Article Text

Abstract
Background Angiosarcoma is a histologically and molecularly heterogeneous vascular neoplasm with aggressive clinical behavior. Emerging data suggests that immune checkpoint blockade (ICB) is efficacious against some angiosarcomas, particularly cutaneous angiosarcoma of the head and neck (CHN).
Methods Patients with histologically confirmed angiosarcoma treated with ICB-based therapy at a comprehensive cancer center were retrospectively identified. Clinical characteristics and the results of targeted exome sequencing, transcriptome sequencing, and immunohistochemistry analyses were examined for correlation with clinical benefit. Durable clinical benefit was defined as a progression-free survival (PFS) of ≥16 weeks.
Results For the 35 patients included in the analyses, median PFS and median overall survival (OS) from the time of first ICB-based treatment were 11.9 (95% CI 7.4 to 31.9) and 42.5 (95% CI 19.6 to 114.2) weeks, respectively. Thirteen patients (37%) had PFS ≥16 weeks. Clinical factors associated with longer PFS and longer OS in multivariate analyses were ICB plus other therapy regimens, CHN disease, and white race. Three of 10 patients with CHN angiosarcoma evaluable for tumor mutational burden (TMB) had a TMB ≥10. Five of six patients with CHN angiosarcoma evaluable for mutational signature analysis had a dominant mutational signature associated with ultraviolet (UV) light. No individual gene or genomic pathway was significantly associated with PFS or OS; neither were TMB or UV signature status. Analyses of whole transcriptomes from nine patient tumor samples found upregulation of angiogenesis, inflammatory response, and KRAS signaling pathways, among others, in patients with PFS ≥16 weeks, as well as higher levels of cytotoxic T cells, dendritic cells, and natural killer cells. Patients with PFS <16 weeks had higher numbers of cancer-associated fibroblasts. Immunohistochemistry findings for 12 patients with baseline samples available suggest that neither PD-L1 expression nor presence of tumor-infiltrating lymphocytes at baseline appears necessary for a response to ICB-based therapy.
Conclusions ICB-based therapy benefits only a subset of angiosarcoma patients. Patients with CHN angiosarcoma are more likely to have PFS ≥16 weeks, a dominant UV mutational signature, and higher TMB than angiosarcomas arising from other primary sites. However, clinical benefit was seen in other angiosarcomas also and was not restricted to tumors with a high TMB, a dominant UV signature, PD-L1 expression, or presence of tumor infiltrating lymphocytes at baseline.
- Immunotherapy
- Sarcoma
- Biomarkers, Tumor
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic
Angiosarcomas can have a high tumor mutational burden and/or a mutational signature of exposure to ultraviolet light. Responses to immune checkpoint blockade-based therapies have been noted anecdotally and in small prospective studies.
What this study adds
In the largest series to date of angiosarcoma patients treated with immune checkpoint blockade, only a minority of patients had a response but approximately one-third of patients derived clinical benefit. Benefit was seen across primary sites of disease, and traditional biomarkers of response (eg, tumor mutational burden, PD-L1, and tumor-infiltrating lymphocytes) were not sufficient to predict responses to immune checkpoint blockade.
How this study might affect research, practice or policy
Future research, ideally using a multicenter collaborative approach, will need to integrate histopathology, genomics, and transcriptomics to identify biomarkers that are predictive of response to immune checkpoint blockade in angiosarcoma.
Introduction
Angiosarcoma is a rare histological subtype of soft tissue sarcoma with an endothelial lineage of differentiation spanning significant clinical and molecular heterogeneity. It can arise in almost any anatomic location but frequently originates in the skin of the head and neck region, in the breast, or in soft tissue.1 Historically, its development has been linked to exposure to ionizing radiation or toxins such as vinyl chloride and to chronic lymphedema,2–4 but most commonly the underlying etiology remains unknown. Recurrent somatic mutations involving angiogenic signaling pathways, including KDR, PTPRB, and PLCG1, have been identified in about 40% of cases,5 6 while MYC amplification is the hallmark of most radiotherapy and chronic lymphedema-associated cases.7
Angiosarcoma typically follows an aggressive clinical course, with an estimated 5-year overall survival (OS) rate of 30%–43% and a median OS of 2.6–3.5 years. Patients with de novo metastatic disease have a median OS of 1 year or less.1 2 The treatment of choice for resectable disease is surgical excision, though local and distant recurrences are common. In patients with advanced disease, cytotoxic chemotherapy can be efficacious but generally does not provide durable clinical benefit. The median PFS and OS of anthracycline-based therapy are 4.9 and 9.9 months, respectively,8 and paclitaxel provides a median PFS and OS of 4 and 8 months, respectively.9
Immune checkpoint blockade (ICB) is a therapeutic modality of interest across various soft tissue sarcomas.10 11 Cutaneous angiosarcomas are known to be infiltrated with lymphocytes and to express the checkpoint proteins programmed cell death protein 1 (PD-1) and Programmed death-ligand 1 (PD-L1). Some studies have reported better prognoes for patients with high expression of these biomarkers, while others have called this association into question.12–18
A number of case reports and small case series have described impressive responses to ICB in patients with angiosarcoma, and a recent phase II study of ipilimumab plus nivolumab reported an overall response rate (ORR) of 25% in a cohort of 16 patients.16 18–22 Cutaneous angiosarcoma of the head and neck (CHN angiosarcoma) may have a high tumor mutational burden (TMB), likely related to a dominant mutational signature of ultraviolet (UV) light, which is hypothesized to explain some of the exceptional responses to ICB.21 Yet, according to some investigators, only a subset of CHN angiosarcomas have a high TMB with immune cell infiltration and not all CHN tumors respond to ICB, highlighting the complex and heterogeneous nature of this disease.23
To describe clinical outcomes of ICB-based therapy in angiosarcoma and to better understand patterns of response and resistance, we performed a retrospective analysis of data from patients treated with various ICB regimens and examined outcomes for correlation with the findings of targeted exome and whole transcriptome sequencing, as well as immunohistochemistry results where available. The objectives of the study were to estimate the clinical benefit of ICB-based therapy for treatment of advanced angiosarcoma and to identify prognostic or predictive biomarkers.
