Article Text

Abstract
Prostate cancer (PC) has previously been established as a cold tumor and develops in an inert immunosuppressive environment. Current research focuses on altering the immune microenvironment of PC from cold to hot; thus, in the present review, the diverse roles of estrogen and estrogen receptor (ER) signaling was examined in the tumor cell and tumor immune microenvironment (TIM). We hypothesized that ERα promotes PC progression and ERβ impedes epithelial-mesenchymal transition in PC cells, while in the TIM, ERβ mediates the immunosuppressive environment, and low levels of ERα is associated with disease development. Selective estrogen receptor modulators (SERMs) or selective ER degraders play diverse roles in the regulation of ER isoforms. Patients with PC may benefit from the use of SERMs, including raloxifene, in combination with anti-PD1/PD-L1 checkpoint immunotherapy, or TGF-β or Wnt antagonists. The present review demonstrated that immunotherapy-based strategies combined with SERMs may be an option for the future of PC-targeting therapy.
- combination drug therapy
- immunotherapy
- immunologic receptors
- urologic neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Prostate cancer and tumor immune microenvironment
Prostate cancer (PC) remains a common health problem, and impacts both society and the economy. According to recent data, it ranks the most common cancer among men in Western countries and possesses the second highest rate of cancer-related death. At present, first-line treatment for PC includes androgen-deprivation therapy, which is used in patients with progressive, recurrent and metastatic PC. However, almost all cases of PC progress from hormone-sensitive PC to castration-resistant PC (CRPC) following treatment.1 2 Notably, numerous epidemiological studies, including meta-analyses, indicate that hormone induction and the tumor immune microenvironment (TIM) are associated with PC progression.3 The TIM of PC plays a critical role in endocrine therapy and immunotherapy targeting tumor progression, metastasis and treatment resistance. Specifically, the prostatic stromal cells associated with immunological cells participate in prostatic pathologies, particularly in cancer, building a complex TIM. Additionally, increased accumulation of pro-inflammatory factors, cancer associated fibroblasts (CAFs) and myofibroblasts generate a progressive and tumor-promotive TIM, in which the stroma cell stimulates the proliferation and migration of PC cells. The diverse activation pathways in CAFs mainly include transforming growth factor-β (TGF-β), platelet-derived growth factor, hepatocyte growth factor, fibroblast growth factor, interleukins, metalloproteinases and reactive oxygen species, which take effect via activating heat shock factor 1 and nuclear factor kappa B (NF-kB).4 The TIM associated with PC is highly immunosuppressive, containing (1) cells, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs); (2) cytokine milieu, secreted by stromal cells and fibroblasts of the TIM; (3) adenosine, produced from prostatic acid phosphatase; and (4) molecular pathways, including TGF-β and Wnt/β-catenin. Both adenosine and TGF-β may serve as potential immunosuppressive molecules or pathways, and act as therapeutic targets5 (figure 1). Moreover, in the TIM, TGF-β and Wnt/β-catenin are frequently activated in advanced PC, and contribute to therapy resistance and metastasis, including resistance to docetaxel, enzalutamide and abiraterone acetate-prednisone.6–8

Communication between prostate cancer (PC) and stromal cells in the tumor immune microenvironment (TIM). These stromal cells include cancer associated fibroblasts (CAF), regulatory T cells (Tregs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), CD8 +T cells, CD4 +T cells, dendritic cells (DCs) and natural killer (NK) cells. A series of immune mediators and their origins are presented in the gray box, containing adenosine, hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), fibroblast growth factor (FGF), interleukins, metalloproteinases, reactive oxygen species, cytokine milieu, heat shock factor 1 (HSF1) and nuclear factor kappa B (NF-kB).
