Article Text

Abstract
Multiple myeloma (MM) is a cancer of plasma cells in the bone marrow (BM) and represents the second most common hematological malignancy in the world. The MM tumor microenvironment (TME) within the BM niche consists of a wide range of elements which play important roles in supporting MM disease progression, survival, proliferation, angiogenesis, as well as drug resistance. Together, the TME fosters an immunosuppressive environment in which immune recognition and response are repressed. Macrophages are a central player in the immune system with diverse functions, and it has been long established that macrophages play a critical role in both inducing direct and indirect immune responses in cancer. Tumor-associated macrophages (TAMs) are a major population of cells in the tumor site. Rather than contributing to the immune response against tumor cells, TAMs in many cancers are found to exhibit protumor properties including supporting chemoresistance, tumor proliferation and survival, angiogenesis, immunosuppression, and metastasis. Targeting TAM represents a novel strategy for cancer immunotherapy, which has potential to indirectly stimulate cytotoxic T cell activation and recruitment, and synergize with checkpoint inhibitors and chemotherapies. In this review, we will provide an updated and comprehensive overview into the current knowledge on the roles of TAMs in MM, as well as the therapeutic targets that are being explored as macrophage-targeted immunotherapy, which may hold key to future therapeutics against MM.
- tumor microenvironment
- macrophages
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The bone marrow (BM) microenvironment plays a key role in the development and progression of multiple myeloma (MM). Tumor-associated macrophages (TAMs) within the BM niche have recently captured the attention of researchers as a potential therapeutic target, given the plasticity and range of functionality inherent to this cell type. Prior reviews focusing on TAMs in MM have discussed mechanisms of action for which they support MM progression, as well as potential treatment options for other forms of malignancies that take advantage of these macrophages.1–4 In this review, we will provide updated and comprehensive insight into the current knowledge on the roles of TAMs, as well as the therapeutic targets that are being explored as macrophage-targeted immunotherapy for MM.
Multiple myeloma
MM is a cancer of plasma cells in the BM and represents the second most common hematological malignancy in the world.5 In the past decade, therapeutic breakthroughs such as proteasome inhibitors (PIs), immunomodulatory drugs, and antibody-based therapeutics have substantially expanded the number of treatment regimens available for patients in all stages of MM.6 However, MM remains to be incurable because almost all patients eventually relapse and/or become refractory to treatment, and relapsed/refractory MM (RRMM) has a lowered median survival of only 5–15 months.7
Development of MM is preceded by two asymptomatic precursor stages—monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM). The transition between healthy cells, MGUS, SMM, and MM, as well as the progression of MM, are heavily influenced by the development of the BM niche.8
The MM tumor microenvironment (TME) within the BM niche consists of a wide range of elements such as hematopoietic stem cells, progenitor cells, mesenchymal stromal cells, endothelial cells, immune cells, osteoblasts, osteoclasts, adipocytes, extracellular matrix proteins, and growth factors.9 10 TME components have been shown to play important roles in supporting MM disease on multiple levels, including MM progression, survival, proliferation, angiogenesis, as well as drug resistance.8 11 12 Together, the TME fosters an immunosuppressive environment in which immune recognition and response are repressed.13–15 Therefore, the BM niche is home to promising therapeutic targets for MM, and targeted reprogramming of protumor TME components may hold key to future therapeutics for MM. This review focuses on the crosstalk between macrophages and MM tumor cells, which have been a particularly promising area of study for MM and other malignancies.
Macrophage polarization and functions
Macrophages are a major component of the innate immune system and can be found across tissue types.16 The origin of tissue macrophages vary; some residential macrophages derive from embryonic yolk sac or fetal liver, while the adult macrophage population are understood to derive from the monocyte precursor through hematopoiesis.17 Mature macrophages in the human body are identifiable by the CD68, CD163, CD16, CD312, and CD115 markers.16 18
Macrophages are recognized for their wide variety of functions19; they are especially well known for their ability to phagocytose pathogens and apoptotic cells, but their contribution to the immunity and homeostasis of the body goes far beyond phagocytosis.20 Macrophages are highly plastic, and can activate and polarize for a specific role depending on environmental cues.21 It is widely accepted that polarized macrophages can be classified on a spectrum of M1 and M2 phenotypes.
