Article Text

Abstract
Surgical removal of tumors remains a front-line therapy for many types of cancer. However, this treatment often fails to eradicate disease due to either recurrence of the original tumor or development of distant micrometastases. To address these challenges, patients are often given non-curative treatments presurgery with the intent of improving surgical outcomes. These treatments, collectively known as neoadjuvant therapies, have traditionally focused on the presurgical use of chemotherapeutics. Recently, however, a variety of immunotherapies have also been identified as potentially effective in the neoadjuvant setting. One of these immunotherapies is oncolytic virotherapy, whose clinical use has exploded with the Food and Drug Administration approval of Talimogene Laherparepvec. This review summarizes both the preclinical and clinical literature examining the use of oncolytic virotherapy in the neoadjuvant setting for different types of cancers and discusses some of the major questions that still need to be addressed in order for this unique use of immunotherapy to become clinically viable.
- Oncolytic Virotherapy
- Oncolytic Viruses
- Adjuvants, Immunologic
- Immunotherapy
- Review
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Despite numerous advances in technology and significant increases in the number of available treatment options, surgery remains a standard of care for the majority of solid cancers. Unfortunately, although surgical techniques have improved over the years,1 2 the efficacy of surgical resection remains limited. For example, despite apparent complete resection (R0/R1) of malignant tissue, only 12% of patients with pancreatic adenocarcinoma survive more than 5 years.3 Similarly, again despite apparent complete resection, more than 25% of patients with non-small cell lung cancer (NSCLC) will develop recurrent disease.4 These failures can be due to several factors. On one hand, virtually all incompletely resected tumors are likely to locally regrow and many apparently localized tumors can form micrometastases prior to surgery. On the other hand, surgery itself is known to contribute to cancer progression by physically disturbing the tumor bed resulting in dissemination of malignant cells5 and accelerating both angiogenesis and the wound-healing response.6–8 Additionally, while surgery often causes general systemic inflammation, it is also known to enhance immune exhaustion.9 10 This exhaustion occurs through multiple mechanisms, including excessive monocyte activation resulting in the overproduction and release of the immunosuppressive prostaglandin PGE2,11 defects in natural killer (NK) cell activity,9 and an increase in neutrophil extracellular traps in the blood.12 Based on the abovementioned challenges, there is an urgent need for methods to improve surgical outcomes.
Neoadjuvant therapy (NAT) is the use of an initial therapeutic treatment designed to shrink a tumor prior to administration of a second more major treatment, such as surgery.13 This type of treatment—most often provided in the form of chemotherapy—is not intended to be curative but is instead designed to improve outcomes of the main intervention. These improvements can come in a variety of forms, the most obvious being directly increasing patient survival after surgery. For example, in a meta-analysis of 2385 patients in 15 randomized controlled trials, it was demonstrated that the addition of standard chemotherapy as a NAT directly improved the 5-year overall survival (OS) of stage I–III NSCLC by 5% (from 40% to 45%) compared with surgery alone.14 In addition to directly improving clinical outcomes, however, NAT can also be used to deescalate the magnitude of a required surgery.15 For example, the expert panel from the 15th St. Gallen International Expert Consensus Conference on the Primary Therapy of Early Breast Cancer recommended that following NAT, surgery could be restricted to only the area still containing residual cancer, not the area comprising the original extent of the cancer.16 Finally, NAT can facilitate the operability of otherwise inoperable tumors such as those found in pancreatic cancer, where the tendency for diagnoses to occur at later stages results in only 15%–20% of newly diagnosed patients being candidates for surgical resection.17 Regardless of the form in which the benefit is realized, there is a robust body of literature demonstrating that the use of NAT prior to surgery can be highly beneficial to some patients.18 19
While the most common form of NAT has historically been chemotherapeutics, the clinical results with these drugs indicate that there remains significant room for improvement. Because of this, the use of various forms of immunotherapies in the neoadjuvant setting is currently being heavily investigated. At the time of this writing, 247 active clinical trials involving immunotherapies in the neoadjuvant setting were listed in ClinicalTrials.gov. The most commonly used type of immunotherapy in these trials is immune checkpoint inhibitors (ICIs), including inhibitors against programmed cell death protein 1 (PD1), programmed death ligand 1 (PD-L1), and cytotoxic T lymphocyte associated protein 4 (CTLA-4). These treatments remove the brakes from the immune system allowing it to more effectively target tumor cells, thereby preventing recurrence.20 Critically, initial results from these studies appear promising. For example, in a trial of patients with head and neck cancer, the use of the PD-1 inhibitor sintilimab prior to surgery or chemotherapy improved overall response rates by 16% (68.4% vs 84.6%), 2-year progression-free survival by 17% (27% vs 44%), and 2-year OS by 9% (61% vs 70%, though this last difference was not significant (p=0.681)21). Based on this and other studies,22 23 ICIs appear to provide some benefit in the neoadjuvant setting, validating the potential use of immunotherapies in this context. However, the incomplete response rates to NAT-ICI suggest that investigation of other immunotherapies might still be warranted.
