Article Text

Abstract
Background Immune checkpoint inhibitor-based combinations have expanded the treatment options for patients with renal cell carcinoma (RCC); however, tolerability remains challenging. The aim of this study was to evaluate the safety and efficacy of the immunostimulatory interleukin-2 cytokine prodrug bempegaldesleukin (BEMPEG) plus nivolumab (NIVO) as first-line therapy in patients with advanced clear-cell RCC.
Methods This was an open-label multicohort, multicenter, single-arm phase 1/2 study; here, we report results from the phase 1/2 first-line RCC cohort (N=49). Patients received BEMPEG 0.006 mg/kg plus NIVO 360 mg intravenously every 3 weeks. The primary objectives were safety and objective response rate (ORR; patients with measurable disease at baseline and at least one postbaseline tumor response assessment). Secondary objectives included overall survival (OS) and progression-free survival (PFS). Exploratory biomarker analyses: association between baseline biomarkers and ORR.
Results At a median follow-up of 32.7 months, the ORR was 34.7% (17/49 patients); 3/49 patients (6.1%) had a complete response. Of the 17 patients with response, 14 remained in response for >6 months, and 6 remained in response for >24 months. Median PFS was 7.7 months (95% CI 3.8 to 13.9), and median OS was not reached (95% CI 37.3 to not reached). Ninety-eight per cent (48/49) of patients experienced ≥1 treatment-related adverse event (TRAE) and 38.8% (19/49) had grade 3/4 TRAEs, most commonly syncope (8.2%; 4/49) and increased lipase (6.1%; 3/49). No association between exploratory biomarkers and ORR was observed. Limitations include the small sample size and single-arm design.
Conclusions BEMPEG plus NIVO showed preliminary antitumor activity as first-line therapy in patients with advanced clear-cell RCC and was well tolerated. These findings warrant further investigation.
- Immunotherapy
- Kidney Neoplasms
- Drug Therapy, Combination
Data availability statement
Data from this study may be shared with qualified researchers, upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic
There remains a need for better-tolerated, immune checkpoint inhibitor-based combination treatments for patients with advanced renal cell carcinoma (RCC).
Bempegaldesleukin (BEMPEG) is an immunostimulatory interleukin (IL)-2 prodrug that has been engineered to deliver a controlled, sustained, and preferential IL-2 pathway signal, with the goal of stimulating an antitumor immune response while minimizing toxicity.
Phase 1 of the PIVOT-02 study showed that combination treatment with BEMPEG plus nivolumab was well tolerated and had encouraging clinical activity in solid tumors, including patients with RCC, warranting further investigation in a dose-expansion cohort.
What this study adds
These data demonstrate the preliminary safety and efficacy of this novel immunotherapy combination in patients with advanced clear-cell RCC.
How this study might affect research
These results provide rationale for the ongoing phase three trial in previously untreated patients with advanced or metastatic RCC (PIVOT 09; NCT03729245).
Introduction
Immunotherapies, particularly immune checkpoint inhibitors (ICIs), have dramatically changed the treatment landscape for patients with advanced renal cell carcinoma (RCC).1 2 Approved first-line treatments for these patients include an ICI combined with a vascular endothelial growth factor receptor (VEGFR)-targeted tyrosine kinase inhibitor (TKI; eg, nivolumab (NIVO) plus cabozantinib3 and pembrolizumab plus axitinib4 5 or lenvatinib6) or ICI combinations (eg, NIVO plus ipilimumab7 8). However, ICI–TKI and ICI–ICI combinations are difficult to tolerate. Rates of grade 3/4 treatment-related adverse events (TRAEs) are high (>48%), 15%–25% of patients experience TRAEs leading to treatment discontinuation, and TRAEs necessitating dose modification are common.3–7 As such, there remains a need for better-tolerated, ICI-based combination treatments that extend treatment benefit to more patients.
The International Metastatic RCC Database Consortium (IMDC) risk assessment criteria were established based on data from patients treated with VEGFR-TKI therapy but they have also been used for ICI-based therapy decision making.9 Historically, patients with IMDC favorable risk have angiogenesis-driven disease and respond well to VEGFR-TKI.10 Identifying which patients will achieve durable responses, and ultimately live longer with ICI-based regimens, is an unmet need.11 Interleukin (IL)-2 is an extensively studied cytokine with immunostimulatory and immunosuppressive effects. Treatment with high-dose IL-2 (aldesleukin) produces durable responses in approximately 5% of patients with metastatic clear-cell RCC,12 and data from a multi-institutional clinical registry (PROCLAIM) of 810 patients demonstrated median overall survival (OS) of 63 months in patients with IMDC favorable risk.13 However, severe toxicity experienced with the administration of high-dose IL-2, such as capillary leak syndrome, which is brought about by the IL-2 mechanism of action along with the pharmacokinetic and pharmacodynamic properties of high-dose IL-2, necessitates inpatient administration at specialized centers.14 High-dose IL-2 treatment, while stimulating the immune system by activating effector T cells and natural killer cells, also promotes the expansion of regulatory T cells, which play an immunosuppressive role in the tumor microenvironment.15 This, combined with the tolerability profile, has led to limited clinical use of high-dose IL-2 and prevented it being combined with other immunotherapies.
