatment could induce stronger tumor-specific humoral immunity compared with the LP, NDV3α-LP, and NDV-LP groups. Compared with LP treatment, iNDV3α-LP, NDV3α-LP, and NDV-LP treatments showed significantly increased titers of IgG antibody both in 4T1-bearing and B16-bearing mouse sera (figure 6I). Moreover, iNDV3α-LP treatment induced better tumor-specific humoral immunity compared with the other three groups, as confirmed by ELISPOT assay, in which more splenocytes secreting tumor-specific IgG were observed in the iNDV3α-LP-treated spleens (figure 6J–K). Consistently, quantitative analysis of IgG1 by ELISA also showed significantly increased IgG titers in the sera from mice treated with iNDV3α-LP, NDV3α-LP, and NDV-LP, with the highest titers observed in mice treated with iNDV3α-LP (figure 6L). Additionally, ELISA analysis showed that the significantly increased titers of IgG subsets were IgG1 and IgG2a (figure 6M). Taken together with the data shown in figure 6E–G, these results suggest that iNDV3α-LP promotes the induction of tumor-specific humoral immunity by stimulating both Th1 and Th2 immune responses.
iNDV3α-LP inhibits tumor vascularization and modulates the tumor microenvironment
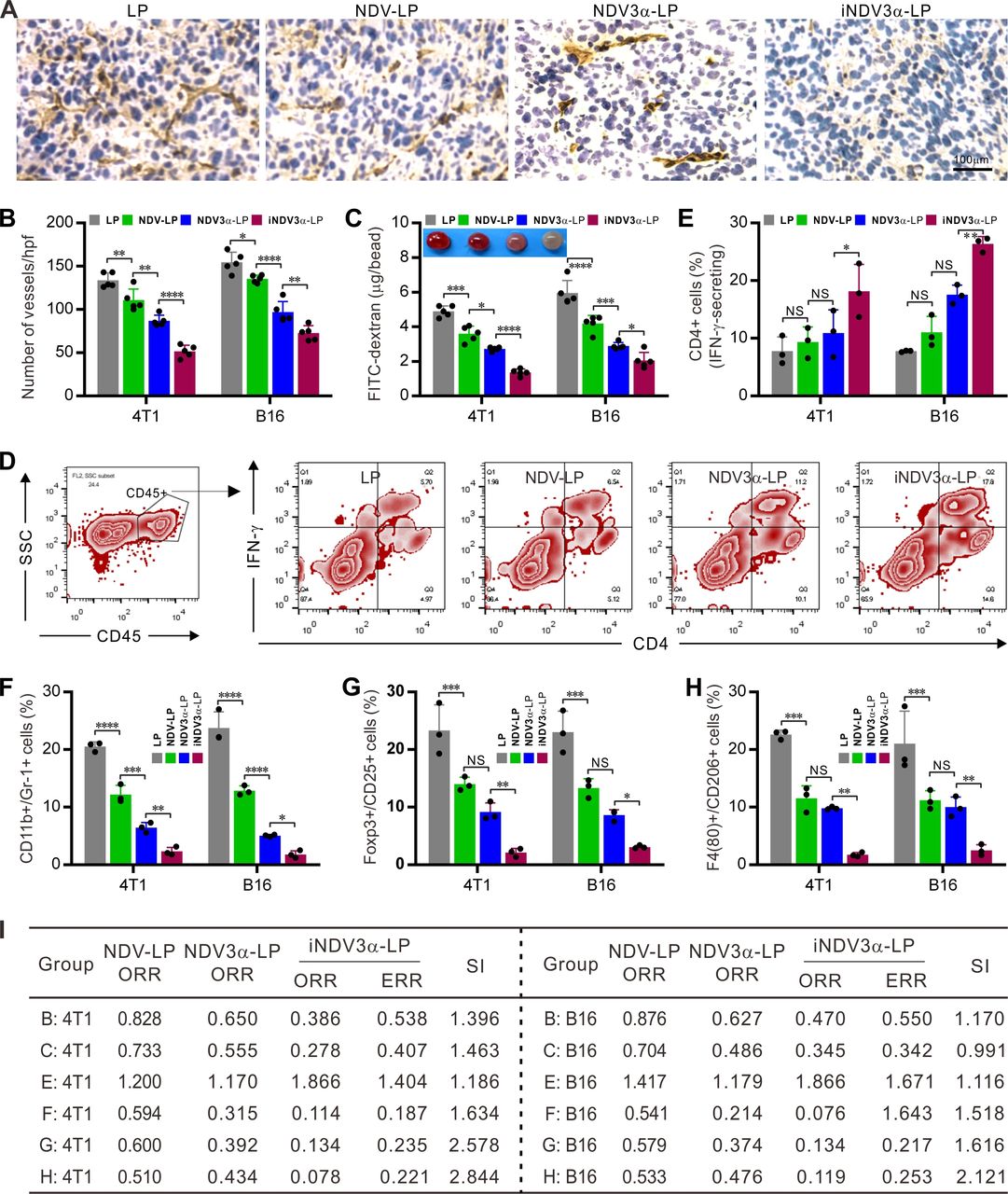
Because iNDV3α-LP targets αvβ3 on vascular endothelial cells, we next observed whether iNDV3α-LP treatment could inhibit tumor angiogenesis in 4T1 and B16 models. Angiogenesis within 4T1 and B16 tumor masses was observed on sections stained with an antibody against CD31, in which inhibition of angiogenesis was observed in iNDV3α-LP-treated sections (figure 7A). Moreover, compared with the LP-treated sections, the mean number of vessels per high-power field was significantly decreased in the iNDV3α-LP-treated, NDV3α-LP-treated, and NDV-LP-treated sections, with the most significant decrease observed in the