Estrogen and ER in PC
Estrogen treatment for PC was verified to be effective in previous clinical trials.9 Estrogen is implicated in the initiation of PC.10 Estrogen-dependent signaling-induced TMPRSS2-ERG expression leads to TMPRSS2-ERG fusion PC.11 It has been demonstrated that estrogen and its active product, 17β-estradiol (E2), originate from androgen metabolism via steroidogenesis, which is associated with PC development.12 In animal models and clinical samples, high levels of estrogen are positively associated with the aggressiveness of PC.13 Moreover, the modulation of estrogen in cancer progression is highlighted due to the activity of estrogen on its receptors ERα and ERβ, which act as transcription factors, contributing to healthy prostate development.14 Results of a previous study demonstrated that treatment with E2 (1 µM) for 24 hours may led to the downregulation of ERs, while treatment with physiological concentrations of E2 (0.1 nM) for 24 hours increased the levels of ERs. E2-induced ERK1/2 activation was not inhibited by ERs antagonists, indicating the involvement of alternative estrogen-regulated mechanisms. Therefore, the molecular mechanism underlying estrogen in the promotion of PC progression is complex.15 ERα activation using the agonist PPT, and ERβ activation using the agonist DPN enhanced the phosphorylation of SRC in PC-3 cells of PC. ER enhanced the tumorigenic potential of PC by inducing rapid responses of SRC and PI3K/AKT.16
Results of a previous study demonstrated that ERα is mainly localized in prostate stroma cells, and estrogen plays regulatory effects on the prostate adenoepithelium using paracrine pathways.17 Results of a previous study further indicated that AR-positive human PC cell lines, including LNCaP, LAPC4 and 22Rv1 did not exhibit ER expression levels. AR-negative human PC cell lines, including DU145 and PC3 did not exhibit ERβ expression levels, while in PC3 cells, weak expression of ERα was determined.18 ERα is crucial in the invasion and migration of PC cells.19 ERα, as the predominant subtype, is associated with high-grade PC.20 21 ERα has oncogenic functions in animal models, and its activation leads to cancer proliferation.22 23
Compared with healthy prostate tissue, ERβ is overexpressed in PC tissues.24 Antiestrogens and selective estrogen receptor modulators (SERMs) are found to reduce the risk of PC development in PC metastases with high levels of ERβ expression.25 26 ERβ expression is associated with poor clinical outcomes in hormone-sensitive PC, and is increased in CRPC.27 The ER isoforms β2 and β5 are associated with poor patient outcomes, and promote PC migration and invasion.28 On the other hand, ERβ is regarded as a tumor suppressor, and ERβ inhibition promoted prostate hyperplasia and tumor development.29–31 ERβ expression is decreased in the process of PC progression.32–34 Therefore, estrogen/ER pathways exhibit diverse and complex roles in PC development.18 The specific mechanisms underlying ER isoforms require further evaluation and exploration.
SERMs and PC
SERMs, including selective estrogen receptor degraders (SERDs), have previously been used for the treatment of breast cancer by targeting ERs.35 These agents include tamoxifen, toremifene and raloxifene.18 SERMs competitively block the binding of estrogen with ERα. SERDs, such as fulvestrant, contribute to ERα downregulation and degradation.36
Notably, ERβ may play a role as a response marker of tamoxifen.37 The majority of clinical trials associated with tamoxifen yielded moderate rates of survival against cancer.38 Targeting ERα with tamoxifen provides a novel therapeutic strategy for the castration-resistant ER-positive subtype of metastatic PC, which is associated with the suppression of phosphatidylinositol-4-phosphate 5-kinase-α/AKT and matrix metalloproteinase (MMP)−9/vascular endothelial growth factor (VEGF) signaling pathways.39 Thus, SERMs may exhibit potential in ER-progressed PC therapy. Results of a phase II clinical trial of CRPC demonstrated that treatment with raloxifene leads to a stable disease status, by inhibiting the growth of tumors in PC xenograft models.40 However, previous studies have also demonstrated the antiproliferative role of ERβ, and a loss of ERβ expression in high-grade tumors. Therefore, treatment with single SERMs targeting ERs may not fully inhibit PC progression.