The classically activated M1 macrophages, which are stimulated by interferon-γ (IFNγ) or lipopolysaccharide, are identifiable by their proinflammatory properties and are involved in the phagocytic and immune response.22 M1 macrophages express inducible nitric oxide synthase and reactive oxygen species, which are both proinflammatory and aid in the killing of pathogens.23 24 These activated macrophages release inflammatory cytokines such as interleukin (IL)-1, IL-6, IL-8, IL-12, and tumor-necrosis factor α (TNFα) that aid in the immune response.21 The M1 proinflammatory macrophages are also known to be professional antigen-presenting cells (APCs), where they phagocytose and present fragments of antigen on its surface major histocompatibility complex (MHC) class II, bridging to the adaptive immune system for a more efficient recognition of pathogens.18
The alternatively activated M2 macrophages are stimulated by IL-4. They are anti-inflammatory and participate in immunosuppression and wound healing. They secrete vascular endothelial growth factor (VEGF) and transforming growth factor-β (TGF-β) which promote angiogenesis and fibroblast activation at the wound site.25 26 Unlike M1 macrophages, M2 macrophages have minimal antigen-presenting ability. Variations in M2 macrophages have been observed in vitro, suggesting that M2 macrophages can be subdivided into M2a, M2b, or M2c subtypes, thoroughly reviewed elsewhere.27
Tumor-associated macrophages
TAMs are an important population of macrophages that reside in the tumor site in large numbers and are heavily influenced by the TME.1 Rather than contributing to the immune response against tumor cells, TAMs are often found to exhibit protumor properties including chemoresistance, tumor proliferation and survival, angiogenesis, immunosuppression, and metastasis.28 29
TAMs originate from circulating monocytes recruited into the tumor site, attracted by cytokines released by the tumor, including VEGF, colony stimulating factor (CSF)-1, CXC motif chemokine ligand (CXCL)-12, and the CC chemokines, particularly CC motif chemokine ligand (CCL)-2.2 3 Common TAM biomarkers include surface markers CD163 and CD206, expression of arginase, as well as production of VEGF, IL-10, and matrix metalloproteinases (MMPs).30 Due to their striking similarity in function and phenotype, TAMs are often paralleled with M2-like macrophages.31 TAMs secrete immunosuppressive cytokines such as IL-10 and TGF-β, which combine to reduce T cell functionality and contribute to the immunosuppressive TME.32
The role of TAMs in MM
TAMs are a significant component in the BM of MM patients, constituting around 10% of the BM.33 The prevalence of TAMs in MM has resulted in heightened attention invested into understanding the interplay between MM cells and TAMs in order to identify novel immunotherapy targets. In this section, we will comprehensively discuss the various ways that TAMs influence MM pathophysiology, including proliferation and survival, angiogenesis, immunosuppression, and drug resistance (figure 1).
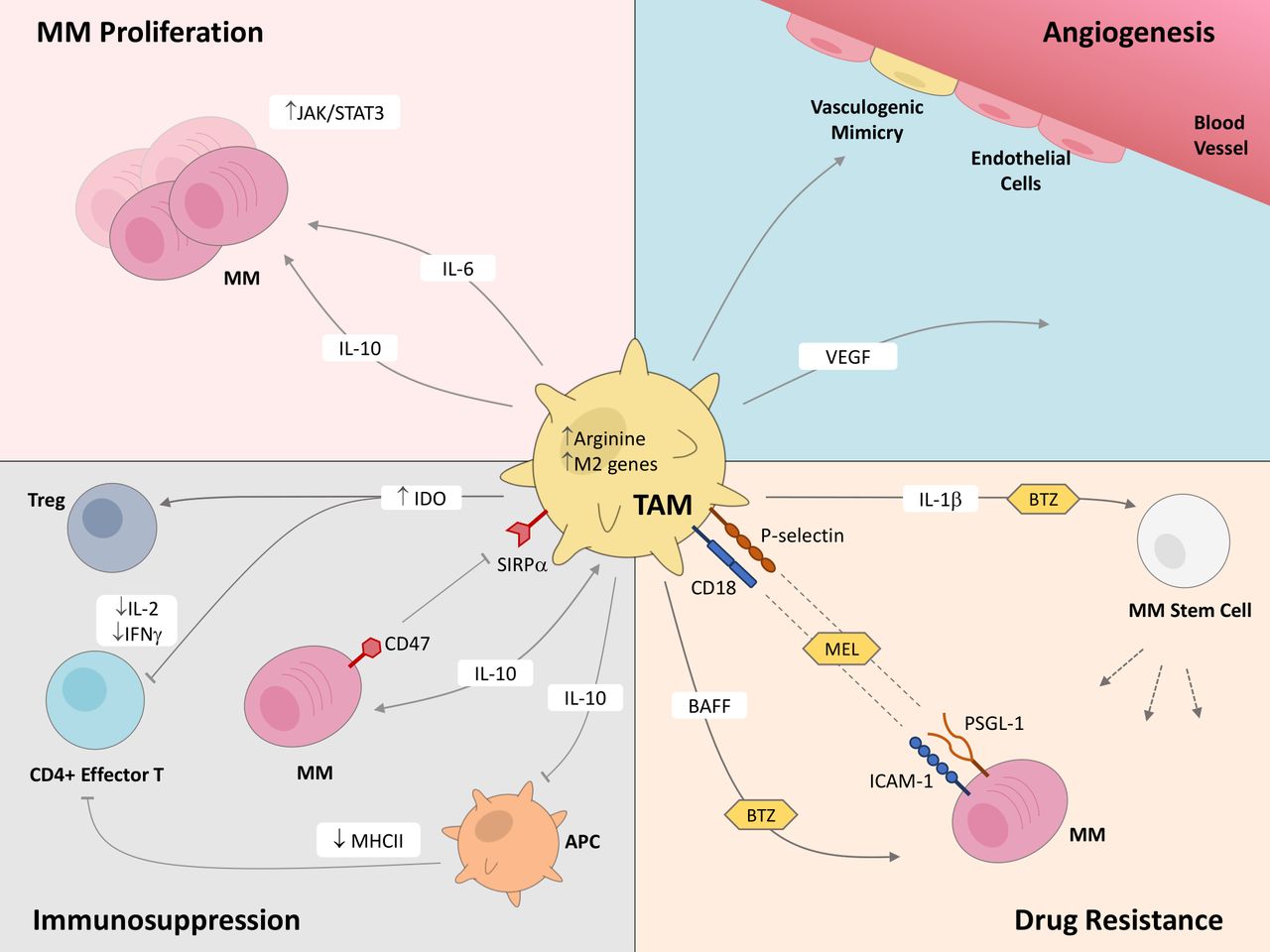
Tumor-associated macrophages (TAMs) play critical roles in multiple myeloma (MM) disease progression. This figure highlights the known effects that TAMs exert on MM cells and cells within the MM tumor microenvironment, through both secretion of molecules and contact-based surface interactions, which support proliferation and survival, angiogenesis, immunosuppression, and drug resistance in MM. APC, antigen presenting cell; BAFF, B-cell activating factor; BTZ, bortezomib; ICAM-1, intercellular adhesion molecule 1; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon-γ; IL, interleukin; JAK/STAT3, Janus kinase/signal transducer and activator of transcription proteins; MEL, melphalan; MHCII, major histocompatibility complex class II; PSGL-1, P-selectin glycoprotein ligand-1; SIRPα, signal-regulatory protein α; VEGF, vascular endothelial growth factor.
MM proliferation and survival