Bempegaldesleukin (BEMPEG, NKTR-214) is an immunostimulatory IL-2 prodrug that has been engineered to deliver a controlled, sustained, and preferential IL-2 pathway signal, with the goal of stimulating an antitumor immune response while minimizing toxicity and allowing for outpatient administration.16–18 The location of the polyethylene glycol chains directs BEMPEG to preferentially bind the dimeric IL-2 receptor βγ complex (predominantly expressed on natural killer cells and CD8+ T cells) over the trimeric IL-2 receptor αβγ complex (predominantly expressed on immunosuppressive regulatory T cells).17 18 In both animal models and patients with advanced solid tumors, BEMPEG has been shown to induce immunological changes,16–19 including preferential expansion and activation of CD8+ T cells and natural killer cells, and limited expansion of regulatory T cells in the tumor microenvironment.20 Treatment with BEMPEG monotherapy was well tolerated in heavily pretreated patients with advanced solid tumors.16 Taken together, these factors support the evaluation of BEMPEG in combination with an ICI.
The phase 1/2 PIVOT-02 study (NCT02983045) established the recommended phase 2 dose of combination treatment with BEMPEG plus NIVO and demonstrated encouraging clinical activity in solid tumors, including RCC.21 22 Data from the phase 2 part of PIVOT-02 has demonstrated encouraging antitumor activity in patients with previously untreated metastatic melanoma19 and advanced/metastatic urothelial carcinoma.23 This regimen is well tolerated with a predictable safety profile related to TRAEs and immune-mediated AEs.19 21 The objective of this study was to evaluate the safety and efficacy of BEMPEG in combination with NIVO as first-line therapy in patients with advanced or metastatic clear-cell RCC.
Methods
Study design and treatment
PIVOT-02 is a non-randomized, open-label, international, multicenter, multi-cohort, phase 1/2 study. In the phase two single-arm dose-expansion cohorts, patients received intravenous BEMPEG 0.006 mg/kg plus NIVO 360 mg administered every 3 weeks for up to 2 years or until documented disease progression, death, unacceptable toxicity, symptomatic deterioration, patient/investigator’s decision to discontinue treatment, patient withdrawal of consent, loss to follow-up, or study termination by the sponsor. Dose reduction of BEMPEG, but not NIVO, was permitted.
The study was conducted in accordance with the Declaration of Helsinki, the US Food and Drug Administration regulations, and the International Council for Harmonization Good Clinical Practice guidelines. All patients provided written informed consent before the conduct of any study procedures.
Patients
Eligible patients had histologically confirmed, advanced (not amenable to surgery) or metastatic RCC (American Joint Committee on cancer stage IV) with a clear-cell component, including tumors with sarcomatoid features. Patients had measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1 and were ≥18 years of age with an Eastern Cooperative Oncology Group performance status of 0 or 1. No prior systemic therapy for advanced or metastatic RCC was allowed.
Patients were required to have archival or fresh tumor tissue (within 6 months) for assessment of programmed death ligand-1 (PD-L1) expression at baseline. Patients were excluded if they had received prior immunotherapy, including IL-2 agents, in any of the neoadjuvant, adjuvant, or advanced settings; had active, known, or suspected autoimmune disease requiring systemic immunosuppressive agents; or had an active infection requiring antimicrobial therapy, active brain metastases, or a known additional malignancy that was progressing or required active treatment.
Study objectives and assessments
The primary objective of the study was to evaluate the safety and objective response rate (ORR) following treatment with BEMPEG plus NIVO. ORR was defined as the percentage of patients with a best response of complete response (CR) or partial response (PR) by RECIST V.1.1. Secondary objectives were to evaluate the efficacy of the combination using OS and progression-free survival (PFS), clinical benefit rate (CBR; a CR, PR, or stable disease ≥7 weeks), and duration of response (DOR).
Safety was evaluated in all patients (safety population) who received at least one dose of study drug, and AEs were categorized per the National Cancer Institute’s Common Terminology Criteria for Adverse Events V.4.03. All efficacy endpoints other than PFS and OS were analyzed in the response-evaluable population (all patients with measurable disease per RECIST V.1.1 at baseline and at least one post-baseline tumor response assessment). Response assessments were performed at screening, every 8 weeks (±7 days), and at end of treatment. Patients with an unconfirmed PR/CR required a confirmatory scan 4 weeks after the previous scan. Long-term follow-up for survival occurred every 3 months. All safety and efficacy endpoints were evaluated by local investigator assessment.