Estrogen/ER on the TIM
Estrogen can induce the activation of M2-type macrophages leading to tumor progression.41 Multiple cytokines secreted by these functional M1-type macrophages, including IL-6, MMP-9, IL-1β and TNFα, are inhibited following activated estrogen/ERα signaling.42–44 Moreover, estrogen may directly or indirectly suppress the cytotoxic activity of NK cells.45 46 As an immune-suppressive regulator, estrogen supports tumor initiation and progression. Therefore, estrogen-targeted therapy may orchestrate an antitumor TIM and guide proimmune surveillance.
Results of a previous study demonstrated that TGF-β and ER signaling interact during cancer development.47 ERα, downstream of TGF-β, plays a dual role in regulating TGF-β signaling and cancer development, via a novel non-genomic mechanism.48–50 The activated ER signaling pathway is associated with TGF-β in the progression of LNCaP PC models.51 Cell migration and invasion induced by ERα are associated with the upregulated activity of MMP-2 and MMP-9, accompanied with the upregulation of a series of epithelial-mesenchymal transition (EMT)-related genes, such as TGF-β1, zinc finger E-box-binding homeobox 1/2 and vimentin.19 In prostate stromal cells, estrogen induces MMP-2 expression via binding ERα to induct TGF-β, and stimulates PC cell invasion in a paracrine manner.52
ERβ is mainly expressed in immune cells, particularly in the inflammatory and hypoxic TIM, compared with ERα,53 which may differentially contribute to the response to anti-estrogen or treatment with SERMs/SERDs.54 Following E2 stimulation, ERβ promotes Treg differentiation, guides the secretion of IL-10 and TGF-β, downregulates CD8 +T cell activity and suppresses tumor immunocytotoxicity induced by T cells.55–57 E2 requires ERβ rather than ERα signaling for increasing the expression levels of Foxp3, GATA3 and CD25 in Treg cells.58 However, results of previous studies have demonstrated that in CD8 +T cells, ERβ signaling increases the activation of T cell receptors and antitumor immunity via a phosphotyrosine switch. ERβ activation promotes IL-1β secretion and enhances neutrophil infiltration in TIM, further suppressing tumor progression and metastasis.59 Therefore, the specific role of ERβ and the phosphotyrosine switch in cancer development is complex, and further studies are required. Recent clinical data indicated that in PC tumor associated stromal cells, a high expression level of ERα was associated with elevated clinical failure-free survival and PC death-free survival, while a high expression of ERβ was associated with decreased biochemical failure-free survival.60 These studies suggested that ERβ may mediate antitumor immunity and play an opposite role to ERα in the TIM.
SERMs on the TIM
Increased evidence based on experimental and clinical research has demonstrated the diverse immunomodulatory roles of SERMs and SERDs.61 Previous studies have explored how SERMs and SERDs act on the TIM. By inhibiting antitumor immunity, Tamoxifen caused a phenotypic shift of CD4 +T cells from Th1-targeting cancer to Th2-tolerating cancer.61 62 Moreover, TGF-β production induced by Tamoxifen and Fulvestrant inhibited the cytotoxic effects of CD8 +T cells, and promoted CD4 +T cell polarization into Foxp3 +Tregs. Tamoxifen and fulvestrant evoked programmed death-ligand 1 (PD-L1) upregulation by blocking Erα, contributing to the cytotoxic T cell evasion.63 Notably, SERMs and SERDs mediated immunosuppressive effects and increased the chance of tumors escaping an immune response.