The MM TAMs in the BM microenvironment have been reported to heavily support MM proliferation and survival. Increased infiltration of macrophages to the tumor site is characteristic in MM patients compared with healthy subjects, and that heightened number of CD163 or CD206 TAMs is a negative prognosis marker in MM patients.34 35 Additionally, in a recent effort to identify the immune landscape in various stages of MM, it was shown that TAMs numbers dramatically increase in patients with an aggressive form of MM.36 It was also revealed that TAMs were among the most correlative informational marker for therapeutic response in patients who underwent standard-of-care chemotherapy treatment; patients not achieving complete remission had higher frequencies of TAMs, and the response to treatment positively correlated with the number of TAMs at time of diagnosis.36
Additionally, soluble CD163 in patient serum was associated with poor survival outcomes; the BM microenvironment had localized production of soluble CD163, further indicating the importance of macrophages in supporting MM proliferation and survival outcomes.37
Mode of action behind TAM-supported MM growth has also been extensively explored under controllable conditions using murine and cell line models. Multiple mechanisms have been revealed regarding TAM supported MM proliferation, especially through prolific secretion and regulation of various cytokines. Most notably, TAMs have heightened IL-6 and IL-10 secretions, and inhibited IL-12 and TNF-α secretions.38
IL-6 is a key cytokine that has pleiotropic functions in both inflammation and immunity. In the context of cancer, IL-6 has been shown to be critical for providing survival benefits to variety of tumor types.39 In MM patients, elevated IL-6 level correlates with adverse prognosis.40 Additionally, IL-6-deficient mice were completely resistance to disease development, showing the essential role of IL-6 in myeloma growth and survival.41 Ex vivo interrogation revealed myeloid cells being the major IL-6 secreting cell type in the TME42; in vitro data demonstrated that TAMs support MM cell survival through activation of the IL-6/JAK/STAT3 pathway.43 Coculture of TAMs with 5T33MM murine MM cells provided survival benefits for the myeloma cells, mediated through activation of STAT3 pathway in 5T33MM cells.44 STAT3 has also been shown to be phosphorylated at a higher level in CD11b+cells of MM-bearing mice compared with naïve mice.45 Thus, the interplay of JAK/STAT activation in both MM and TAMs relates to MM survival and pathogenesis.
Furthermore, it has been reported that IL-6 leads to the production of IL-10, which also plays an important role in the survival and proliferation of MM.38 46 In MM patients, high IL-10 serum level correlates with disease prognosis,47 suggesting IL-10’s clinical significance in MM pathogenesis and progression.
Angiogenesis
Proximity to vasculature is necessary for proliferation and survival of MM by providing oxygen and nutrients. BM neovascularization in MM supports disease progression, and more evidence has been recovered to suggest that TAMs in the BM microenvironment play a role through angiogenic and vasculogenic activities.48 CD206 +Tie2+macrophages are found to correlate with increased proangiogenic cytokines and microvessel density in an MM mice model.49 IL-10 secreted by MM-associated TAMs positively correlates with angiogenic cytokines and proliferation markers.50
The main player in angiogenesis is VEGF. On its own, MM can direct angiogenesis through its expression of VEGF and secretion of the corresponding protein.51 On the other hand, macrophages can also express and secrete angiogenic factors, such as VEGF, CCLs, and MMPs.29
It was reported that TAMs and MM cells could synergistically promote human umbilical vein endothelial cells (HUVECs) proliferation, migration, and tube formation in vitro, and that VEGFA released from both cell types was crucial to HUVEC tube formation ability.52 Additionally, MM-associated macrophages exposed to VEGF and basic fibroblast growth factor began to mimic vasculature by acquiring endothelial cell markers and forming capillary-like vessels.53 Thus, macrophages aid in angiogenesis in MM through both direct and indirect means.