Analyses between dichotomous biomarker categories measured in baseline tumor biopsies and ORR were explored. Tumor biomarkers associated with response to ICIs were selected for analyses and assessed at baseline (including PD-L1 expression on tumor cells by immunohistochemistry (IHC) (by 28–8 PharmDx assay, Dako, an Agilent Technologies, Inc. company, Santa Clara, California, USA), interferon gamma (IFN-γ) gene-expression profile (GEP), CD8+ tumor-infiltrating lymphocytes (TIL) by IHC, and tumor mutational burden (TMB); see online supplemental information) for additional methods24–26). Tumors were defined as PD-L1 negative (<1% tumor cell expression), positive (≥1% tumor cell expression), or unknown (indeterminate/non-evaluable).
Supplemental material
Statistical analysis
The study was designed to enroll a minimum of 26 patients in the phase 2 cohort at the recommended phase 2 dose of BEMPEG plus NIVO. Analyses reported here also include patients with RCC treated at the recommended phase 2 dose of BEMPEG plus NIVO during the dose escalation period in phase 1 of the study. Baseline characteristics and safety data were summarized by descriptive statistics and frequency tables. The Clopper–Pearson method was used to calculate response rates with two-sided 95% confidence intervals (CIs). DOR, PFS, and OS were evaluated by the Kaplan-Meier method. DOR and PFS data for patients without disease progression (per RECIST V.1.1) and alive, or with unknown status, were censored at the time of the last tumor assessment prior to new anti-cancer therapy. Statistical analyses for biomarker evaluations included calculation of the difference in response rates with 95% CIs between biomarker-defined groups. For the primary analysis, all efficacy and safety endpoints were analyzed up to and including the cut-off date (January 8, 2021). Statistical analyses were performed with SAS (V.9.4) and Prism V.8.0 (GraphPad Software, San Diego, California, USA).
Results
Patients
Between April 3, 2017, and February 26, 2018, 49 patients (phase 1, n=11; phase 2, n=38) were enrolled at 10 sites across the US and Italy. Baseline demographic characteristics are shown in table 1. All patients were evaluable for safety and response.
Baseline characteristics of all enrolled patients
At data cut-off (January 8, 2021), all patients had ended treatment. BEMPEG was discontinued for the following reasons: progressive disease by RECIST (n=31); AE (n=6); maximum 2-year treatment duration reached (n=7); patient decision (n=1); clinical progression (n=1); and physician decision (n=3). Reasons for discontinuation from NIVO are shown in online supplemental figure 1.
The median duration of exposure to BEMPEG was 7.2 months (range, 0–25.8), with a median of 9.0 (range, 1–35) treatment cycles. The median duration of exposure to NIVO was 6.3 months (range, 0–25.8), with a median of 9.0 (range, 1–35) treatment cycles.
Primary analysis: safety
Ninety-eight per cent (48/49) of patients experienced at least one TRAE during treatment (table 2), and 38.8% (19/49) of patients experienced grade 3/4 TRAEs, with the most frequent events (occurring in at least one patient) being syncope (8.2%; 4/49), increased lipase (6.1%; 3/49), fatigue (4.1%; 2/49), hyponatremia (4.1%; 2/49), and hypotension (4.1%; 2/49) (table 2). All AEs, regardless of causation, are included in online supplemental table 1.
Incidence of TRAEs*
TRAEs leading to discontinuation of either study drug occurred in 12.2% (6/49) of patients (6 patients: pneumonitis (n=2), increased alanine aminotransferase, increased aspartate transaminase, cerebrovascular accident, deep vein thrombosis, peripheral edema, peripheral sensory neuropathy, and pulmonary embolism (n=1 each)). TRAEs leading to any dose reduction of BEMPEG were observed in seven patients (14.3%; 7/49).
Serious TRAEs occurred in 22.4% (11/49) of patients (online supplemental table 2), with the most frequent serious TRAEs (occurring in more than one patient) being fever (6.1% (3/49 patients)) and cerebrovascular accident (4.1% (2/49 patients)). Immune-mediated AEs are shown in online supplemental table 3. There were no treatment-related deaths.
Primary analysis: ORR
After a median duration of follow-up of 32.7 months, investigator-assessed ORR (response-evaluable population) was 34.7% (17/49; 95% CI 21.7 to 49.6) (table 3), including 3/49 CRs (6.1%) and 14/49 PRs (28.6%; figure 1). Of the 17 patients with response, 14 patients continued to respond for over 6 months, 12 patients continued to respond for over 12 months, and 6 patients continued to respond for at least 24 months. The median time to response was 4.0 months (range 1.3–8.8).