On the other hand, SERMs also enhance the anticancer immunity and cancer immunogenicity. Tamoxifen and Fulvestrant inhibit cancer via targeted immunotherapy, without impacting healthy tissue.64 Tamoxifen increased the cytotoxicity of NK cells, leading to tumor cell lysis.65 Toremifene bridges the immunological synapses between cancer and NK cells by upregulating the expression levels of intercellular adhesion molecule-1 (ICAM-1).66 Tamoxifen abolished estrogen-induced cancer-immune tolerance though suppressing FasL and blocking chemokine (C-C motif) ligand 2(CCL-2)/CCL-5.67 68 Depletion of TAMs in combination with tamoxifen increased the inhibitory effects of tamoxifen on aggressive PC.39 Fulvestrant reduced MDSCs and Tregs, and increased the infiltration of DCs, CD8 +and CD4+T cells in a xenograft tumor model. The effects of SERDs on TIM markedly improved the efficiency of PD-L1-targeted immunotherapy.69 SERMs/SERDs-mediated anticancer immunity along with immune check-point blockers (ICBs) increases the potential of PD-L1 inhibition. Moreover, tamoxifen and toremifene were also found to inhibit JNK activation and enhance activated T cells.70
Pharmacodynamic evaluation of SERMs
Certain SERMs exert the same pharmacodynamic effects on both ERα and ERβ, including 4-hydroxytamoxifen (IC50; 3.3 nM), raloxifene (IC50; 2.9–5.7 nM) and toremifene (IC50; 1000 nM), while bazedeoxifene, lasofoxifene and SERD fulvestrant exhibit differential effects on each. Notably, bazedeoxifene exerts differential effects on ERα (IC50; 26 nM) and ERβ (IC50; 99 nM); lasofoxifene exerts differential effects on ERα (IC50; 1.08 nM) and ERβ (IC50; 4.41 nM) and fulvestrant exerts differential effects on ERα (IC50; 0.47 nM) and ERβ (IC50; 3.8 nM).18 Based on these pharmacodynamic analyses, we concluded that SERMs exert inhibitory activities on both ERα and ERβ isoforms. Moreover, 4-hydroxytamoxifen, toremifene and raloxifene exert the same inhibitory activities on both isoforms. Compared with the effects on ERβ, bazedeoxifene, lasofoxifene and fulvestrant (SERD) exert increased inhibitory activities on ERα. Moreover, in PC cells, raloxifene induced apoptosis via ERβ activation and ERα inhibition.71
Hypotheses and results
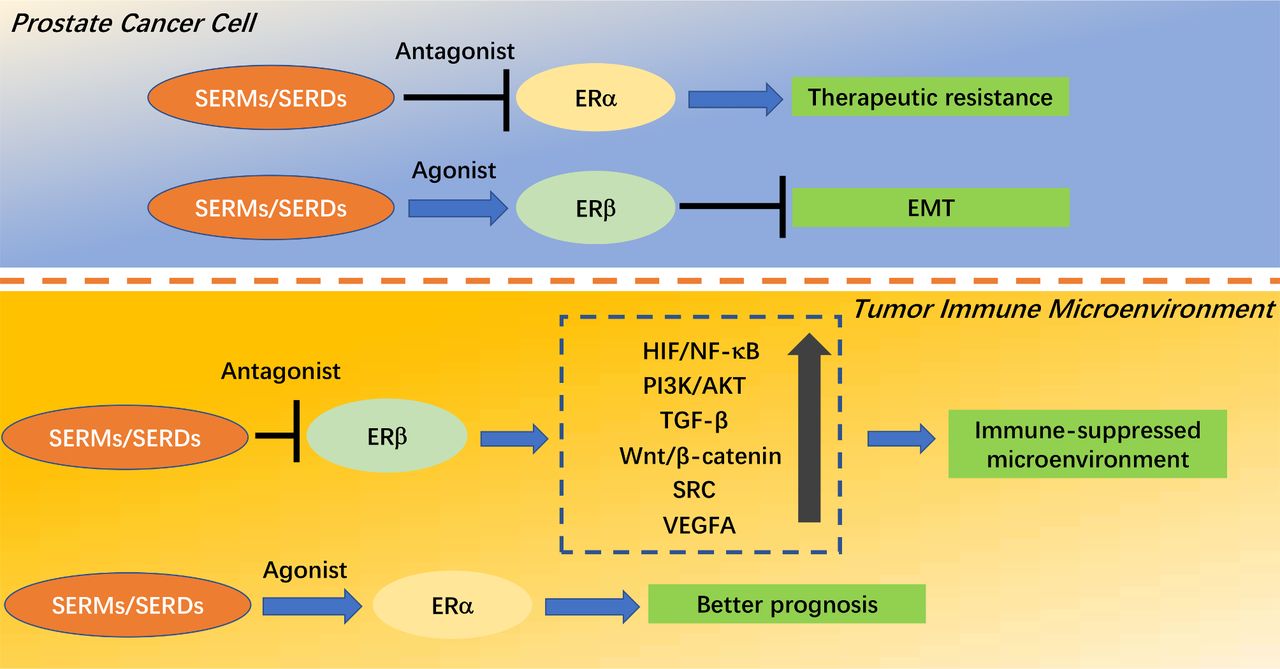
In PC cells, ERα activation and ERβ inactivation may lead to therapeutic resistance and EMT, respectively. A SERM that antagonizes ERα may reverse resistance, while an agonist of ERβ activity may reverse EMT.