Immunosuppression
In addition to promoting tumor progression and angiogenesis, TAMs are also known to directly influence the development of an immunosuppressive TME in many cancer types. As a member of the innate immune system, macrophages are essential in coordinating with the adaptive immune system. Evidence has been reported that TAMs participate in modifying immune cells to increase presence of immunosuppressive cell types while decreasing antitumor cell types.54 In MM, a few mechanisms have been reported, mainly pertaining to suppression of effector T cell activity.
MM-primed macrophages decreased T cell proliferation and activation, through downregulation of IFN-γ secretion.55 In a recent effort investigating BM immune landscape changes in MM disease stages compared with healthy donors using single-cell RNA sequencing, it was revealed that dysregulation of MHC class II in CD14 +monocytes conferred T cell suppression.56
IL-10 is a key immunosuppressive cytokine mainly secreted by myeloma-associated macrophages that participates in an array of tumor supportive activities, such as tumor growth, angiogenesis, and disease progression.50 57 IL-10 is known to inhibit expression of MHC class II and production of proinflammatory cytokines in APCs, which in turn limit effector T cell functions.32
Indoleamine 2,3-dioxygenase (IDO) is an enzyme that degrades the essential amino acid tryptophan into kynurenine. IDO is known to inhibit effector T cells and promote differentiation of T regulatory cells (Tregs), thus inhibiting the immune response. In MM, MM secreted IL-32 increases production of IDO in macrophages, which inhibits immunogenic response through inhibition of CD4+ T cell growth, IL-2, IFNγ, and TNFα production.58
Finally, macrophages play a direct part in immune evasion via the macrophage immune checkpoint CD47-SIRPα. It has been shown that many solid and hematological malignancies overexpress the CD47 protein on the surface as a protective ‘self-marker’.59 Binding of CD47 to signal-regulatory protein alpha (SIRPα) receptor on the surface of macrophages leads to downstream signaling within the macrophages, resulting in inhibition of phagocytosis activity, thus the CD47-SIRPα interaction is also known as the ‘don’t-eat-me’ signal.60 In MM, CD47 is overexpressed in CD138+ primary tumor cells compared with normal BM cells, and positively correlates with the stage of the disease,61–63 which lead to impaired immune recognition.
In summary, MM-associated macrophages secrete signaling molecules that consequently suppress T cell functions, in addition to actively participate in the CD47-SIRPα checkpoint.
MM drug resistance
One of the greatest difficulties with MM from a clinical standpoint is its tendency to develop resistance to treatment and relapse. In a clinical study, high TAM frequencies in the patient MM BM was found to negatively correlate with patient survival outcome with dexamethasone-containing chemotherapy.64 An increasing amount of research has been dedicated to seeking resistance mechanisms toward anti-MM drugs with contributions from MM TAMs.
Macrophages enable drug resistance in MM through both signaling and molecular contact based mechanisms. One mechanism for resilience toward bortezomib is through TAM secreted IL-1β, which resulted in increased number of MM-tumor-initiating cells. It was found that there was also an increase in the total number of proinflammatory macrophages (CD68+/CCR2+ in human and F4/80+/CD11c+ in mice) within the BM following bortezomib exposure.65 Another mechanism for bortezomib resistance was identified to be expression of B-cell activating factor (BAFF) by MM-influenced macrophages. BAFF was shown to activate MM survival through classical and alternative NF-κB pathways, which prevented bortezomib induced apoptosis.66
Additionally, contact-based mechanisms have also been reported. Myeloma/macrophage interaction pairs, P-selectin glycoprotein ligand-1(PSGL-1)/P-selectin and ICAM-1/CD18, were reported to mediate resistance to melphalan.67
Furthermore, macrophages also appear to respond to factors from the TME to help MM resist drug treatment. For instance, the CCL2 chemokine not only recruits macrophages and triggers their polarization to the M2 phenotype, but also stimulates macrophages to express the monocyte chemoattractant protein (MCP)-1-induced protein (MCPIP1), which improves protection of myeloma from bortezomib-induced apoptosis.68
Immunotherapeutic strategies targeting TAMs in MM
Cancer immunotherapies targeting immune cells or the interplay of immune and cancer cells have taken center stage in oncology research. T cell-targeted immunotherapies have transformed the treatment landscape for many cancers; however, these therapies are often limited by variable patient responses.