Methods
Patient selection and study design
The study subjects were patients with histologically confirmed angiosarcoma who were treated with ICB at Memorial Sloan Kettering prior to January 2021 and had clinical data available for review. Demographic, pathological, and clinical data were retrieved from the medical record of each patient. The following variables were included in the analysis: age, sex, race, ethnicity, date of diagnosis, primary site of disease, extent of disease at diagnosis (localized vs metastatic), date of ICB initiation, date of ICB cessation, reason for cessation of ICB, number of systemic therapies prior to ICB, vital status, and date of death or last follow-up. Primary disease was categorized by site of origin or presumed underlying etiology, such as prior exposure to ionizing radiation or the presence of chronic lymphedema at the site of primary disease. The cut-off date for clinical follow-up was March 8, 2021. To minimize potential biases associated with retrospective studies, we included all patients who received at least one dose of ICB, regardless of clinical outcome after treatment.
ICB regimens were grouped into three principal categories: ICB monotherapy (anti-PD-1 or anti-PD-L1 therapy alone), ICB combination therapy (anti-CTLA-4 plus anti-PD-1 therapy), and ICB plus other (anti-PD-1 or anti-PD-L1 agent plus a novel immunomodulatory therapy). ‘Other’ therapies, which had been studied in combination with ICB in various clinical trials, were bempegaldesleukin (pegylated interleukin-2 agonist), talimogene laherparepvec (T-VEC; oncolytic herpesvirus), epacadostat (indoleamine 2,3-dioxygenase-1 inhibitor), and tiragolumab (anti-TIGIT antibody). For patients who were treated on more than one clinical trial of ICB, only the first ICB regimen received was used in the analysis of clinical outcome.
MSK-IMPACT assay
Some of the patients included in the study had provided informed written consent to participate in a prospective tumor sequencing initiative using the Memorial Sloan Kettering Integrated Molecular Profiling of Actionable Cancer Targets assay (MSK-IMPACT).24 25 MSK-IMPACT is a hybridization capture-based next-generation sequencing platform of 341–505 exons and select introns, depending on the assay version. Genomic alterations were annotated using the OncoKB precision oncology knowledge base to identify functionally relevant variants.26 Variants of unknown significance (variants without annotation of predicted, likely, or known oncogenic relevance) were excluded from the analysis unless a gene was altered in the patient population at a high frequency (>15% of patients). Although not annotated as functionally relevant previously, we hypothesized that such genes may be relevant in this population of patients with a rare sarcoma.
For each sample, TMB was computed as total number of nonsynonymous mutations divided by total number of base pairs sequenced. Fraction of genome altered (FGA) was calculated as the sum of absolute log2 copy number occurrences >0.2 divided by the size of the genome profiled for copy number. Mutational signatures were extracted for samples with ≥15 single nucleotide polymorphisms using the COSMIC V.3 catalog of exome reference signatures and default parameters (https://github.com/mskcc/tempoSig).27 Detectable UV signatures were identified using the threshold of p<0.05 and a minimum of 1 observed mutation attributed to the signature, where the number of observed mutations was defined as the observed mutational signature fraction multiplied by the number of single nucleotide polymorphisms per sample. Pathway analyses were performed using previously curated signaling pathway definitions.28 These were extended to include DNA damage repair and epigenetic modifier pathways.29–31
Immunohistochemistry
On the clinical trials of pembrolizumab plus T-VEC and pembrolizumab plus epacadostat, paraffin-embedded tumor specimens were analyzed by QualTek Molecular Laboratories for PD-L1 and tumor infiltrating lymphocytes (TIL), as described previously.32 On the clinical trial of pembrolizumab plus bempegaldesleukin, PD-L1 expression and CD8 expression were examined by Mosaic Laboratories using the anti-PD-L1 rabbit clone antibody 28–8 and anti-CD8 mouse clone antibody C8/144B, and the percent positive cells in each sample was determined. In addition to the above samples from clinical trials, all patients were screened for availability of tissue for further immunohistochemistry analyses. For patients with such availability, the tissue samples were evaluated by an in-house expert soft tissue pathologist (CRA) for expression of CD8 +TIL. Samples were classified as ‘immune deserts’ if there was complete absence of TIL, ‘immune excluded’ if TIL were present exclusively at the tumor-stroma border, and ‘immune infiltrated’ if intratumoral lymphocytes were present.
RNA sequencing
Quantification and quality control of total RNA from nine samples was performed using the Agilent Bioanalyzer. Seven were baseline samples, and two were on-treatment samples. Selection of polyadenylated RNA and creation of a TruSeq library for these samples were performed according to instructions provided by Illumina (TruSeq Stranded mRNA Library Prep Kit; catalog no. RS-122-2102), with 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 System using a run of paired-end 100 base pair reads (HiSeq 3000/4000 SBS Kit; Illumina). On average, 41 million paired reads were generated per sample. Ribosomal reads represented 0.9%–20% of the total reads generated, and mRNA bases averaged 68%. RNA sequencing FASTQ files were aligned using STAR (V.2.7.0f)33 and Ensembl (V.75)34 software as part of an in-house RNA sequencing pipeline. Picard (V.2.22.0)35 software was used for quality control of the resulting BAM files. RNA expression was then quantified using the Kallisto (V.0.46.2)36 tool, and gene level expression was summarized using Ensembl (V.75). The number of normalized transcripts per million was calculated using the Sleuth (V.0.30) package (sleuth to matrix).37 The function HCPC in the FactoMineR package was used to compute hierarchical clustering on principal components to determine different clusters.38
To identify genes differentially expressed between patients with high PFS (≥16 weeks) and patients with low PFS (<16 weeks), we examined baseline samples as well as on-treatment samples from patients who did not have baseline samples available for analysis. Linear models with gene expression as the dependent variable, PFS as the independent variable, and sequencing batch effect and time point (baseline/on treatment) as covariates were applied to identify significantly expressed genes. Pathway enrichment analysis was performed using the pathEnrich function of the splineTimeR package in R39 and Hallmark datasets for 50 pathways from the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/). We used a list of genes differentially expressed between the high-PFS group and the low-PFS group to determine pathway enrichment.