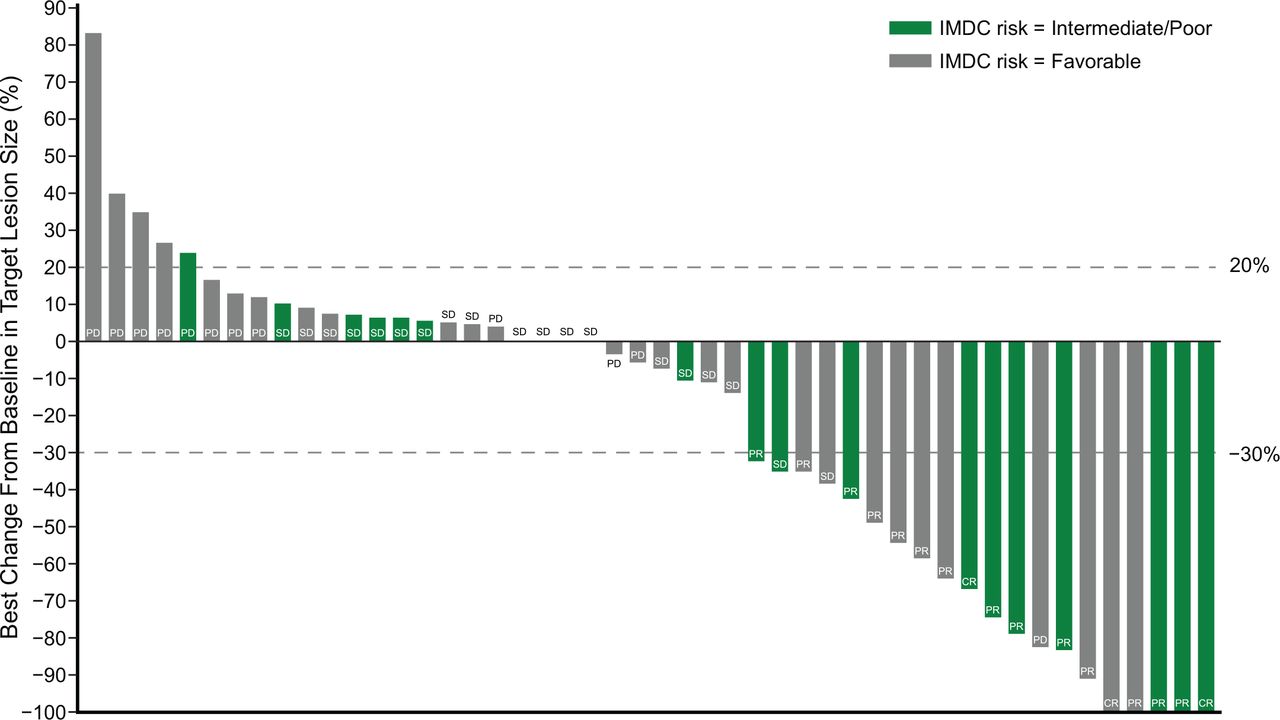
Efficacy of BEMPEG plus NIVO (investigator assessment). Waterfall plot of maximum change in tumor size from baseline (response-evaluable population*; n=49). Data cut-off: January 8, 2021. *Response-evaluable population includes patients who have measurable disease (per RECIST V.1.1) at baseline and have at least one postbaseline assessment of tumor response. BEMPEG, bempegaldesleukin; IMDC, International Metastatic RCC Database Consortium; NIVO, nivolumab; RECIST, Response Evaluation Criteria in Solid Tumors.
Objective response per RECIST V.1.1 by investigator review by IMDC risk score (response-evaluable population; n=49*)
ORR by IMDC risk score was 52.9% (9/17) in favorable-risk patients, 31.8% (7/22) among intermediate-risk patients, and 10.0% (1/10) in poor-risk patients. When combining the intermediate-risk and poor-risk patient groups, the ORR was 25.0% (8/32) (table 3).
Secondary analysis: DOR, CBR
Among responding patients, the median DOR was 26.1 months (95% CI 8.2 to not reached); Kaplan-Meier estimates of response are summarized in table 3.
CBR was 73.5% (36/49) in the overall RCC population (response-evaluable), 94.1% (16/17) in favorable-risk patients, 68.2% (15/22) in intermediate-risk patients, and 50.0% (5/10) in poor-risk patients (table 3).