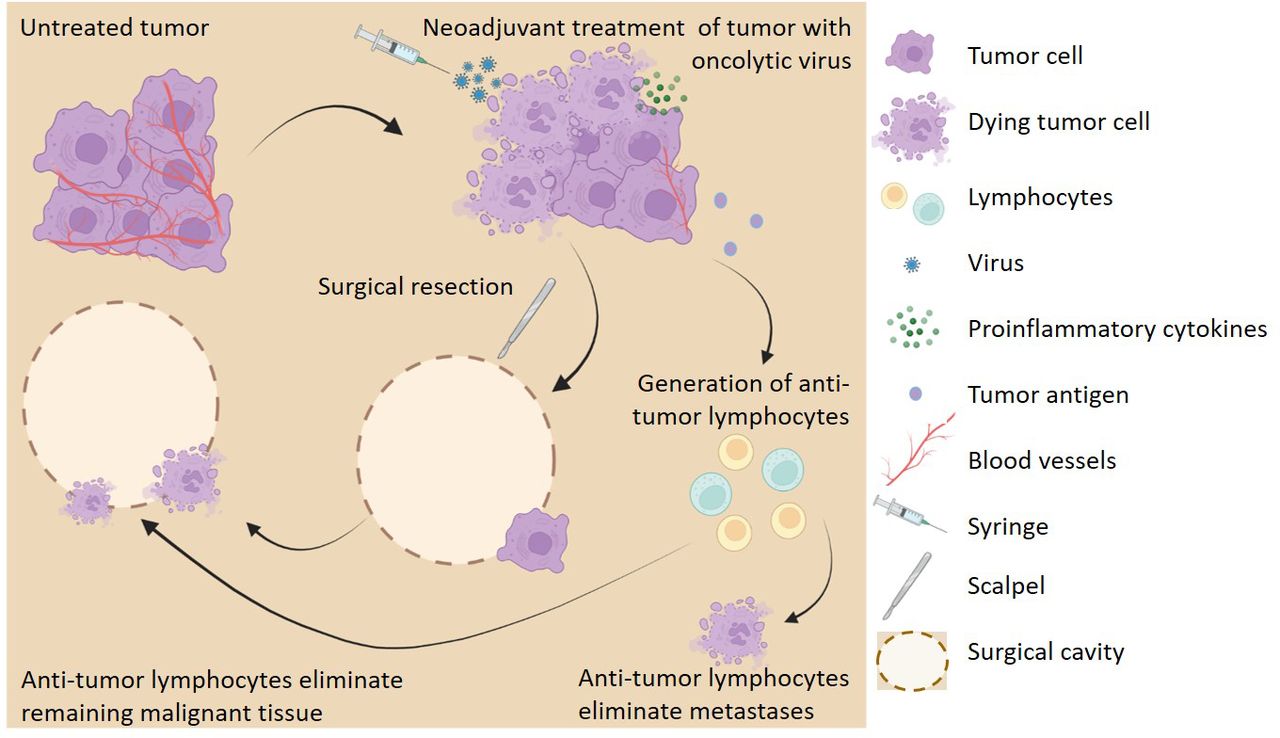
Recently, oncolytic virotherapy (OVT) has been presented as a novel strategy to address the challenges that accompany using surgical resection as the primary treatment for cancers (figure 1). OVT is an attractive NAT candidate due to its ability to provide long-term therapeutic benefits when applied as an acute treatment.24 This is largely an effect of OVT’s ability to generate antitumor immunity, which allows for elimination of remaining tumor cells even after the virus itself is cleared.25 26 This effect synergizes well with NAT since the therapeutic benefit of OVT can continue following surgical removal of the treated tumor mass. Additionally, localized OVT has been shown to be effective against distant metastases and disseminated disease. This allows for localized OVT of a single tumor mass which will be surgically removed to cause the destruction of micrometastases that cannot be detected or removed using surgical resection.27 28 Finally, OVT has been shown to directly alleviate several of the problems caused by surgical resection including angiogenesis.29–32 However, although the properties of OVT appear to make it an attractive immunotherapy to use in the neoadjuvant setting, there is only a limited body of literature examining this topic. Notably, while the first published work investigating OVT as a NAT was published in 200333 no follow-up studies were published until 10 years later.34 Interest in this concept appears to have finally gained traction, however, with a steady stream of manuscripts being published since the beginning of 2018 and six active clinical trials. The purpose of this review is to summarize the existing body of literature on the use of OVT as a NAT and highlight some of the major questions that remain to be addressed.