Fluorescence in situ hybridization
Fluorescence in situ hybridization on interphase nuclei of tissue sections was performed by applying custom probes from bacterial artificial chromosomes covering MYC (RP11-440N18; 8q24.21:128 596 756-128 777 986)40 and CRKL (RP11-1058B20 and RP11-958H20; 22q11.21). Bacterial artificial chromosome DNA was isolated and labeled with different fluorochromes in a nick translation reaction. The slides were pretreated, denatured, and hybridized by probes. After overnight incubation, the slides were serially washed and mounted with 4′,6-diamidino-2-phenylindole in an antifade solution. Two hundred successive nuclei were manually scored using a Zeiss Axioplan fluorescence microscope (Zeiss, Oberkochen, Germany), controlled by Isis 5 software (MetaSystems, Newton, Mass, USA). MYC or CRKL amplification was defined as >10% of tumor cells with MYC or CRKL (to control for a centromeric probe ratio of >10) or as presence of tightly clustered signals characteristic of homogeneously staining regions.
Survival
A PFS of ≥16 weeks was categorized as indicative of durable clinical benefit.41 42 PFS was defined as the time from the first dose of ICB until the date of progression (in clinical trial patients this was determined by RECIST V.1.1 [http://recist.eortc.org]; in nontrial patients this was determined by the treating clinician), the date of treatment cessation due to toxicity, or death. Patients who stopped ICB for any reason other than clinical or radiological progression, toxicity, or death, were censored. OS was defined as the time from ICB initiation until the date of death or last contact.
Statistical analyses
Patients were divided into subgroups based on the primary site, presence of an oncogenic or a likely oncogenic genomic alteration, presence of a detectable UV signature, TMB, and FGA. TMB and FGA were tested both as continuous variables and by using the median as a cut-off. A cut-off of ≥10 was also used for TMB, previously defined as high TMB in angiosarcoma.21 Median, range, and IQR were used to describe continuous variables; count and percent were used for categorical variables. To compare variables across groups, the Fisher exact test was used for categorical variables and the Wilcoxon rank-sum test was used for continuous variables. Significant gene expression was determined using the Wald test. For assessment of differential gene expression, the Benjamini-Hochberg correction was used to adjust for the false discovery rate (Q value).43 Survival was analyzed using the Kaplan-Meier method, and log-rank p values are reported. Univariate and multivariate analyses were performed with Cox proportional hazard regression models. Statistical analyses were performed using R software (V.3.6.3). All tests were two sided, and p<0.05 was considered significant.
Results
Patient characteristics
Thirty-five patients with angiosarcoma were diagnosed between July 2010 and February 2020 and treated with ICB-based therapy between August 2014 and October 2020. Patient characteristics are listed in table 1. The median age at diagnosis was 69 years (IQR 56–73). Most patients identified as white (83%) and female (54%). The most common primary disease sites were CHN (n=14, 40%) and soft tissue or visceral (n=10, 29%). Five patients (14%) had prior radiotherapy at the primary site (two in the breast and one each in the bladder, pleura, and larynx), three (9%) had primary breast angiosarcoma without prior radiotherapy, and two (6%) had lymphedema-associated angiosarcoma (also known as Stewart-Treves syndrome.3 One patient had a history of Klippel-Trenaunay syndrome44 and a cutaneous primary tumor of the upper back. Nearly one-third of patients had metastatic disease at diagnosis. Thirty-two patients (91%) had received prior systemic therapy; the mean number of prior regimens was 2.6 (range 0–6).
Patient characteristics (n=35)
Nine patients (26%) received ICB monotherapy (pembrolizumab or durvalumab), nine received nivolumab plus ipilimumab, nine received nivolumab plus bempegaldesleukin, five (14%) received pembrolizumab plus T-VEC, two (6%) received pembrolizumab plus epacadostat, and one (3%) received atezolizumab plus tiragolumab. Of the 19 patients (54%) treated on a clinical trial, nine received nivolumab plus bempegaldesleukin, five received pembrolizumab plus T-VEC, two received pembrolizumab plus epacadostat, one received ipilimumab plus nivolumab, one received durvalumab, and one received atezolizumab plus tiragolumab.