Secondary analysis: survival
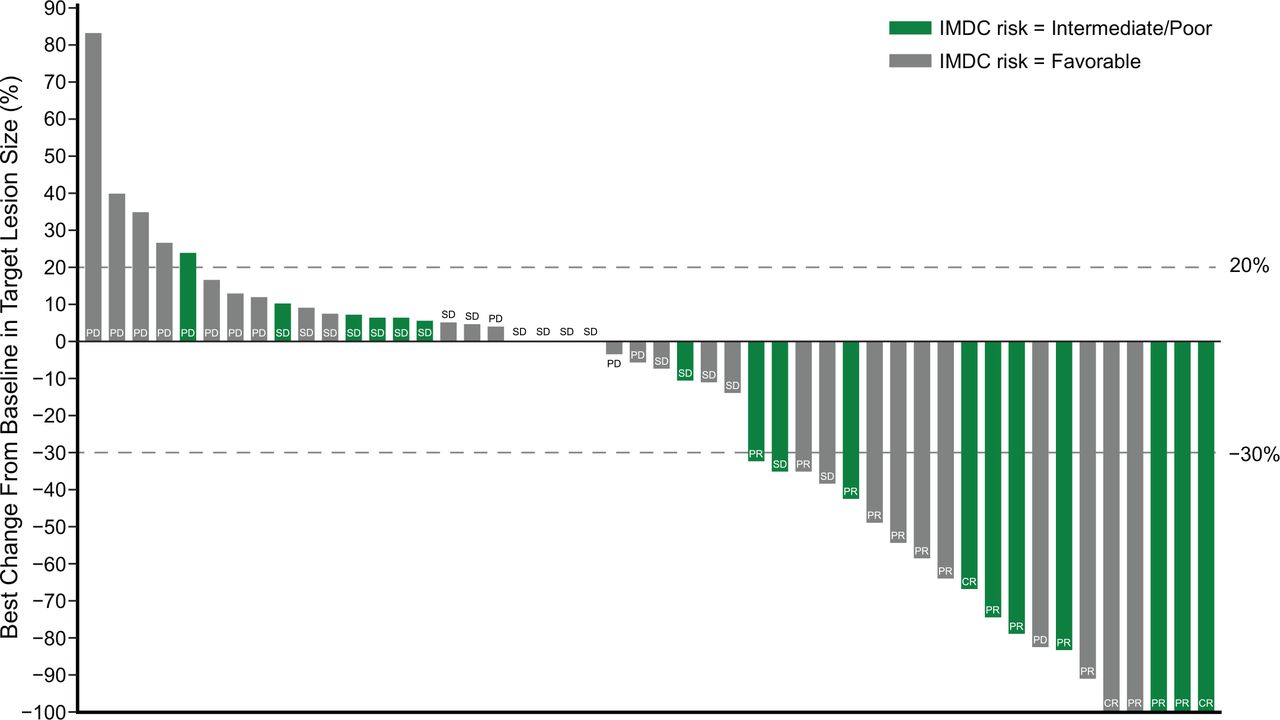
The median PFS was 7.7 months (95% CI 3.8 to 13.9; figure 2A; safety population). PFS probabilities were 39.4% (95% CI 25.6%% to 52.9%) at 12 months and 24.1% (95% CI 13.0% to 37.0%) at 24 months (online supplemental table 4).

{kind=link}
{kind=link}
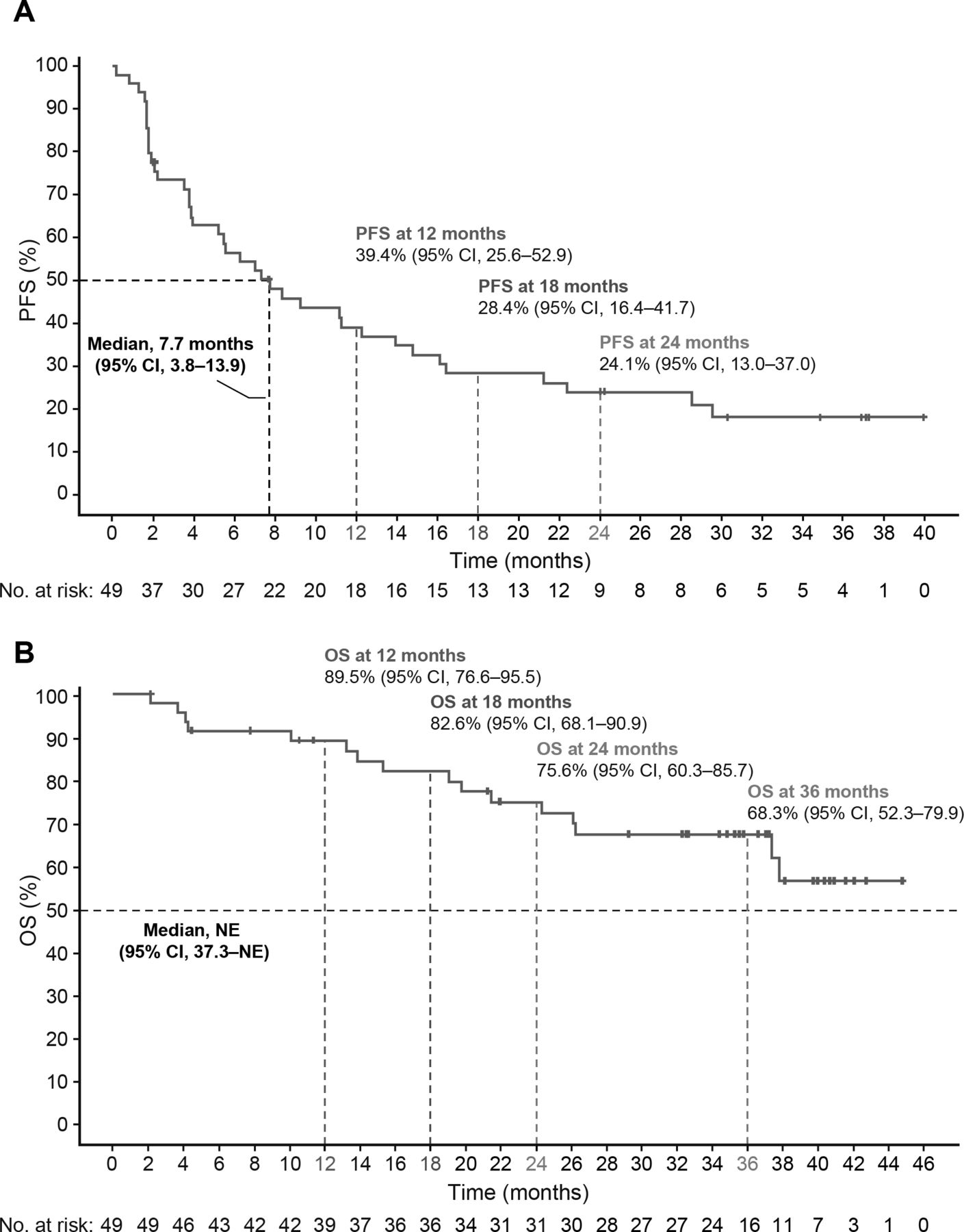
Efficacy of BEMPEG plus NIVO. (A) Kaplan-Meier estimates of progression-free survival by investigator assessment and (B) overall survival (safety population; n=49). Data cut-off: January 8, 2021. BEMPEG, bempegaldesleukin; NIVO, nivolumab; NE, not estimable; OS, overall survival; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Median PFS among IMDC favorable-risk patients was 12.2 months (95% CI 5.2 to 16.4), for intermediate-risk patients was 7.1 months (95% CI 1.8 to 21.3), and for poor-risk patients was 2.9 months (95% CI 0.8 to not reached) (online supplemental table 4).
Median OS was not reached (95% CI 37.3 to not reached; figure 2B and online supplemental table 5).
Exploratory biomarker analyses
In the exploratory biomarker univariate analyses (data cut-off May 15, 2020), patients were grouped by baseline PD-L1 expression in tumor cells≥1% vs <1% group (n=46), CD8+ TIL ≥73 cells/mm2 (median of n=43 patients) vs <73 cells/mm2, TMB ≥3.51 mutations per megabase (median of n=19) vs <3.51 mutations per megabase, or IFN-γ GEP ≥0.59 (median of n=37) vs <0.59; no statistical difference was observed in ORR between the two groups based on each biomarker (online supplemental figure 2).
Discussion