{kind=link}
Neoadjuvant oncolytic virotherapy (OVT) improves surgical outcomes. Vascularized tumor treated with OVT results in tumor cell death, localized inflammation, decreased vasculature, release of tumor antigens, and generation of tumor cell-specific lymphocytes. Surgical resection of the tumor results in incomplete margins and tumor cell dissemination. Tumor-specific lymphocytes eliminate remaining malignant cells preventing local recurrence and metastases. Figure created with Biorender.com.
Efficacy of OVT as a neoadjuvant therapy in preclinical models
A variety of oncolytic viruses have been shown to be effective as NATs in preclinical models. In particular, several studies have demonstrated the potential therapeutic use of OVT in the neoadjuvant setting for breast cancer including one study on the Maraba rhabdovirus35 and a second study comparing the efficacy of vesicular stomatitis virus (VSV), herpes simplex virus (HSV), adenovirus and reovirus. All of these studies involved a rechallenge model in which OVT was applied to a primary 4T1 tumor as a NAT followed by surgical resection of this tumor and secondary rechallenge of animals with a new bolus of 4T1 cells. The results demonstrated that all viruses except HSV decreased the volume of the directly treated primary tumors prior to surgery. Similarly, all viruses except reovirus delayed the growth of secondary tumors implanted postresection of the primary tumor. Interestingly, this included HSV which did not display any beneficial effects during treatment of the primary tumor. Critically, all the tested viruses except reovirus also conferred increased survival in these studies. Outside of breast cancer, another study examined the use of unmodified parapoxvirus ovis (ORFV) in a murine model of surgical stress.34 In this model, B16LacZ cells were administered intravenously followed 10 min later by abdominal nephrectomy, which was shown to dramatically increase the number of lung metastases formed. Using this model, the authors demonstrated that the surgically enhanced rate of metastasis to lungs was decreased by the neoadjuvant use of ORFV. Similar results were observed using ORFV in a second model of surgical stress using breast cancer cells.34 Taken together, these data suggest that there are a variety of options available when considering a viral backbone to develop for use as a NAT (table 1).
Preclinical studies of neoadjuvant oncolytic virotherapy
While unmodified oncolytic viruses often display efficacy in preclinical models, a significant body of literature has shown that this efficacy can be dramatically improved by genetic “arming” of the viral backbone.36 It is therefore not surprising that several studies have examined the potential of improving OVT as a NAT by using recombinant, armed viruses. Indeed, the first published work investigating the use of OVT as a NAT used an HSV armed with interleukin 12 (IL-12). In this study, hepatic malignancy in rats was treated with either unarmed HSV or HSV encoding IL-12 followed by surgical resection. Interestingly, while both viruses decreased the volume of treated tumors before surgery, treatment with HSV-IL12 decreased the size of these tumors to a greater extent than treatment with unarmed HSV. Additionally, only treatment with HSV-IL12 led to a significant decrease in the volume of secondary tumors, which were introduced through intraportal rechallenge after hepatectomy.33 In addition to HSV, another study investigated the use of vaccinia virus (VACV) armed with IL-12. VVΔTKΔN1L-mIL12 was generated from the Lister 15 strain of VACV by deletion of the genes encoding thymidine kinase (TK) and protein N1 (N1L) and inclusion of IL-12. When used in the neoadjuvant setting, this virus prevented both local recurrence and the development of distant metastases after surgical removal of primary tumors in murine models of Lewis lung cancer (LLC), breast cancer (LY2), HNC (HCPC1), and pancreatic cancer (DT6606) compared with surgery alone. The unarmed parental virus (VVΔTKΔN1L) also improved outcomes when used as a NAT in the LY2 and HCPC-1 models; however, this improvement was not as pronounced.25 Outside of IL-12, an attenuated VACV that contains the immune-modulating gene granulocyte macrophage colony-stimulating factor (GM-CSF), JX- 594, has also displayed efficacy as a NAT in the previously mentioned model of abdominal nephrectomy.34 However, this study did not directly address the role of the added GM-CSF by comparing efficacy to unarmed VACV.