Clinical outcome
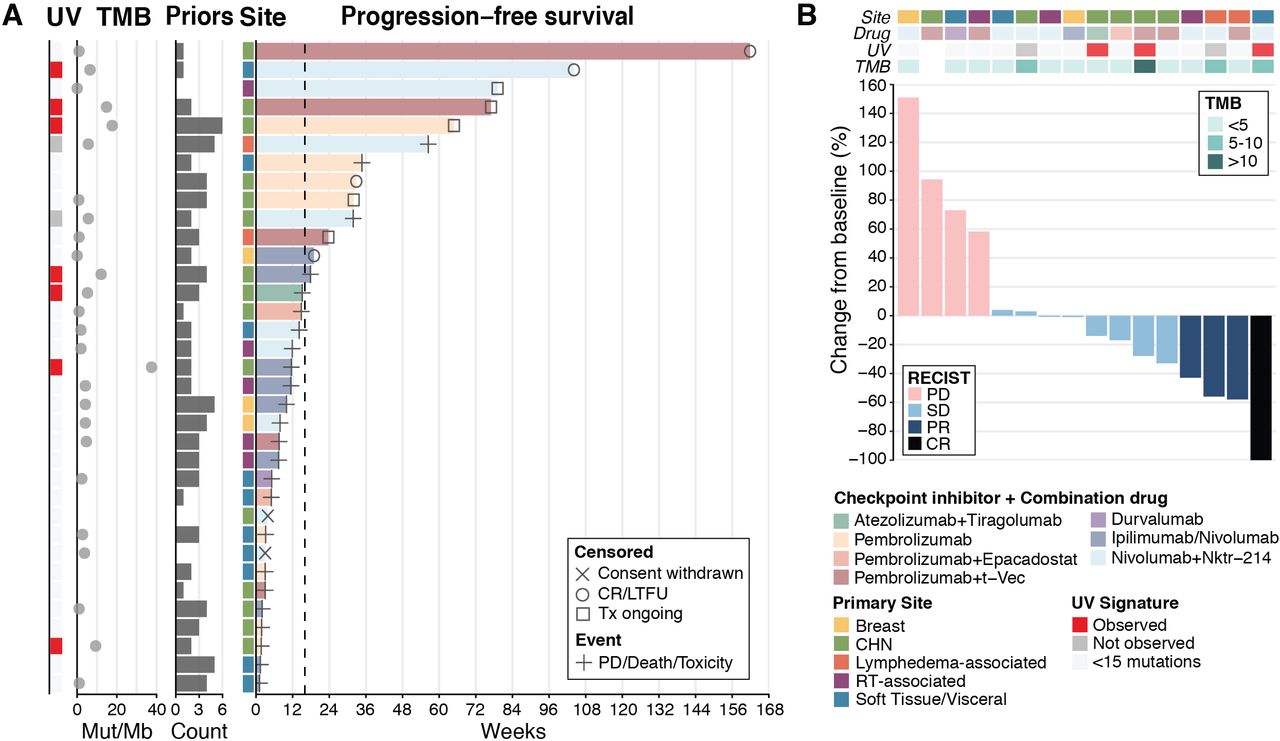
The median follow-up time for survivors was 33.5 months from the time of diagnosis and 8.2 months from the time of ICB initiation. The median PFS and OS from the time of first ICB-based treatment across all patients were 11.9 (95% CI 7.4 to 31.9) and 42.5 (95% CI 19.6 to 114.2) weeks, respectively (online supplemental figure S1A,B). Ten patients (29%) received only one dose of ICB because of rapid progression, clinical deterioration, death, or withdrawal of consent from a clinical trial. Among those who received more than one dose of ICB, median PFS and OS were 17.9 (95% CI 11.6 to not reached) and 52.4 (95% CI 41.1 to not reached) weeks, respectively (online supplemental figure S1C,D). Thirteen patients (37%) had durable clinical benefit, defined as PFS ≥16 weeks: six with a CHN primary tumor, two with a lymphedema-associated tumor, two with soft tissue or visceral disease (one in the vaginal wall and one in an unknown location), one with a breast tumor, one with a radiotherapy-associated tumor, and one with a cutaneous primary tumor of the upper back (figure 1A).
Supplemental material

Clinical outcomes in patients with angiosarcoma treated with ICB. (A) PFS (n=35). (B) Best objective response by RECIST 1.1 criteria in 16 patients treated on a clinical trial of ICB. CR, complete response; ICB, immune checkpoint blockade; LTFU, lost to follow-up; Nktr-214, bempegaldesleukin; Mut, mutations; PD, progression of disease; PFS, progression-free survival; PR, partial response; Priors, regimens received before ICB; RT, radiotherapy; SD, stable disease; TMB, tumor mutational burden; Tx, treatment; UV, ultraviolet.
Of the 19 patients treated on a clinical trial, 16 were evaluable for response by RECIST V.1.1 (2 withdrew consent and one had rapid disease progression). Four (25%) of the 16 patients had complete (n=1) or partial (n=3) response, eight (50%) had stable disease, and four (25%) had disease progression (figure 1B). One patient with stable disease had an unconfirmed partial response (33% tumor regression). Three of four patients with an objective response received nivolumab plus bempegaldesleukin. Clinical benefit by RECIST V.1.1 (objective response or stable disease) was seen across various primary sites and across various treatment regimens.
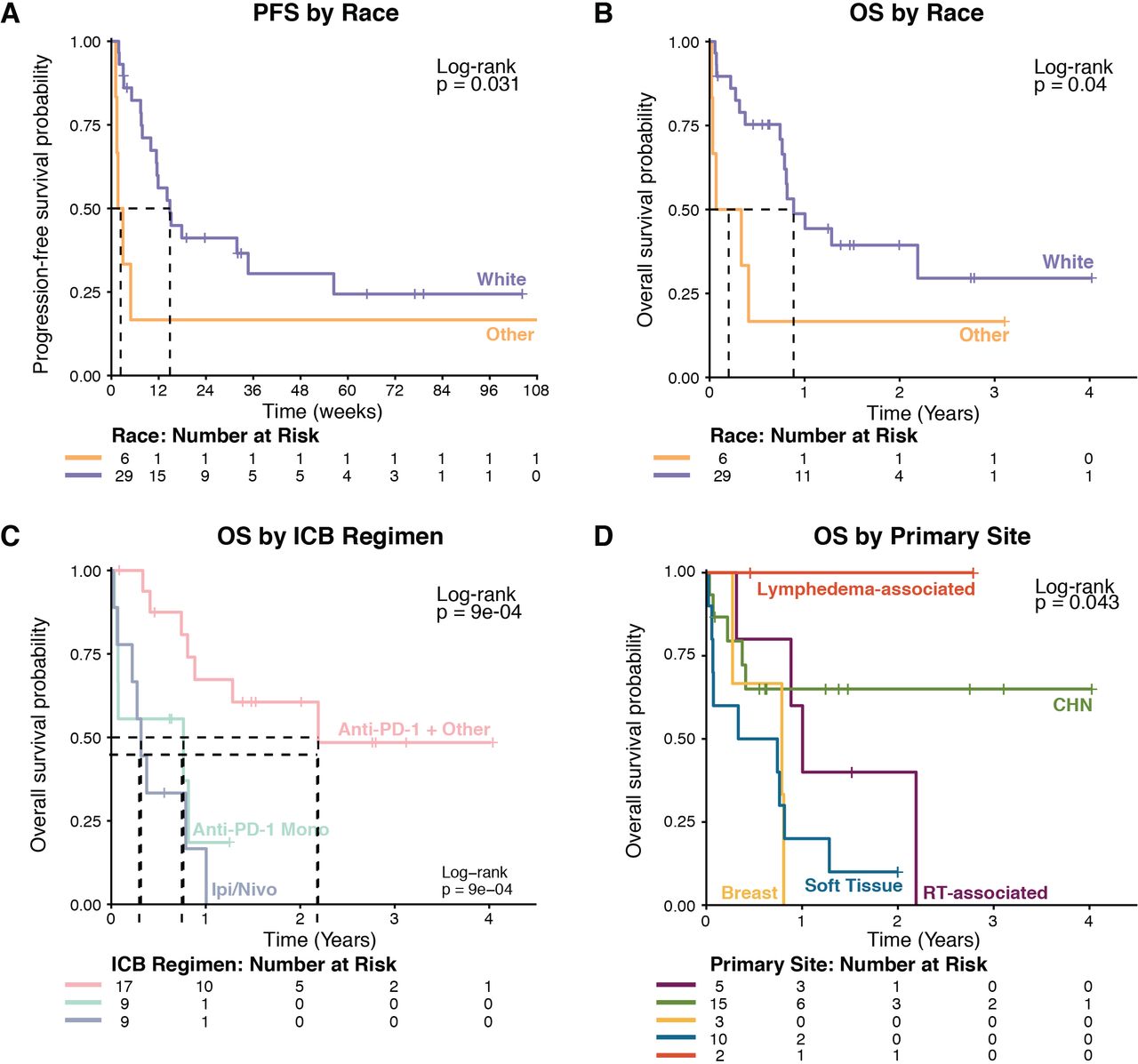
Univariate analysis was used to measure the association of clinical variables (primary site, ICB regimen, sex, race, and metastatic disease at diagnosis) with outcome in the whole cohort of 35 patients. Primary tumors were classified as CHN, soft tissue or visceral, radiotherapy associated, lymphedema associated, or breast. One patient with cutaneous disease of the upper back that was not associated with radiotherapy or lymphedema was grouped with CHN