The international, multicenter PIVOT-02 trial showed that the novel immunostimulatory IL-2 cytokine/ICI combination of BEMPEG plus NIVO produced durable responses in some patients (response >12 months in 71% of responding patients) with advanced, favorable-risk and intermediate-risk clear-cell RCC. The safety profile of the combination treatment was predictable, consistent with that of the individual compounds,21 27 and consistent with the safety profile reported in other PIVOT-02 cohorts.19 23 No new safety signals were identified. BEMPEG has been engineered as a prodrug to deliver a controlled and sustained IL-2 pathway signal and thereby overcome some of the challenges of high-dose IL-2 therapy, namely high toxicity requiring inpatient administration and intensive monitoring at centers with extensive experience administering this treatment. Capillary leak syndrome, a serious AE associated with high-dose IL-2 therapy, was not observed in this cohort. The most frequently observed AEs with BEMPEG plus NIVO were mostly grade 1 or 2 in severity and included flu-like symptoms (fatigue, fever, chills, and influenza-like illness) and pruritus that were mostly transient and manageable using standard treatment protocols. Thyroid dysfunction is the most common immune-mediated AE observed with IL-2 therapy,28 and in this cohort, the most common immune-mediated AE with BEMPEG plus NIVO was hypothyroidism (10 of 49 (20%); all grade 1 or 2; these rates are consistent with those reported in other PIVOT-02 cohorts.19 23 The rate of grade 3/4 TRAEs with the combination reported here (39%) compares favorably with reported rates from large randomized trials of other combination treatments in this patient population, including NIVO plus ipilimumab (48%),7 NIVO plus cabozantinib (61%),3 pembrolizumab plus axitinib (63%),4 and pembrolizumab plus lenvatinib (72%).6 The rate of AEs leading to discontinuation of combination treatment reported here (12%) is lower than that reported with ICI–TKI and ICI–ICI combinations (15%–25%3–7).