In the TIM, ERβ activation and ERα inactivation will result in immunosuppression and poor prognosis, respectively. A SERM that antagonizes ERβ may cause a shift from an immunosuppressed TIM to an immune-activated TIM, while an agonist of ERα activity may improve prognosis (figure 2).
ER isoforms are expressed in different locations in PC. ERs exert different roles in PC cells and the TIM, for example, an ERα-dominant phenotype in PC cells contributes to therapeutic resistance, while an ERβ-dominant phenotype in the TIM induces the suppression of immunotherapies.
Patients with PC may benefit from the use of SERMs, including raloxifene, in combination with anti-PD1/PD-L1 checkpoint immunotherapy, or TGF-β or Wnt antagonists.
Uncovering the optimal activity profile of SERMs should include (1) engineering a novel SERM that is easily attached to or highly selective to PC; (2) designing highly selective SERMs for ERα and ERβ; and (3) developing a targeted delivery system to exert downstream effects.
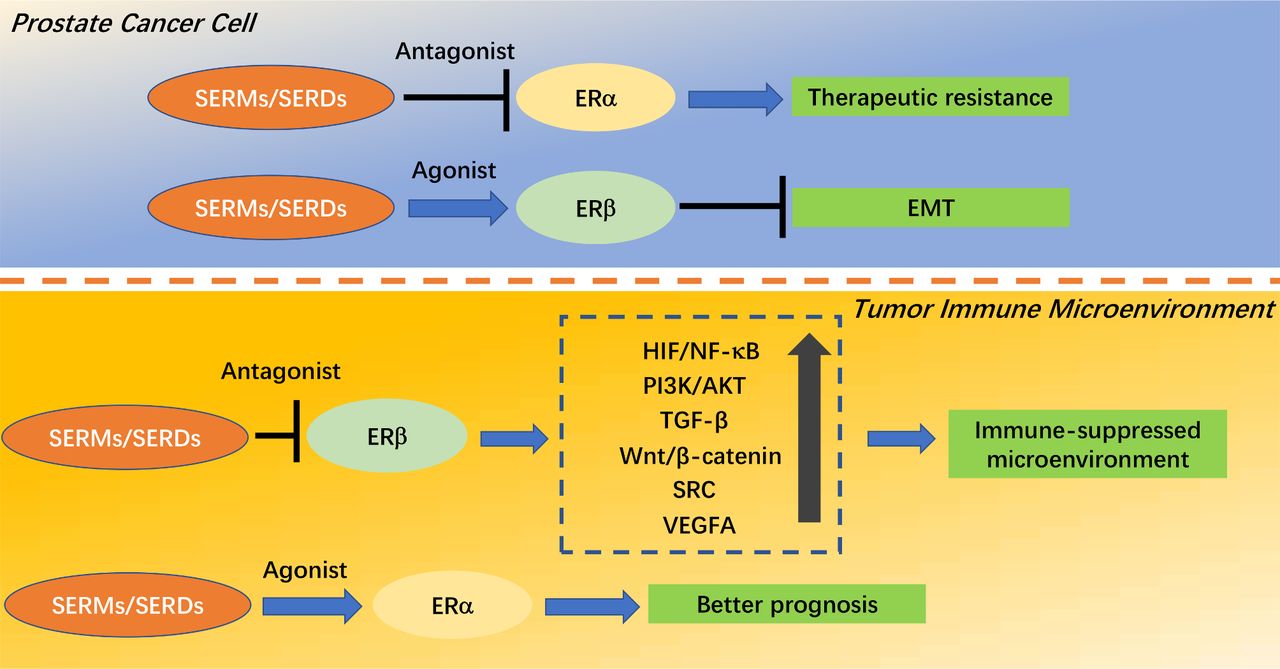
Downstream effects of estrogen receptors (ERs) and regulatory modes of selective estrogen receptor modulators (SERMs)/selective estrogen receptor degraders (SERDs) in prostate cancer (PC). In the top panel, SERM/SERDs which antagonize ERα would reverse resistance, while an agonist of ERβ activity would reverse epithelial-mesenchymal transition (EMT) in PC cells. In the bottom panel, SERMs/SERDs which antagonize ERβ would shift the immunosuppressed tumor immune microenvironment (TIM) to an immune-activated TIM, while an agonist of ERα activity would improve prognosis. VEGFA, vascular endothelial growth factor A.
Discussion
ERs promote PC development/progression
ERs may promote PC development and progression, whereas exogenous estrogen is a treatment option currently used to inhibit the development of PC. Multiple signaling pathways are involved in the process. Both ERα and ERβ activation promotes migration, invasion and anchorage-independent growth of PC-3 cells. Their activation upregulates β-catenin expression and contributes to cell proliferation. These