patients. Race was the only variable significantly associated with PFS (table 2, figure 2A). Race, ICB regimen, and primary disease site were significantly associated with OS (table 2, figure 2B–D). In multivariate analysis, ICB plus other regimens (when compared with ipilimumab plus nivolumab), cutaneous primary site, and white race were significantly associated with longer PFS. ICB plus other regimens, white race, and cutaneous primary site were significantly associated with longer OS (table 3).
Univariate analyses of PFS and OS from the time of initiation of ICB-based therapy

Kaplan-Meier survival curves with stratification by race, ICB regimen, and primary disease site. ICB, immune checkpoint blockade; IPI, ipilimumab; Nivo, nivolumab; OS, overall survival; PFS, progression-free survival; RT, radiotherapy.
Multivariate analyses of PFS and OS from the time of initiation of ICB-based therapy
Molecular analyses
MSK-IMPACT data were available for 26 patients. Sixteen (62%) of the 26 samples were from primary tumors, and the remaining samples were from metastatic foci. The most common oncogenic or likely oncogenic abnormalities were TP53 alterations (27% of patients), MYC amplifications (23%), and ATRX alterations (15%) (figure 3A). In addition, the CRKL gene on 22q11.21 was amplified in 27% of patients; this amplification is classified as a variant of unknown significance by OncoKB.26 Frequent genomic alterations were found in the epigenetic (38% of patients), TP53 (31%), RTK-RAS (27%), and DNA damage repair (23%) pathways across various primary sites (figure 3B). RTK-RAS pathway alterations were identified only in cutaneous tumors (online supplemental figure S2A).
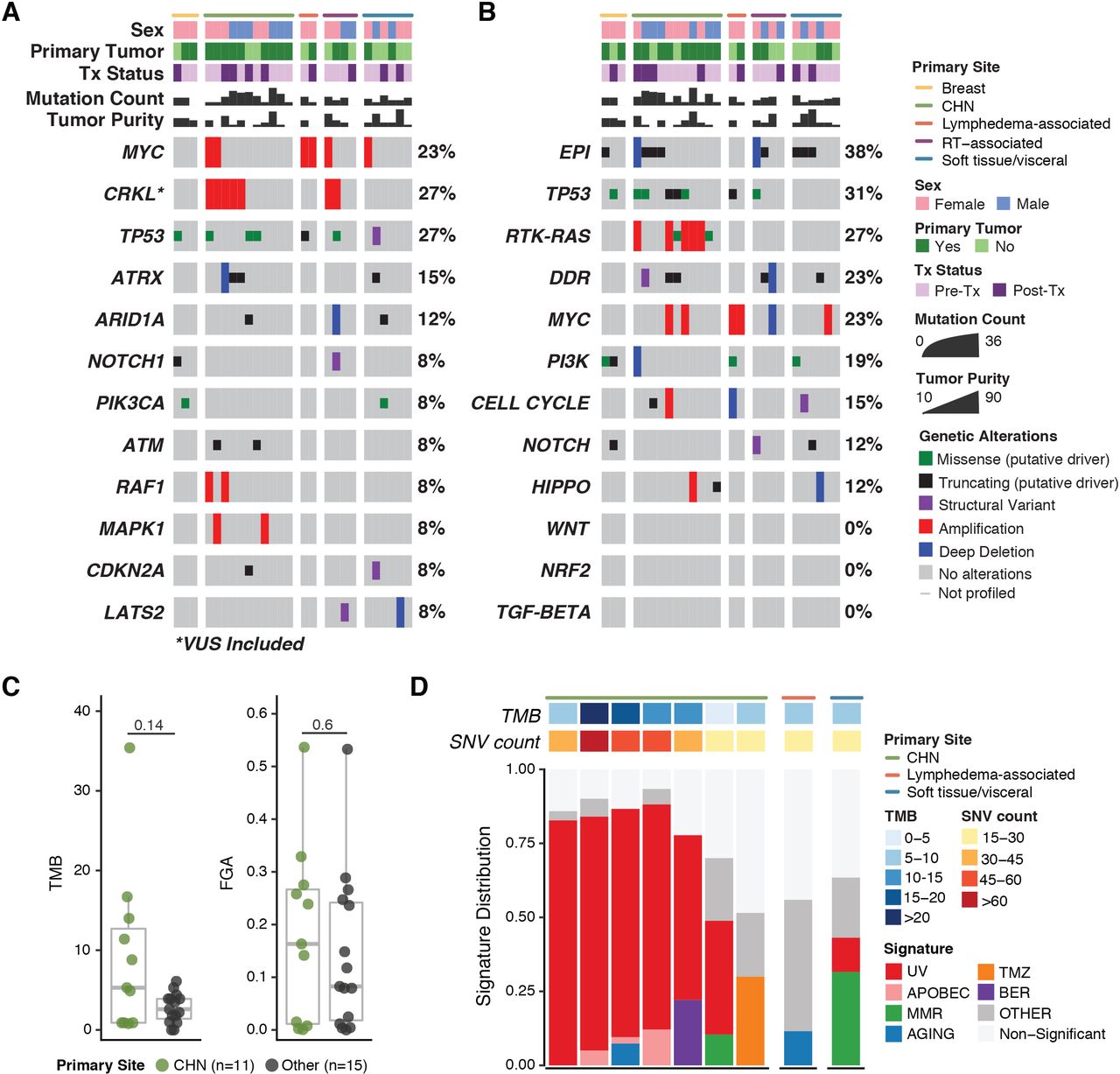
OncoPrints of targeted somatic next-generation genomic sequencing (MSK-IMPACT) data demonstrating alterations in individual genes (A) and alterations by pathway (B).(C)Median TMB and FGA. (D) Mutational signature analysis of samples with ≥15 single nucleotide variants (SNVs). CHN, cutaneous angiosarcoma of the head and neck; DDR, DNA damage repair; EpI, epigenetic; FGA, fraction of genome altered; RT, radiotherapy; TMB, tumor mutational burden; Tx, treatment; UV, ultraviolet.
The median TMB was 3.7 mutations per megabase (range 0–35.4) (figure 3C). Four patients (15%) had a high TMB: three with CHN tumors and one with a cutaneous tumor of the upper back. The median FGA, a measure of copy number alteration burden, was 0.12955 (range 0.0001–0.5365). No samples were identified as microsatellite instability high. A dominant UV signature was detected in six patients: five with CHN and one with a cutaneous tumor of the upper back (figure 3D).