Thirty-five per cent of patients achieved an objective response with BEMPEG plus NIVO in this small phase 2 study. This ORR is similar to that reported with ICI monotherapy (pembrolizumab, 34%29 ; NIVO, 32%30) but notably lower than ORRs with ICI–TKI combinations (NIVO plus cabozantinib, 56%3 ; and pembrolizumab plus axitinib, 60%5 or lenvatinib, 71%6) and ICI–ICI combinations (NIVO plus ipilimumab, 39%7). Responses were durable in patients achieving a response with BEMPEG plus NIVO with a median DOR of 26 months, and 71% of responding patients having a response that lasted more than 12 months. These results, while preliminary and based on a small sample size, are consistent with median response durations reported in recent studies with ICI–TKI combinations, ranging from 20 to 26 months.3 5 6
The median PFS observed with BEMPEG plus NIVO in this trial, 7.7 months, was lower than reported with other ICI based therapies in phase three trials.3 5–7 Although median OS was not yet reached in the overall population or favorable-risk population in this PIVOT-02 cohort, median OS was encouraging in intermediate- (38 months) and poor-risk (26 months) patients.
In PIVOT-02, objective responses were observed across all IMDC risk groups, with patients in the favorable-risk category treated with BEMPEG plus NIVO demonstrating higher ORR (53% vs 32% vs 10%) and longer median PFS (12.2 vs 7.1 vs 2.9 months) than those in the intermediate- and poor-risk categories, respectively. Data from the PROCLAIM database has shown that favorable-risk/intermediate-risk RCC patients treated with high-dose IL-2 had better outcomes than poor-risk patients; CBR were higher, and OS duration was longer in favorable-risk/intermediate-risk patients, with the most durable OS observed in the favorable-risk group.13 In patients with favorable-risk disease in CheckMate 214, the ORR with NIVO plus ipilimumab was 30% (CR, 12%) compared with 52% (CR, 6%) in the sunitinib arm.7 The 53% ORR and 12% CR rate in the favorable-risk group with BEMPEG plus NIVO is encouraging in this risk group and provides support for investigating the combination of an ICI with an agent such as BEMPEG that modulates the tumor microenvironment. For these favorable-risk patients, novel treatment combinations with comparable or better efficacy than anti-VEGF therapy, but with an improved tolerability profile, are desirable. The lower ORR observed in the 10 patients with poor-risk features is consistent with RCC literature,31 although the limited number of poor-risk patients should be considered when interpreting the findings. A better understanding of how to identify patients who will benefit from ICI-based combinations, across risk-groups, is needed.
Exploratory biomarker analysis suggested that the ORR was not associated with any of the baseline biomarkers assessed, including baseline PD-L1 status. The association between baseline PD-L1 status, as well as other biomarkers, and clinical outcomes will be further explored in ongoing studies.
The small sample size of this cohort, and the lack of a comparator arm, are limitations of the trial, as is the potential for selection bias as patients were enrolled from a small number of academic specialty centers. DOR data should be interpreted with caution, given that the results are derived from Kaplan-Meier estimates in the 17 patients with a response. However, this study had a relatively long duration of follow-up (32 months). Rates of previous nephrectomy in this study were high (97%) and minimally higher than reported in recent studies in metastatic RCC (~70%–80%).3 4 6 Ongoing studies will help elucidate the clinical benefits of adding BEMPEG to NIVO as first-line therapy in patients with advanced or metastatic RCC.
Conclusions
In conclusion, our results provide preliminary evidence of the safety and efficacy of BEMPEG plus NIVO in patients with advanced or metastatic RCC with a clear-cell component. The findings reported here are currently undergoing validation in an ongoing randomized, registrational, phase three trial in previously untreated patients with advanced or metastatic RCC (PIVOT 09; NCT03729245). The combination of BEMPEG plus NIVO is also being investigated as part of a triplet regimen in combination with a TKI (cabozantinib or axitinib) in a phase 1/2 trial in patients with previously untreated advanced or metastatic RCC (PIVOT IO 011; NCT04540705).