iNDV3α-LP-treated sections (figure 7B). Additionally, an alginate-bead encapsulation assay was adapted to quantify the angiogenesis by measuring the uptake of FITC-dextran into the tumor cell encapsulated alginate beads.15 16 Consistently, compared with LP treatment, iNDV3α-LP, NDV3α-LP, or NDV-LP treatment resulted in clear inhibition of angiogenesis in both 4T1-encapsulated and B16-encapsulated beards, but the most evident inhibition was found in the iNDV3α-LP-treated beads (figure 7C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
iNDV3α-LP treatment inhibits tumor angiogenesis and reverses tumor immunosuppressive microenvironment. (A and B) Sections of frozen B16 tumor tissues from mice treated with the indicated formulations were stained with anti-CD31 antibody to show the tumor microvessels (A) and determine the vessel density by counting the number of microvessels per high-power field (B). (C) FITC-dextran uptake in alginate beads encapsulated with 2×105 B16 or 4T1 cells treated with the indicated formulations. (D–H) CD45+ immune cells were gated from the total cell population from the tumor tissues treated with the indicated formulations. Representative flow cytometry images (D, also see online supplemental figure S3), and the mean percentage of TILs with the following features in the B16-bearing or 4T1-bearing mice treated with the indicated formulations: IFN-γ-secreting CD4+ lymphocytes (E), CD11b and GR-1 double positive MDSCs (F), Poxp3 and CD25 double positive Tregs (G), and F4/80 and CD206 double positive M2-type tumor-associated macrophages (H). (I) The synergistic indexes (SIs) were calculated with the mean percentage of TIL cell subtypes in (E–H). iNDV3α-LP, iRGD-liposome loaded with NDV expressing MIP-3α; MDSCs, myeloid-derived suppressor cells; TILs, tumor-infiltrating lymphocytes; Tregs:, regulatory T cells.