As more evidence uncovers the crucial role of macrophages in MM pathogenesis, efforts have been made to target MM-associated macrophages as a therapeutic approach to MM. A great number of strategies have been explored to target TAMs in a variety of cancers to overcome immunosuppressive barriers.69 70 Increasing numbers of preclinical studies are directed at blocking protumor functions of TAMs and/or promoting their antitumor activities.71 72
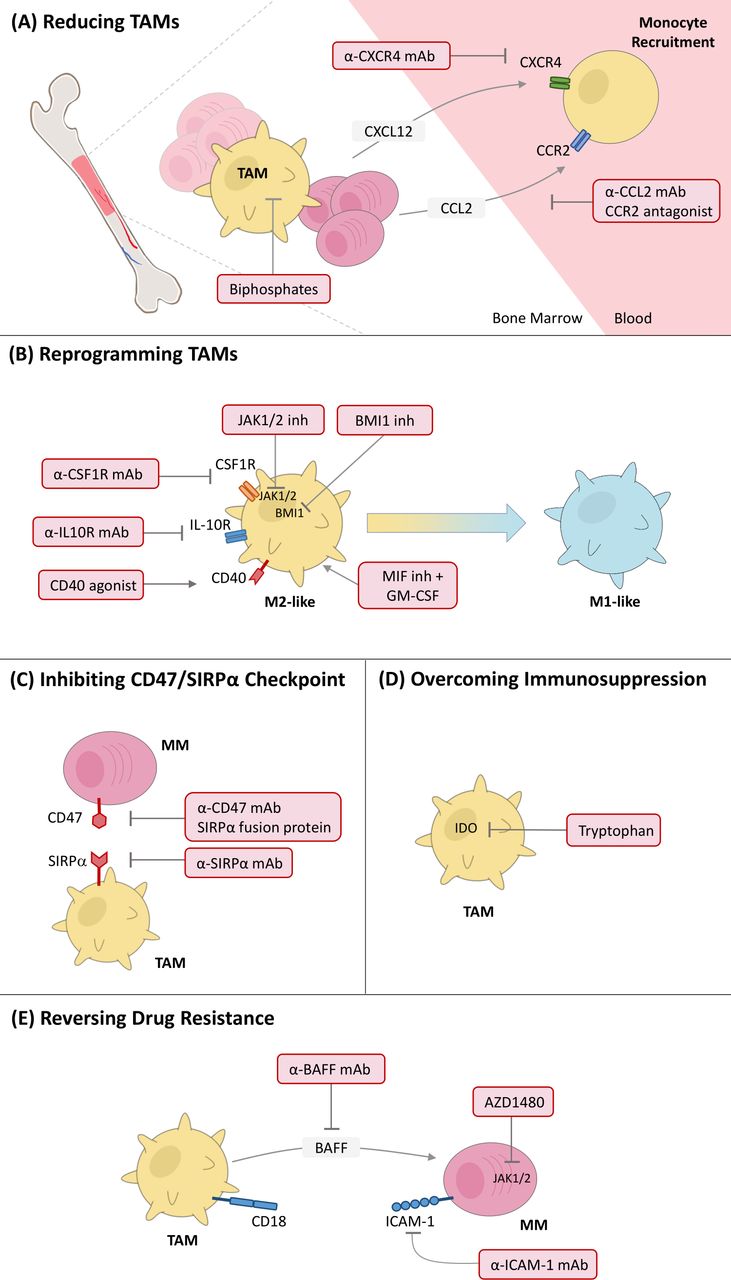
In this section, we will discuss different strategies used to counteract macrophages’ supportive role in progression of MM, including reducing TAMs, reprogramming TAMs, inhibiting CD47/SIRPα checkpoint, overcoming immunosuppression, and reversing drug resistance (figure 2).
{kind=link}
{kind=link}
Targeting TAMs as promising immunotherapy for MM. Currently reported preclinical strategies include (A) reducing TAMs, (B) reprogramming TAMs, (C) inhibiting CD47/SIRPα checkpoint, (D) overcoming immunosuppression, and (E) reversing drug resistance. CCL2, CC motif chemokine ligand 2; CCR2, CC motif chemokine receptor 2; CXCL12, CXC motif chemokine ligand 12; CXCR4, CXC chemokine receptor type 4; CSF1R, colony-stimulating factor-1 receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; mAb, monoclonal antibody; MIF, macrophage migration inhibitory factor; MM, multiple myeloma; TAMs, tumor-associated macrophages.
Reducing TAMs
Reducing the number of macrophages by direct killing is one strategy to reduce macrophage tumor support. Direct depletion of BM resident macrophages by clodronate-liposome administration before cancer cell inoculation resulted in impaired MM cell homing and tumor development.73 Moreover, a single dose of the clodronate-liposome led to a significant reduction of tumor burden in a C57BL/KaLwRijHsd murine model, suggesting that direct depletion of BM macrophages is a promising strategy for treatment of MM.
In contrast to direct killing of TAMs, controlling monocyte recruitment to the tumor and therefore reducing TAM replenishment is more extensively explored. The recruitment of monocytes/macrophages into tumors is primarily regulated by cytokines, chemokines, and growth factors that are derived from tumor and stromal cells in the TME.