To increase the number of patients with data available for molecular analysis, we performed fluorescence in situ hybridization for MYC and CRKL amplifications in patients who lacked MSK-IMPACT data and had sufficient tissue available. Both radiotherapy-associated breast tumors and both lymphedema-associated tumors had MYC amplification, as expected based on prior reports.7 40 MYC amplification was rare in primary tumors at other sites and was not detected in radiotherapy-associated tumors that did not arise from breast tissue, in agreement with a recent report.45 CRKL amplification was present in 50% of CHN tumors and was also found in two radiotherapy-associated tumors and two soft tissue/visceral tumors (online supplemental figure S2).
Genomic correlates of outcome
As CHN tumors were the largest group and had the highest TMB, we compared genomic alterations between them and primary tumors at other sites. No individual gene of interest (defined as an oncogenic or likely oncogenic alteration or CRKL amplification) was significantly enriched in CHN tumors versus primary tumors at other sites (online supplemental figure S3). In contrast, RTK-RAS pathway alterations were enriched in CHN tumors (adjusted p=0.005; Fisher’s exact test). While median TMB and FGA were higher in CHN tumors than in tumors at other sites, the differences were not significant.
No gene or pathway was significantly associated with PFS or OS in univariate analysis of clinical outcome in the 26 patients with MSK-IMPACT results available. Alteration in a Notch pathway gene (three patients) was of borderline significance for association with PFS: patients with an oncogenic or likely oncogenic alteration in this pathway had a median PFS of 7.9 (95% CI 5.1 to not reached) compared with 17.9 weeks in patients without such alteration (10.0 to not reached) (p=0.052). OS did not differ significantly with respect to alteration in Notch (p=0.274) (online supplemental figure S4). TMB (high vs low) and UV signature status were also not significantly associated with PFS or OS (online supplemental figure S5).
Transcriptomic correlates of outcome
Whole transcriptome sequencing was performed on nine unique-patient samples collected on one of three clinical trials: nivolumab plus bempegaldesleukin, pembrolizumab plus T-VEC, or pembrolizumab plus epacadostat. Seven samples were obtained before treatment (baseline), and two were obtained during the course of protocol therapy. In comparing tumor samples from patients who had clinical benefit (PFS ≥16 weeks; n=4) and patients who did not (n=5), 528 genes (176 upregulated, 352 downregulated) were found to be differentially expressed (online supplemental figure S6 and online supplemental table S1). Gene set enrichment analysis identified the apical junction, angiogenesis, inflammatory response, coagulation, and KRAS signaling pathways were upregulated in patients with PFS ≥16 weeks, while fatty acid metabolism was downregulated (online supplemental table S2).