Supplemental material
To investigate whether iNDV3α-LP treatment could modulate the immunosuppressive microenvironment, single cells were generated from 4T1 or B16 tumor masses. The CD45+ immune cells were gated out from the total cell population, and their subtypes, including IFN-γ-secreting CD4+, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs), were analyzed by FCM (figure 7D and online supplemental figure S3A–G). The results showed that the percentage of IFN-γ-expressing CD4+ lymphocytes was significantly increased in the iNDV3α-LP-treated tumor tissues (figure 7D–E and online supplemental figure S3A). However, the percentages of CD11c+ and Gr-1+ MDSCs (figure 7F and online supplemental figure S3B,C), Foxp3+ and CD25+ Tregs (figure 7G and online supplemental figure S3E), and F4/80+ and CD206+ M2 TAMs (figure 7H and online supplemental figure S3 F–G) were significantly decreased in iNDV3α-LP-treated tumor tissues. Taken together with the results shown in figure 6E–G, these results suggest that the immunosuppressive microenvironment is effectively reversed by iNDV3α-LP treatment.
To understand whether iNDV3α-LP treatment can effectively inhibit tumor vascularization and modulate the tumor microenvironment, we used the previous data to calculate the SIs. As predicted, almost all SIs calculated from the data shown in figure 7 were >1, and some were even >2 (figure 7I). With the SIs shown in figure 6H, which were calculated from the data shown in figure 6E–G, these findings also indicate that combination therapy induced by iNDV3α-LP enhances inhibition of tumor vascularization and modulation of the tumor microenvironment.
Discussion
Oncolytic immunotherapy is a paradigm-shifting treatment modality for cancers, which is primarily based on virus-mediated oncolysis and the subsequent induction of tumor-specific immune responses. Several lines of evidence from clinical trials of currently available oncolytic immunotherapeutic indicate that robust tumor-specific immunity leads to remarkable and long-term responses in a subset of patients.1 8 12 25 In this study, we engineered a tumor neovasculature-targeted liposome iNDV3α-LP coated with iRGD, which specifically binds to the αvβ3 receptor on endothelial cells in the tumor neovasculature. The DC chemokine gene MIP-3α was encapsulated in the liposome iNDV3α-LP, as this chemokine has been shown to promote systemic antitumor immunity in several murine tumor models.24 Successful construction of iNDV3α-LP and the three control liposomes, NDV3α-LP, NDV-LP, and LP, was evidenced by electron microscopy observation, laser-based analysis, and encapsulation-efficiency detection of NDVs. In contrast to the results of our previous study, which showed no significant difference in viral replication and tumor lysis between recombinant NDV3α and wild-type NDV, iNDV3α-LP promoted greater viral replication and tumor and HUVEC lysis compared with NDV-LP, although NDV3α-LP also induced significant virus replication and tumor and HUVEC lysis compared with NDV-LP. Detection of ICD-marker DAMP molecules also showed that iNDV3α-LP-treated B16 and 4T1 produced or secreted more DAMP molecules. As predicted, iNDV3α-LP could specifically bind to αvβ3-expressing 4T1 and HUVECs in vitro and to the neovasculature in the tumor masses in vivo. Moreover, the tumor growth observed in B16 and 4T1 murine models was remarkably suppressed, and the survival was longer in iNDV3α-LP-treated mice. The mechanism study performed using several immunological techniques showed that iNDV3α-LP treatment induced the strongest tumor-specific cellular and humoral immunity of all tested liposomes. Surprisingly, iNDV3α-LP treatment could concurrently suppress tumor angiogenesis and reverse the tumor immune-suppressive microenvironment in both B16 and 4T1 model mice. Taken together, these data suggest that iNDV3α-LP that targets both tumor and its neovasculature is a novel highly efficient strategy for oncolytic immunotherapy.