CXCL12-CXCR4 signaling is an important macrophage recruitment mechanism to the MM BM.74 Research has shown that MM cells highly express the chemokine CXCL12; the CXCR4/CXCL12 axis not only contributes to MM cell adhesion and migration,75 but also promotes monocyte recruitment and differentiation toward a proangiogenic and immunosuppressive M2-like phenotype with heightened CD206 expression and IL-10 production.55 Inhibition of CXCR4 with a neutralizing antibody significantly suppressed monocyte recruitment to the BM.55
The CC chemokines are demonstrated to be crucial for macrophage infiltration in various types of cancers.76 CCL2, also well known as MCP-1, is a major player in driving chemotaxis of myeloid and lymphoid cells. It also maintains a scope of functions in various diseases beyond migration, making it an attractive therapeutic target.77 In MM, evidence shows that CCL2 is responsible for macrophage homing to the BM, as well as proliferation and polarization.78 Recently, it was demonstrated that MM-secreted CCL2 in the microenvironment elicits immunosuppressive MCP-1-induced protein (MCPIP1) expression in macrophages via the JAK2-STAT3 pathway, which promotes polarization toward the M2-like phenotype and protects MM cells from chemotherapy-induced apoptosis. Inhibition of CCR2 receptor using a monoclonal antibody (mAb) disrupted this protective effect.68 Targeting the CCR2/CCL2 axis has shown significant promise, preclinically and clinically, for blocking TAM recruitment in many cancer types including liver, pancreatic, and bladder cancers.79–81 Therefore, agents such as an anti-CCL2 mAb or CCR2 antagonist could be promising for disrupting macrophage recruitment in MM.
Reprogramming TAMs
Rather than depletion of TAMs, more targeted therapies are directed at blocking the protumor functions of TAMs, while promoting antitumor activities. Reprogramming TAMs to reduce their immunosuppressive M2 phenotype and promote their immunostimulatory M1 phenotype, represents a popular new field in MM treatment.
Gutiérrez-González et al reported a double treatment strategy in which they introduced the pro-M1 cytokine granulocyte–macrophage CSF (GM-CSF) while simultaneously blocking the pro-M2 cytokine macrophage migration inhibitory factor with an inhibitor.82 This dual treatment induced macrophage M1 genes and remarkable antitumor effects in vitro, performing better than GM-CSF treatment alone. Furthermore, this combination treatment resulted in macrophage-dependent therapeutic responses in a subcutaneous human MM cell line xenograft mouse model, in which TAMs isolated from treated mice had upregulated M1 and downregulated M2 markers compared with control animals. These results prove that fine tuning of TAMs polarization toward antitumor phenotype is a promising strategy for treatment of MM.82
In another report, Wang et al studied the potential of treating MM by targeting macrophages using CSF 1 receptor (CSF1R)-blocking mAbs.83 In vivo, CSF1R blockade was able to inhibit MM growth by partially depleting MM-associated macrophages and polarizing them to the M1 phenotype, as well as inducing a tumor-specific CD4 +T cell response. Moreover, the combination of CSF1R blockade and bortezomib or melphalan chemotherapy displayed additive therapeutic efficacy. These results suggest that targeting macrophages with anti-CSF1R mAbs may be a promising method to repolarize them to promote anti-myeloma immune and chemotherapy responses in MM patients.
Additionally, a JAK1/2 inhibitor Ruxolitinib has shown promise in suppressing the M2 phenotype in macrophages through reducing Tribbles homolog 1 protein kinase expression. In this preclinical study, treatment with ruoxolitinib resulted in decreased M2 and increased M1 polarization, both in vitro and in vivo, and was shown to overcome resistance to lenalidomide.84
Targeting CD40 is another popular strategy to activate macrophages in cancer. CD40 is a cell surface costimulatory protein found on APCs and is essential for their activation.85 Agonistic CD40 antibodies is a promising treatment of cancer patients through stimulating strong activation of innate and adaptive immunity, which are under clinical investigation in the solid tumor space.86 In myeloma, a preclinical study reported a macrophage repolarizing effect using sequential CD40 activation and TLR ligation. The immunotherapy with an agonistic anti-CD40 antibody in combination with TLR agonist CpG successfully elicited innate immune response toward MM ex vivo and in vivo.87
Lastly, targeting the underlying mechanism by which MM influence the development of TAMs represent a novel therapy. Several recent studies focused on elucidating the MM-derived elements which mediate protumor functions of MM-associated macrophages. Zhang et al showed that BMI1 protein to be a critical regulator in macrophages under MM influence.88 Sonic hedgehog secretion by myeloma was identified to be critical for BMI1 upregulation though the Hedgehog-Myc axis, and inhibitors for Hedgehog signaling attenuated BMI1 expression in macrophages. BMI1 was further demonstrated to promote macrophage proliferation and confer various promyeloma functions including angiogenesis, chemoresistance, and myeloma growth. Finally, in a 5T murine myeloma model, a BMI1 inhibitor PTC596 was able to decrease tumor burden and prolong mice survival through depletion of MM-macrophages.