Supplemental material
Data availability statement
Data from this study may be shared with qualified researchers, upon request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by WIRB IRB registration number: IRB00000533CGIRB. IRB registration number: IRB00001313. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We want to thank all patients, their families, and the investigators who participated in this study. We also thank Dako for the collaborative development of the PD-L1 immunohistochemistry 28–8 PharmDx assay (Bristol Myers Squibb, Princeton, NJ, USA). This study was sponsored by Nektar Therapeutics, San Francisco, CA, USA. Medical writing assistance was provided by Alison Lovibond Ph.D. and Suzanne Patel Ph.D. from BOLDSCIENCE Inc. and was funded by Nektar Therapeutics.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NMT accepts full responsibility for the work and/or the conduct of the study, had full access to all the data in the study, and takes responsibility for the integrity of the data analysis and controlled the decision to publish. NMT, AOS-R, AD, DCC, UH, SLC, MAT, JZ, and MEH were responsible for the study’s concept and design. NMT, DCC, AD, MS, MAB, AVB, GG, MEH, and AOS-R conducted patient recruitment and data acquisition. LT, AC, and DT performed the statistical analysis. All authors contributed to the analysis and interpretation of data, drafting of the final manuscript for publication, the critical revision of the manuscript for important intellectual content.
Funding This trial was designed and funded by the sponsor (Nektar Therapeutics). The sponsor and their representatives collected and analyzed the data. All authors had full access to study data, and the corresponding author had final responsibility for the decision to submit for publication.
Competing interests AOS-R received research funding from BioClin Therapeutics, Bristol Myers Squibb, Janssen, Merck Sharp & Dohme, Michael and Sherry Sutton Fund for Urothelial Cancer, Nektar Therapeutics, US National Institutes of Health, and Takeda. She has also served as an advisor/consultant to AstraZeneca, Bavarian Nordic, BioClin Therapeutics, Bristol Myers Squibb, EMD Serono, Genentech, Inovio Pharmaceuticals, Janssen, Merck, US National Comprehensive Cancer Network, Nektar Therapeutics, and Seattle Genetics. DCC has received consulting fees from Pfizer, Nectar, Torque, and Puretech. AD has been an advisory board member for Nektar Therapeutics. MS has served as a consultant/advisor for Genentech-Roche, Bristol Myers Squibb, AstraZeneca/MedImmune, Pfizer, Novartis, Kyowa-Kirin, Amgen, Merus, Seattle Genetics, Immune Design, Prometheus, Anaeropharma, Astellas-Agensys, Immunova, Nektar Therapeutics, Neostem, Pierre-Fabre, Eli Lilly, Symphogen, Lion Biotechnologies, Amphivena, and Adaptive Biotechnologies. MAB has acted as a paid consultant for, and/or as a member of the advisory boards of, Exelixis, Bayer, BMS, Eisai, Pfizer, AstraZeneca, Janssen, Genomic Health, Nektar Therapeutics, and Sanofi and has received grants to his institution from Xencor, Bayer, Bristol Myers Squibb, Genentech/Roche, Seattle Genetics, Incyte, Nektar Therapeutics, AstraZeneca, Tricon Pharmaceuticals, Peleton Therapeutics, and Pfizer for work performed as outside of the current study. AVB has received institutional research funding from AstraZeneca/MedImmune, F Hoffmann–La Roche/Genentech, Merck, and Seattle Genetics; and honoraria from AstraZeneca/MedImmune, F Hoffmann–La Roche/Genentech and Merck. He has also served as an advisor/consultant to AstraZeneca/MedImmune, Cerulean Pharma, F Hoffmann–La Roche/Genentech, Incyte, Merck, Nektar Therapeutics, Pfizer/EMD Serono, and Seattle Genetics/Astellas. GG has received grants and personal fees from PharmaMar, grants from Novartis, and personal fees from Lilly, Pfizer, Bayer, and Eisai, outside the submitted work. EP, LT, DC, UH, AC, and DY are employees of, and have ownership interest (eg, stock) in, Nektar Therapeutics. SLC is a former employee of, and has ownership interest (eg, stock) in, Nektar Therapeutics. MAT is the chief medical officer at Nektar Therapeutics and has ownership interest (eg, stock) in the company. JZ is the chief research and development officer at Nektar Therapeutics and has ownership interest (eg, stock) in the company. MEH has had a consulting or advisory Role with Nektar Therapeutics, Janssen, Crispr Therapeutics, Bristol Myers Squibb/Celgene, and Exelixis; and has received research funding from Apexigen, Astellas Pharma, AstraZeneca/MedImmune, Bayer, Bristol Myers Squibb, Corvus Pharmaceuticals, Lilly, Endocyte, Genentech, Genmab, Innocrin Pharma, Iovance Biotherapeutics, Merck, Nektar Therapeutics, Novartis, Pfizer, Progenics, Sanofi/Aventis, Seattle Genetics, Torque, Unum Therapeutics, and Achilles Therapeutics. NMT has received grant support, consulting fees, and honoraria from Bristol Myers Squibb; grant support from Epizyme and Mirati Therapeutics; grant support, consulting fees, and honoraria from, and served on an advisory board for, Exelixis and Novartis; received consulting fees and honoraria from Argos Therapeutics and Pfizer; and received consulting fees and honoraria from, and served on an advisory board for, Eisai, Nektar Therapeutics, and Oncorena.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.