In addition to the genetic arming of oncolytic backbones, another well-established method to improve the efficacy of OVT is to combine it with additional immune modifiers, particularly ICIs.37 Since ICIs have also shown efficacy as NATs, it is not surprising that groups have examined the combined use of both OVT and ICI in the neoadjuvant setting. Bourgeois-Daigneault et al examined the potential to combine NAT-Maraba virus with secondary ICI given postsurgery in several murine models of triple-negative breast cancer (TNBC). In this study, administration of PD-L1 and CTLA-4 blocking antibodies after NAT-OVT resulted in a significant decrease in the growth of secondary tumors implanted after surgical resection. This delay was not observed in mice treated with ICI’s in the absence of NAT-Maraba virus.35
While the majority of studies investigating the efficacy of OVT in the neoadjuvant setting have been conducted in surgical models, Yamada et al recently published a study using radiofrequency ablation (RFA) as the primary treatment. This study used HSV-G47Δ as a NAT prior to RFA and also examined the combination of this therapy with an ICI in a contralateral model of neuroblastoma (Neuro2a). The results demonstrated that treatment of a single tumor with NAT-OVT improved control of both the treated and non-treated tumors following RFA. All irradiated tumors were eradicated and no local recurrence occurred by day 26 while untreated tumors showed decreased/delayed tumor growth compared with control. Additionally, when mice were rechallenged with secondary tumors on the day of RFA, a significant decrease was observed in the engraftment rate in mice treated with NAT-HSV-G74Δ compared with RFA alone (4/7 vs 5/5, respectively). As in the previously mentioned studies, addition of anti-PD-L1 antibody improved the efficacy of NAT-HSV-G74Δ treatment, as demonstrated by delayed/decreased tumor growth of untreated contralateral tumors in this group compared with those receiving no NAT or anti-PD-L1 antibody alone.26 Taken together, these data suggest that numerous oncolytic viruses possess potential to be effective as NAT and that methods to improve this efficacy might also exist.
Clinical studies of OVT as a neoadjuvant therapy
Several clinical trials have demonstrated the efficacy of OVT in the neoadjuvant setting, with more trials currently in progress and awaiting results. While the majority of these trials are investigating the use of the Food and Drug Administration (FDA)-approved Talimogene Laherparepvec (T-VEC) to treat melanoma via intratumoral injection, other viruses, cancer types, and modes of administration are also being studied. Additionally, trials have sought to evaluate potential effects of combining chemotherapy or ICIs with OVT in the neoadjuvant setting (table 2).
Clinical trials of oncolytic virotherapy as a neoadjuvant
A phase 2 trial investigating T-VEC as a NAT studied its efficacy in stage IIIB–IVM1a melanoma as an intratumoral injection. A total of 150 patients were treated with either T-VEC followed by surgery or surgery alone. The NAT arm demonstrated improved estimates for 2-year relapse-free survival (RFS) (29.5% vs 16.5%), OS (88.9% vs 77.4%), and 3-year RFS (28.1% vs 16.9%). Combined, a 25% reduction in risk of disease recurrence was estimated for the group receiving T-VEC and surgery compared with the group receiving surgery alone.38 39
While T-VEC is FDA approved for the treatment of unresectable metastatic stage IIIB/C–IVM1a melanoma, a phase 1/2 trial also sought to evaluate its use (in combination with paclitaxel) as a NAT in patients with non-metastatic stage II–III TNBC. In this one-armed trial, the end point was a pathological complete response rate (pCR) defined as the disappearance of histopathological evidence of malignant cells in breast and axillary lymph nodes for up to 3 years. Patients demonstrated a pCR of 43.24% at the maximum-tolerated dose, which was determined in the preceding phase 1 trial.40 For comparison, a meta-analysis of patients with stage II–III TNBC receiving NAT involving platinum-based chemotherapy demonstrated a pCR of only 31%.41 Results on RFS and OS rates are expected to be collected for this trial by September 2023.42
A different oncolytic HSV, G207, with deletion of both γ134.5 loci and insertional inactivation of UL39 was tested for safety in a phase 1b trial. Six patients with resectable recurrent malignant glioma were administered G207 by a stereotactically placed catheter, which delivered 13% of the final viral load 2–5 days before surgery. At the time of en bloc resection, the remainder of the virus was inoculated into the surgical cavity. While efficacy could not be determined for this small cohort, the safety outcomes were encouraging. No patients left the study due to adverse effects and no patients demonstrated encephalitis or required acyclovir administration. Additionally, none of the peripheral samples (saliva, urine, conjunctiva, or serum) were positive for the LacZ insert gene, while excised tumor tissues from all patients were LacZ positive, demonstrating the feasibility of this delivery method for NAT-OVT.43