We recently reported on the critical role of the IL-10/IL-10R pathway in MM-TAM interaction. IL-10 secretion from MM cells polarized macrophages toward heightened M2 phenotype, and these macrophages in turn supported MM proliferation and drug resistance. Inhibition of IL-10/IL-10R signaling between the two cell types using an IL-10R blocking antibody robustly reprogramed TAMs to lose their M2 phenotype, in vitro, in vivo, and ex vivo. Moreover, this resulted in the reversal of TAM supported MM proliferation and overcame drug resistance toward lenalidomide and dexamethasone.89
Inhibiting CD47/SIRPα checkpoint
Recently, immune checkpoints have been receiving heightened attention in the field of cancer therapy. Immune checkpoints are inhibitory mechanisms that cancer cells use to escape from recognition and killing by immune cells, effectively putting ‘breaks’ on the immune system.90 While T cell immune checkpoint therapies targeting PD-1/PD-L1 and CTLA-4 have shown progress in various cancers, it has not been effective in treating MM.91 Hence, targeting immune checkpoints in other immune cells such as macrophages has been a topic of discussion.
We and others have explored targeting CD47 using anti-CD47 mAbs to reverse immune suppression and enhance macrophage mediated phagocytosis and killing in MM.63 92 93 A plethora of CD47 targeting agents are under clinical investigations, including anti-CD47 mAbs and SIRPα fusion proteins.94
TTI-621 is a SIRPα-IgG1 Fc fusion protein being investigated in hematological malignancies in a phase 1b clinical trial (NCT02663518). In a preclinical study, TTI-621 effectively triggered macrophage-mediated phagocytosis of MM cells. In an MM xenograft model, it showed antitumor effects and further improved efficacy when combined with PI drugs bortezomib or carfilzomib.95
Similarly, the TTI-622 SIRPα-IgG4 Fc fusion protein is also under clinical investigation in phase 1a/1b study for advanced relapsed or refractory lymphoma or myeloma (NCT03530683). In MM, TTI-622 is being investigated as monotherapy and as combination therapy with carfilzomib and dexamethasone. Preliminary data in lymphoma patients show the agent is well-tolerated and has dose-dependent binding, leading to cases of durable responses.96
AO-176 is a humanized IgG2 anti-CD47 mAb that showed preclinical anti-MM activity in MM xenograft models,93 and the efficacy was further demonstrated to have combination effect with other anti-MM therapies including bortezomib, daratumumab, lenalidomide, or pomalidomide. Currently, a phase 1/2 clinical study is underway to evaluate AO-176 as monotherapy and as combination with bortezomib/dexamethasone in MM patients (NCT04445701).
Another phase 1a/1b clinical trial is underway to study SRF231, a fully human anti-CD47 mAb, as a monotherapy in patients with advanced solid and hematological malignancies (NCT03512340).
Additionally, a novel microRNA-based method in controlling CD47 has been reported using miR-155, a direct regulator of CD47. MiR-155 overexpression suppressed CD47 expression on myeloma cells and induced macrophage phagocytosis, which also reversed bortezomib drug resistance in cell lines.97
In summary, inhibition of the CD47-SIRPα axis represents a promising strategy in boosting macrophage immune surveillance activity, providing a great tool to combat the immunosuppressive environment of the MM BM niche.
Overcoming immunosuppression
Recently, preclinical strategies have been explored to relieve the immunosuppressive environment through targeting macrophage-related molecules. It was reported that MM cells mediate the production of immunosuppressive IDO in macrophages, specifically through binding to proteinase 3 (PR3) found on macrophages and activation of STAT3 and NF-κB pathways.58 Knockdown of PR3 or inhibition of STAT3 and NF-κB pathways in macrophages all reduced the capacity for IDO production in vitro. Moreover, inhibition of IDO restored CD4 +T cell proliferation and anti-inflammatory cytokine production. Hence targeting pro-IDO mediators such as PR3 may be a novel treatment to overcome macrophage mediated immunosuppression. Another study directly targeted IDO with a chemical inhibitor D,L-1-methyl-tryptophan in patient primary cells, which reverted Tregs expansion and T helper type 1 inhibition that were resulted by IDO.98
Reversing drug resistance
Much evidence has been published to indicate that macrophages contribute to myeloma cell survival and resistance to chemotherapeutic drugs such as melphalan and bortezomib. In addition to previously described TAM reprogramming methods to overcome M2 TAM supported drug resistance, a few strategies have been explored to target the crosstalk between TAMs and MM to overcome drug resistance.
ICAM-1 on MM directly interacts with CD18 macrophages and is important for conferring drug resistance to MM cells.67 99 100 As a result, ICAM-1 has been explored as a candidate for immunotherapy in MM. BI-505 is a mAb developed against ICAM-1 to enhance macrophage activation.101 In preclinical studies, BI-505 decreased cell growth and bone damage in the SCID-hu model. The in vivo efficacy of BI-505 was macrophage dependent with pronounced recruitment of monocyte/macrophages to the diseased BM.101 In a phase 1 dose-escalation study (NCT01025206), BI-505 was overall tolerable in RRMM patients.102 However, to date, BI-505 has shown limited clinical efficacy. Phase 2 clinical trials (NCT01838369) in patients with SMM showed no clinical relevant response,103 and another phase 2 trial evaluating BI-505 in conjunction with autologous stem cell transplantation (ACST) was put on hold by the U.S. Food and Drug Administration due to cardiovascular events (NCT02756728).