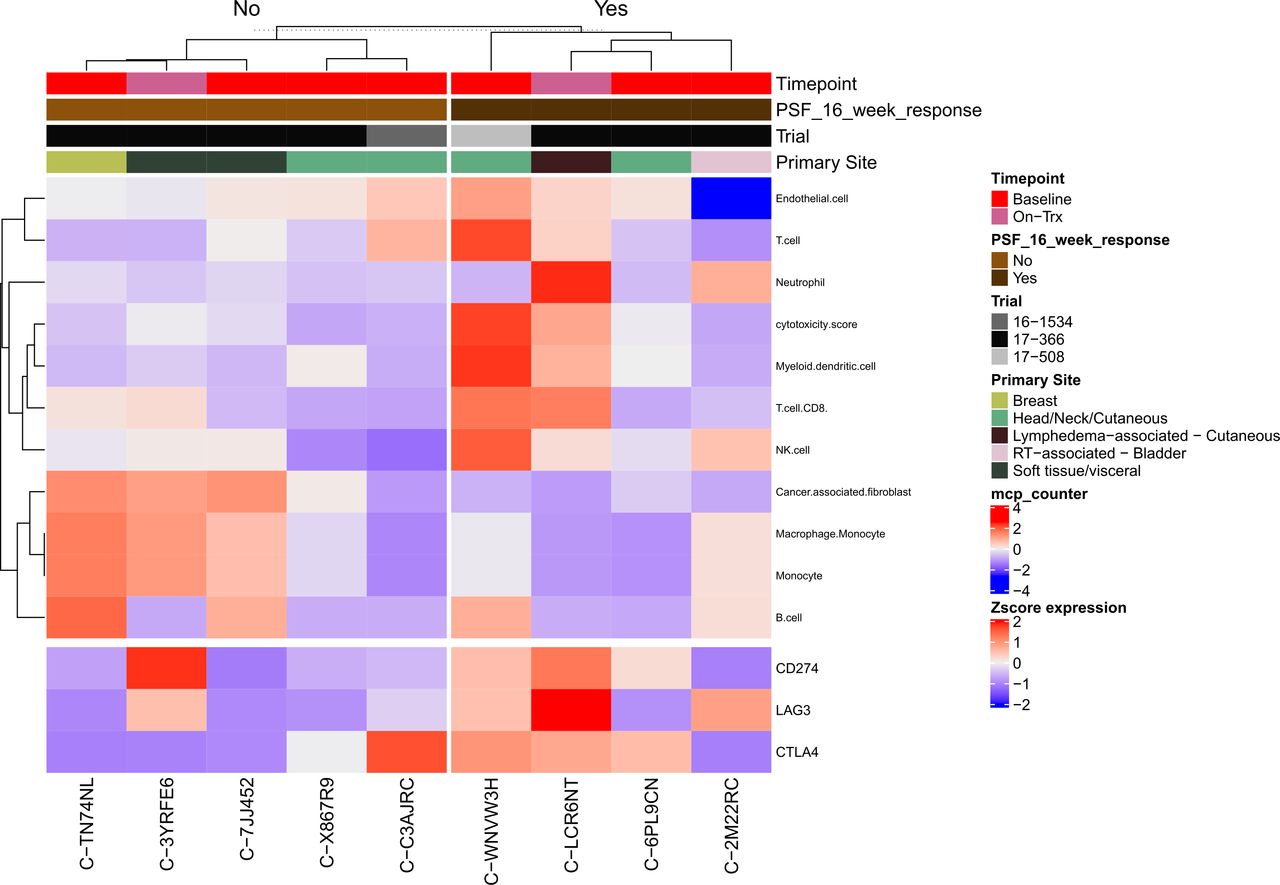
Microenvironment Cell Populations (MCP)-counter46 was used to investigate the tumor immune and non-immune stromal cell populations in patients with PFS ≥16 weeks and compared with patients with PFS <16 weeks (figure 4). Patients who had PFS ≥16 weeks had more cytotoxic T cells (p=0.06), myeloid dendritic cells (p=0.05), and NK cells (p=0.03), while cancer-associated fibroblasts were increased in the PFS <16 group (p=0.05). On correction for multiple hypothesis testing, none of the differences between groups approached or met statistical significance. Unsupervised hierarchical clustering of MCP-counter cell populations47 distinguished four distinct clusters: an RT-associated bladder tumor with few endothelial cells; two soft tissue tumors with high numbers of cancer-associated fibroblasts, monocytes, and macrophages; five cutaneous tumors with few monocytes and macrophages; and a breast primary tumor with high T cells, myeloid dendritic cells, and NK cells (online supplemental figure S7 and online supplemental table S3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Heatmap of immune and stromal cell deconvolution by PFS (≥16 or <16 weeks), including select immune checkpoint genes of interest. PFS, progression-free survival.
Immunohistochemistry
Immunohistochemistry to assess PD-L1 expression and/or TIL in baseline tumor samples was performed for 12 patients (online supplemental table S4). Two of seven patient samples assessable for PD-L1 had expression in ≥1% of cells. Four patients had paired tumor samples (baseline and on treatment) assessable; three of them had increased PD-L1 expression after initiation of ICB-based therapy (from 0% at baseline to 5% on treatment in patient 9, from 25% to 35% in patient 11, and from 1% to 60% in patient 12). Clinical benefit was seen in patients with and without PD-L1 expression at baseline. Ten patients had baseline tumor tissue available for TIL analysis. Nearly all (9 of 10) had some degree of lymphocyte infiltration. Four had paired biopsy samples assessable for TIL; two of them had a demonstrable increase in TIL and partial response to ICB-based therapy. PD-L1 expression and presence of TIL were not restricted to one specific primary disease site.
Discussion
In this retrospective analysis of the largest cohort to date of patients with angiosarcoma treated with ICB-based therapy, many of whom were treated prospectively on a clinical trial and most of whom had received prior systemic therapy, the median PFS was approximately 12 weeks. Although most patients failed to achieve clinical benefit after ICB-based treatment, approximately 40% had PFS of ≥16 weeks. Clinical benefit was seen across angiosarcoma primary sites and was not restricted to patients with cutaneous disease. We included every patient treated at our institution with at least one dose of ICB in this analysis, regardless of performance status or number of prior therapies. For patients who received more than one ICB dose, the median PFS was approximately 18 weeks.