It is generally accepted that oncolytic immunotherapy is not a ‘magic bullet’ for cancer therapy, and the activity of monotherapy is modest for most agents.31 To enhance antitumor activities, oncolytic immunotherapy combined with other treatment modalities, including immune checkpoint inhibitors, immune modulators, cancer vaccines, chemotherapy, radiation therapy, or even photothermal therapy by nanoparticles, has been explored in various preclinical and clinical trials.5 7 9 32 However, oncolytic immunotherapy has various problems that still require further study. Therefore, novel combination strategies to redirect the dominant immune response toward cancer cells are warranted. The successful combination of T-VEC-expressing GM-CSF has proved that OVs have an additional advantage of delivering various transgenes to enhance antitumor activities. We have previously demonstrated that a recombinant oncolytic Newcastle virus expressing MIP-3α (NDV3α) can remarkably promote systemic antitumor immunity in several murine models.24 In this study, we hypothesized that targeting recombinant NDV3α to both the tumor and its neovasculature by encapsulation of NDV3α in an iRGD-coated liposome (iNDV3α-LP) will induce combinatory anti-tumor effects. We considered that iNDV3α-LP will function to directly lyse both tumor and blood vessels and may additionally enhance the production of specific antitumor immunities as a result of MIP-3α directing DCs to tumor masses and increasing the presentation of tumor antigen. Our results showed that iNDV3α-LP, but not NDV3α-LP, directly induced the lysis of both tumor cells and HUVECs. Moreover, iNDV3α-LP could also bind to tumor neovasculature in vivo and secrete DC chemokine MIP-3α. Therefore, the iNDV3α-LP treatment could function as a combination of tumor and vessel lysis, and MIP-3α immunotherapy, NDV3α-LP treatment as tumor lysis and MIP-3α immunotherapy, NDV-LP treatment as only tumor lysis, and LP treatment as control. In this study, we calculated the SIs using the tumor volume, tumor angiogenesis, and percentage of TIL subtypes to evaluate whether the iNDV3α-LP treatment induced combinatory antitumor activities.15 Our results show that iNDV3α-LP treatment induces significant lysis of both tumor and endothelial cells in vitro and inhibition of B16 and 4T1 tumor growth, as evidenced by SIs >1 at most time points. Moreover, we also found that most of the SIs calculated using the data of angiogenesis and TIL were >1 (figure 7I). Taken together, these data indicate that the iNDV3α-LP treatment strategy induces combinatory antitumor activities by concurrent lysis of tumor and endothelial cells, suppression of tumor angiogenesis, and systemic modulation of the tumor immunosuppressive microenvironment.
In our study, the results demonstrated that NDV-LP only reserved the function of wild-type NDV (tumor lysis and induction of immunogenic cell death), and NDV3α-LP added immune reinforcing function due to the recombinant MIP-3α chemokine; however, iNDV3α-LP had all the functions of both NDV-LP and NDV3α-LP. Therefore, the therapy with iNDV3α-LP could be considered as a treatment modality that combines NDV-LP with NDV3α-LP. Herein, synergy index (SI) analysis, an accepted method in the literature, was applied to determine the synergistic effect induced by iNDV3α-LP. SI>1 indicates a synergistic effect. Unfortunately, the results indicated that some SI was less than 1, which mainly appeared in figures 5D and 6H. With an in-depth analysis of the samples, we found that there were only 10 and 3 samples in the figures 5D and 6H, respectively, and there were extreme values appearing in every time point when SI was less than 1. We thought that the extreme values may be the major reason for SI<1. Therefore, it is necessary to increase the number of samples to further certify whether iNDV3α-LP immunotherapy induced a really synergistic effect in future study.
The formation of neovasculature, or angiogenesis, is a necessary prerequisite for tumor generation and metastasis. Several lines of evidence have shown that certain OVs have the capability to selectively target the tumor neovasculature. For example, the vesicular stomatitis virus can cause thrombosis in the tumor neovessels by selective infection of endothelial cells in the tumor microenvironment.33 Herpes simplex virus (HSV) and vaccinia virus replicate more effectively in the tumor microenvironment with high levels of vascular endothelial growth factor and fibroblast growth factor.33–35 Thus, following successful infection, HSV and vaccinia virus can damage the tumor endothelium.33–35 Additionally, OVs can be genetically engineered to express antiangiogenic factors, such as VEGF inhibitors.36 37 Until now, no study has provided evidence of the ability of NDV to selectively damage the tumor endothelium. This phenomenon was proven by our results in that neither NDV3α-LP nor NDV-LP could target HUVECs in vitro or the tumor neovasculature in vivo and could not significantly lyse HUVECs in vitro. In contrast, iNDV3α-LP could bind to HUVECs in vitro and the tumor neovasculature in vivo and caused increased HUVEC lysis compared with the three other groups. Moreover, the most remarkable antiangiogenesis activities were found in the tumors from iNDV3α-LP-treated mice. Thus, we believe that the induction of the specific antitumor immunity by iNDV3α-LP may be of relevance to iRGD-directed binding and subsequent damage to the neovasculature in tumor-bearing mice.