effects are blocked by PKF 118–310, a compound disrupting the complex β-catenin/TCF/LEF. PKF 118–310 also inhibited the upregulation of ERs-induced VEGF A.72 ERs activate both the SRC and PI3K/AKT signaling pathways, enhancing the tumorigenic potential, cell proliferation, migration, invasion and tumor formation of PC.16 Moreover, ERs can also intersect with the HIF/NF-kB signaling pathway in PC.73 ERs-mediated long non-coding RNAs, including NEAT1, H19, MALAT1 and HOTAIR to promote PC development and the acquisition of the CRPC phenotype.74 The progressive emergence of the ERα combined with a series of genes that are regulated by ERα during CRPC progression, including progesterone receptor, PS2, TMPRSS2-ERG fusion and NEAT1 indicates that ER signaling can bypass the AR for tumor growth.14
Immunogenic potential of SERMs/SERDs in PC
PC is considered a cold tumor due to the limited number of tumor-associated antigens, a suppressive TIM and resistance to ICBs.75 PC grows slowly compared with other types of cancers and becomes a potential target for immunotherapy. However, previous clinical trials associated with various types of immunotherapies achieved minimal effects in metastatic CRPC. These limitations included an immunosuppressive TIM leading to limited efficacy of immunotherapy. The TIM associated with PC decreases the number of tumor-infiltrating immune cells and increases the number of immunosuppressive cells, including TAM, Tregs and MDSCs. Moreover, a low tumor mutation burden, PD-L1 expression and less somatic mutations induce fewer responses to ICBs or PD-1/PD-L1 inhibitors. The upregulated expression of TGF-β is also a limitation, as blocking TGF-β combined with ICBs increases Th1 and CD8 T cells, and promotes the regression of metastatic CRPC.76 Thus, shifting from an immunosuppressed TIM to an immune-activated TIM-based immunotherapy is key for the successful treatment of PC. Immunotherapies may also be combined with ADT, radiotherapy, chemotherapy and immunogenic agents, highlighting that personalized and combination therapy may exhibit potential for the treatment of PC.