Within the past 5 years, several groups have highlighted the efficacy of ICB against angiosarcoma, often in patients with CHN angiosarcoma. Florou et al reported five objective responses in a group of seven patients with angiosarcoma treated with ICB-based therapy.16 Painter et al described six patients with angiosarcoma treated with ICB, two of whom had exceptional responses.21 Most recently, Wagner et al reported an ORR of 25% to ipilimumab and nivolumab in a phase II study of patients with advanced angiosarcoma.22 Interestingly, the 25% ORR among clinical trial patients in our study mirrors those results. Wagner et al reported responses in three of five patients with CHN disease; a patient with radiotherapy-associated angiosarcoma of the breast also had a response. A similar pattern of responses (in patients with CHN angiosarcoma and radiotherapy-associated angiosarcoma of the breast) was seen in the Florou et al series.16
Our multivariate analysis found that patients who received ICB plus another novel immune modulator had longer survival rates than patients who received either ipilimumab plus nivolumab or ICB monotherapy. While this analysis is limited by its retrospective nature and the heterogeneity of the cohort, the data imply that novel combinatorial strategies can lead to improved outcomes compared with ICB monotherapy. Ipilimumab plus nivolumab did not appear superior to ICB monotherapy nor were outcomes comparable to ICB plus other novel agents. However, nearly all ipilimumab-treated patients in our cohort were treated outside of a clinical study and these findings are not in line with the recently published prospective data.
A unique finding from our cohort is that both patients with lymphedema-associated angiosarcoma had prolonged clinical benefit from ICB, suggesting that this angiosarcoma subtype may be particularly sensitive to ICB. Although only 2 of 10 patients with soft tissue or visceral disease had PFS ≥16 weeks, one had a complete response to treatment. These responses underscore the need to explore the use of ICB in patients with non-CHN angiosarcoma. Additionally, white race was found to be independently associated with a longer survival. Nearly all CHN tumors occurred in white patients. Perhaps like melanoma,48 decreased skin pigmentation increases the risk for UV-related damage and development of CHN angiosarcoma. Yet, white race portended a favorable prognosis after ICB treatment independent of primary site, potentially indicating that other clinical or biological factors contributed to the difference in outcome between the racial groups in this cohort.
Our findings confirm what Painter et al21 first described: the presence of a dominant mutational signature attributable to UV light and a relatively high TMB in patients with CHN angiosarcoma. While this molecular phenotype may increase the likelihood of clinical benefit from ICB-based therapy, neither TMB nor a UV signature appears to be a necessary or sufficient biomarker of response to ICB across angiosarcomas. Additional research is needed to better understand the immune context of angiosarcoma and to differentiate between the different primary sites of disease. At baseline, tumor lymphocyte infiltration was more common in our cohort than expression of PD-L1. While there was no readily apparent trend linking expression of one of these biomarkers to clinical benefit, a larger analysis of tumor specimens in patients treated with ICB is needed to reach a more definitive conclusion regarding their utility as biomarkers of response.
Targeted exome sequencing showed angiosarcoma to be a highly heterogenous disease not easily classifiable on a molecular level. Frequent mutations in the MAP kinase pathway were previously noted by Murali et al49 in an analysis of 34 angiosarcoma samples. Similarly, we found significant enrichment of alterations in the RTK-RAS pathway (which includes BRAF, RAF1, and MAPK1), but specifically in patients with CHN angiosarcoma, as well as frequent amplifications of CRKL, a gene located on the same cytoband as MAPK1. CRKL is an adaptor protein with an array of reported functions involving human development, immune cell migration, and signal transduction pathways.50 Overexpression of CRKL has been reported in various cancers and is associated with a poor prognosis.51 52 In laryngeal squamous cell carcinoma, CRKL amplification is associated with increased nuclear expression of CRKL, a requirement for tumor cell viability.53 In non-small cell lung cancer, CRKL amplification is a noted oncogene that activates the RAS and SRC pathways and is associated with resistance to EGFR inhibition.54 Although our analysis did not show an impact of CRKL amplification on outcome of ICB-based treatment, it may be prognostic of outcome in a larger dataset of angiosarcoma. The function of CRKL amplification in angiosarcoma warrants further study, especially as this protein becomes a possible therapeutic target.50
Our transcriptomic analyses confirmed a high degree of heterogeneity even within the tumor immune and stromal cell compartments of this small cohort. This analysis is constrained by a limited number of samples for analysis; yet, we identified the same number of immune clusters (four) as the recent report by Espejo-Freire et al.55 Cancer-associated fibroblasts, a stromal population not included in the Petitprez et al sarcoma immune classification,47 are upregulated in a subset of angiosarcomas.55 We identified a trend toward inferior PFS in these patients, potentially indicating a mechanism of resistance to ICB, a finding confirmed in select carcinomas.56 Additionally, gene set enrichment analysis found that upregulated KRAS signaling, a pathway that we found to be significantly altered in CHN angiosarcoma, was associated with PFS ≥16 weeks after ICB. These signals of response and resistance to ICB warrant further exploration .
The major strength of this study is its size relative to other published series of patients with angiosarcoma treated with ICB. Key limitations include the retrospective study design, the heterogeneity of ICB-based treatments administered, and the limited number of tissue samples available for correlative analysis. Other novel immune modulators, such as T-VEC or bempegaldesleukin, may have contributed to the antitumor activity of anti-PD-1 therapy, which confounds our analysis of outcomes after ICB. Prospective studies with larger cohorts of patients treated with the same therapy will be invaluable in confirming the efficacy of ICB in angiosarcoma. Future multi-institutional collaborations that can pool resources and patient data, as exemplified by the Angiosarcoma Project,21 will be critical in building larger datasets that can incorporate a broader set of tools, such as multiomic analyses, to deepen our understanding of this rare and multifaceted disease.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Institutional Review Board, Memorial Sloan Kettering Cancer Center, protocol 16-1101. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We acknowledge the patients and their families, the Integrated Genomics Operation Core (funded by the NCI Cancer Center Support Grant P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ER and CRA contributed equally.
Contributors All authors made substantive contributions to data analysis and writing the manuscript. As corresponding author, E.R. accepts full responsibility for the quality of the work and conduct of the study.
Funding Memorial Sloan Kettering Cancer Center is supported in part by the National Institutes of Health/National Cancer Institute Core Grant P30 CA008748.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.