The STAT3 pathway is activated within MM cells when cocultured with IL-6 producing M2-polarized macrophages. This leads to protection of MM cells from apoptosis resulted by decreased cleavage of caspase-3.44 AZD1480, a potent and competitive small‐molecule inhibitor of JAK1/2 kinases, was used as a strategy to break this survival benefit. AZD1480 treatment in vitro was effective on both MM cell and myeloid cell populations, and abrogated the TAM-mediated MM cell survival by partially inhibiting resistance to bortezomib. Significant reduction of tumor load was observed with AZD1480 and bortezomib combination treatment in the murine 5T33MM model, and no significant killing of TAM populations were seen in vivo.44
To overcome macrophage-supported bortezomib resistance in myeloma, BAFF targeting by neutralizing antibody was investigated compared with bortezomib alone in a subcutaneous xenograft model. Anti-BAFF antibody in combination with bortezomib resulted in significantly delayed tumor growth compared with bortezomib alone, indicating BAFF as a favorable target for reversing bortezomib resistance in MM.66
Future directions and conclusions
In recent years, immunotherapy has taken center stage in the field of cancer treatment. Modulation of the immune system to induce a durable response represents an exciting idea with potential to elicit superior efficacy than traditional therapies. Much of the current attention focuses on the engagement of T cells, with PD-1/PD-L1 checkpoint inhibition being explored in over 3000 clinical trials.104 While there has been considerable success in T cell-targeted therapies, variable response and low T cell infiltration presents a road block.
Expanded investigation toward other immune players poses a novel area of research. Macrophages are crucial players of the innate immune system and present in high numbers in the TME. Thus, heightened attention is directed toward studying the role of macrophages in cancer. Increased amount of evidence reports prevalence of TAMs in various malignancies, and their tumor-supportive characteristics in promoting progression and drug resistance. Therefore, unearthing novel insights into mechanisms allowing TAMs’ tumor-promoting roles will provide new clues for future macrophage-targeted tumor therapy.
In this review, we provided an overview of the current knowledge of TAMs’ contribution in MM, including their role in proliferation, angiogenesis, immunosuppression, and drug resistance. We also reviewed classes of preclinical strategies targeting TAMs for MM immunotherapy, with many showing promising results. However, no clinical trials are in progress for immunotherapy strategy aside from those focusing on the CD47 checkpoint (table 1).
Clinical investigation of immunotherapeutic strategies targeting TAMs in MM
A number of key questions remain to be answered. For example, most of what is known regarding the role of macrophages in generating an immunosuppressive environment is about its effect on CD4 +effector T cells. In the future, more studies are warranted to investigate its effect on additional antitumor immune cell populations, such as CD8 +cytotoxic T cells, B cells, and NK cells, as well as other immunosuppressive populations such as regulatory T cells.
ACST remains to be a critical standard of care for younger myeloma patients. However, limited clinical data is available regarding the role of macrophages in efficacy of such therapy. In a clinical study investigating TAM numbers and response to therapy in MM patients, the response rate was not significantly different between high-TAM and low-TAM groups that received chemotherapy and ACST.35 In another study, a preclinical murine model of ACST showed the CSF-1R expression level on BM macrophages 6 weeks after ACST was significantly higher in animals that relapsed compared with those that were progression-free.105 More investigation is warranted for the correlation between macrophage population, including accumulation and phenotype, to the patient’s prognostic outcome.
In addition, cellular therapy is on the rise to take center stage in immunotherapy. While no genetically engineered macrophages have been reported in myeloma, such methods are being explored in the realm of solid tumors with considerable progress.106 107 In MM, one interesting approach was reported to use myeloid cells as cellular carriers of oncolytic for treatment of myeloma. The myeloid cells successfully localized to tumors and transferred infection to myeloma cells, prolonging survival in an MM xenograft model.108
Lastly, synergistic combination of TAM targeting strategies with popular T cell-targeted immunotherapies could be promising, potentially inducing robust improvements in the efficacy of checkpoint inhibition, T cell engagers, as well as CAR-T therapies by removing TAM-mediated immunosuppression.109
In conclusion, TAM-targeting therapy represents a promising treatment for cancer patients. This class of therapy could supplement current T cell therapies, impacting the current treatment regimen by overcoming unresponsiveness and drug resistance. More research is warranted to elucidate underlying molecular processes between macrophages, MM cells, and subpopulations of the TME, and strategies need to be verified in a clinical setting.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @kareemazab
Contributors JS conceptualization, writing, visualization; CP: writing, visualization; NG, SG, LZ, BL, OA, HB, YC, MM, and BM: writing; AKA: conceptualization, supervision. All authors read and agreed to the published version of the manuscript.
Funding This research was supported by an award from the National Institutes of Health (NIH) and the National Cancer Institute of the NIH (U54CA199092), as well as the Paula C. and Rodger O. Riney Blood Cancer Research Initiative Fund. JS was supported by the Spencer T. and Ann W. Olin Fellowship for Women in Graduate Study at the Washington University in St. Louis.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.