Immunogenic cell death (ICD) can activate DCs or other immune cells to trigger specific immunity against targeted cells.38 The induction of ICD changes ‘cold’ tumors to ‘hot’ tumors, which are more visible to the immune system, especially to DCs, which phagocytose the dying tumor cells, process and cross-present the tumor antigen and initiate tumor-specific immunity.39–41 During the ICD process, cancer cells induce endoplasmic reticulum (ER) stress, which is evidenced by the translocation of the ER-associated protein disulfide isomerase ERp57 from the ER lumen to the membranes of dying cells.42 During ER stress, CRT from the ER translocates to the cell surface, forming surface-expressed CRT (ecto-CRT). Additionally, cancer cells undergoing ICD express or secrete high levels of other DAMP molecules, such as extracellularly secreted ATP and passively released chromatin-binding protein HMGB1.43 The detection of these DAMP molecules helps to confirm existing ICD in tumor cells. In this study, compared with both NDV3α-LP- and NDV-LP-treated cells, the results of western blot and ELISA showed remarkably increased levels of secreted HMGB1 (sHMGB1) in the supernatant of iNDV3α-LP-treated αvβ3-expressing 4T1 cells, but not in B16 cells. Although the sHMGB1 titer significantly increased in iNDV3α-LP-treated B16 cells, there were no significant differences among iNDV3α-LP-treated, NDV3α-LP-treated, and NDV-LP-treated B16 cells. The results of ATP and ecto-CRT were similar to that of sHMGB1. Additionally, several parameters, including cell lysis, neovasculature-targeting, tumor volume, cellular and humoral immunity, and antiangiogenesis, were only superior in iNDV3α-LP-treated 4T1 cells or mice. These data indicate that iNDV3α-LP treatment leads to increased production of ICD-related DAMPs, which suggests that the induction of better antitumor effects by iNDV3α-LP treatment is related to its neovasculature targeting the αvβ3 positive endothelium.
Conclusion
In this study, we engineered an iRGD-liposome encapsulating the recombinant NDV, which expressed the DC chemokine MIP-3α (named as iNDV3α-LP), to explore its antitumor activities. The results show that iNDV3α-LP increased viral replication, tumor and HUVEC cell lysis, stronger tumor ICD, and a more remarkable antitumor immunity. The mechanism investigations show that iNDV3α-LP bound to αvβ3-expressing tumor and HUVECs in vitro and to the neovasculature in the tumor masses in vivo. Additionally, iNDV3α-LP treatment triggered stronger tumor-specific cellular and humoral immunity. Moreover, iNDV3α-LP treatment significantly suppressed tumor angiogenesis and reversed the tumor immunosuppressive microenvironment. These findings offer a strong rationale for further clinical investigation into a combination strategy using multifunctional recombinant OV as a tool, such as the formulation iNDV3α-LP in this study.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
J-YW, HC, S-ZD and F-YH contributed equally.
Contributors G-HT, F-YH, W-PZ, and LL designed experiments and analyzed and interpreted the data. J-YW, HC, S-ZD, F-YH, Y-YL, and C-CW performed the experiments. G-HT and F-YH analyzed the FCM data. W-PZ and LL offered some of the experiment resources and technical support. G-HT and F-YH directed the project and wrote the manuscript. All authors approved the final version of the manuscript. G-HT is the guarantor responsible for the overall content.
Funding This work was supported by the National Natural Science Foundation of China (81760634, 81 860 650 and 81960547, 82060639), and by the Hainan Provincial National Natural Science Foundation (2019RC217, 2019RC234 and ZDKJ202003). We would like to thank the Project supported by Hainan Province Clinical Medical Center.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.