As previously described, SERMs and SERDs have been used to enhance the immunogenicity of BC and improve the antitumor immunity. SERMs and SERDs effectively promote the expression of α-lactalbumin, c-erbB-2 and ICAM-1, which boost the cytotoxicity of NK cells, bridge and form the immunological synapses, reduce the number of MDSCs and Tregs, and increase the infiltration of DCs, and CD8 +and CD4+T cells.64–66 69 Tamoxifen and toremifene upregulate the expression levels of IFN-related genes and TNF-R2, which inhibit the activation of JNK and promote T cell proliferation.70 77 SERMs and SERDs abolish estrogen-induced tumor immunetolerance and improve the efficiency of anti-PD-L1 therapy.67 68 Numerous clinical trials evaluating the efficiency of ICBs in combination with SERMs and SERDs in BCs are ongoing.54 These agents, in combination with immunotherapeutics, are the current focus of research for the successful treatment of PC. Moreover, SERMs and SERDs may enhance immunogenicity by improving the expression of immunotherapeutic targets; SERMs and SERDs may increase the infiltration of NK cells, neutrophils, DCs, CD8 + and CD4+T cells, and reduce the counts of MDSCs and Tregs; and SERMs and SERDs may upregulate the expression levels of ICAM-1, IFN and TNF-R2, and inhibit the activation of JNK to promote cellular immune responses. Moreover, SERMs and SERDs may abolish tumor immunetolerance in combination with anti-PD-L1 therapy and ICBs. Results of a previous study demonstrated that fulvestrant downregulated AR expression and diminished androgenic responses in PC LNCaP cells, by the direct repression of AR gene transcription.78
Conclusion and perspective
Androgen-targeted endocrine therapy- and chemotherapy-based drug treatment models is not largely enough for impeding PC progression and could not bring overall benefits for patients. An increasing number of ER-targeted therapies and immunotherapies targeting PC and adverse effects followed by first-line therapies have entered clinical trials. A summary of 14 clinical trials on SERMs and SERDs focusing on PC prevention and treatment are summarized in table 1. A total of 192 clinical trials focusing on the use of immunotherapy for the treatment of PC were found in a keyword search on https://clinicaltrialsgov. According to our hypotheses, the ER signaling pathway may regulate the TIM, and immunotherapy-based strategies combined with selective SERMs may be the future direction for adjunctive therapy of PC. Further clinical trials based on immunotherapies and SERMs are required.
Updated clinical trials associated with the application of SERMs/SERDs, combined with other agents in treatment and prevention of PC or adverse effects followed by first-line therapy
Changing the immune microenvironment of PC from cold to hot remains an ongoing problem. The majority of clinical trials that aim to develop a single immunosuppressive target result in limited therapy efficacy.79 According to the stromal status of the TIM, PC may be divided into immune-activated and immunosuppressive subtypes. Moreover, the WNT/TGF-β signaling pathway and the extracellular matrix were highly concentrated in the immunosuppressive group. Therefore, immunosuppressive patients may benefit from ICB therapy in combination with TGF-β inhibitors.80 The present review indicated that a combination of immunotherapy-based strategies with SERMs, TGF-β or Wnt antagonists requires further investigation for the development of future treatment options for PC (figure 3).

{kind=link}
{kind=link}
{kind=link}
Schematic diagram of a novel immunotherapy-based comprehensive strategy combined with SERMs/SERDs, transforming growth factor-β (TGF-β) or Wnt antagonists for PC-targeting therapy. The SERMs/SERDs increase immune cell infiltrations, including natural killer (NK) cells, CD8 +T cells, CD4 +T cells, dendritic cells (DCs) and neutrophils, and decrease immunosuppressive cancer associated fibroblasts (ICAFs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) by altering the activity of downstream pathways and synergizing with other agents.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors DT wrote and reviewed the MS.
Funding This work was supported by DT, Chongqing Science and Technology Commission (Basic and Frontier Research Project; grant no, cstc2018jcyjAX0645) and Chongqing Municipal Health and Health Committee (Science and Health Combined Medical Research Project; grant no, 2018QNXM041).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.