In addition to these completed studies, several ongoing trials are also examining the use of T-VEC as a NAT. A single-arm, phase 2 study investigating biomarkers for response to T-VEC as a NAT in high-risk early melanoma is currently in progress with the goal of developing protocols to predict responders and non-responders. The primary outcome measures are pathological response at the time of surgery based on immune response and molecular changes in residual tumors; RFS and OS will also be measured. The estimated time for study completion is May 2024.44 Another single-arm, phase 2 study is investigating T-VEC in combination with nivolumab as a NAT for early metastatic stage IIIB/C/D–IV M1a melanoma. The primary outcome measure is pathological response, with the secondary outcomes being rate of delay of surgery, rate of failure to perform surgery, RFS, safety, and analysis of tissues for biomarker research. This study is expected to be completed in August 2023.45 Finally, another single-arm, phase 2 study is investigating T-VEC injected directly into palpable lymph nodes in combination with pembrolizumab prior to lymph node dissection in stage III melanoma. The primary outcome measured will be pathological response rate in the regional nodal basin after lymph node resection, while the secondary outcome will be safety and tolerability of the combination treatment prior to surgery. It is estimated that this study will be completed on March 1, 2022.46
Outside of T-VEC, a completed phase 2 trial examined IT-delivered HSV HF10 in combination with intravenous nivolumab prior to resection of stage IIIB, IIIC, IVM1a melanoma. The primary end point was recurrence after surgery which was determined by both radiological scans and biopsy. Death within the follow-up period was also considered recurrence. RFS was measured for up to 2 years and was accomplished in two of six patients (33.3%). OS and pCR were seen in five of six patients (83.3%). Critically, grade 3–5 adverse effects related to HF10 were experienced by only one of seven patients (14.3%) treated in this trial.47
While administration of OVT is generally accomplished via intratumoral injection, Samson et al demonstrated the therapeutic potential of intravenous administration in delivering OVT to the brain in a window-of-opportunity clinical study. This study investigated intravenous administration of reovirus to patients with recurrent high-grade gliomas or brain metastases (EudraCT 2011-005635-10). Presurgical administration of virus resulted in expression of viral capsid σ3 mRNA and protein as well as increased abundance of both CD3 and CD8 proteins in tumors.48 While no efficacy data were provided, the demonstration of the possibility of intravenous administration to brain tumors is encouraging. Another phase 2a study also investigated the effects of intravenous or intratumoral administration of JX-594 in the neoadjuvant setting in patients with metastatic colorectal tumors within the liver. The study was completed in March of 2013, but efficacy results are not available, although changes to the peripheral blood mononuclear cells collected as part of the trial are described below.49
In addition to intratumoral and intravenous administration, intravesical administration is also being studied for the delivery of virus directly into the bladder. A phase 1 study in which virus was administered to patients with bladder carcinoma who were scheduled for subsequent cystectomy sought to assess the safety and feasibility of intravesical administration of two recombinant fowlpox (rF) viruses, as well as to obtain correlative data on the efficiency of viral infection and gene function and host immune response to treatment. Patients received either rF-GM-CSF or rF-TRICOM, which encodes B7.1, ICAM-1, and LFA-3. The potential for functional transgene delivery was demonstrated by positivity for the LacZ gene in all six tested patients. Systemic immunity was demonstrated by increased sera antibodies to LacZ and fowlpox compared with pretreatment in all tested patient sera.50 51
Finally, an ongoing phase 1 study is investigating an attenuated intravesical Measles virus encoding the thyroidal sodium iodide symporter delivered directly into the bladder of patients with urothelial carcinoma. The primary outcomes measured are safety and toxicity of the virus prior to surgery, while the secondary outcome is pathological staging at the time of surgery, as a preliminary measure of the OVT efficacy. The estimated study completion date is December 2022.52 Obviously, there remains much work to do to enhance the use of OVT as a NAT; however, these existing clinical results generally support that this concept is safe for patients and worth additional exploration.
Mechanism(s) for OVT as a neoadjuvant therapy
OVT is known to function through numerous mechanisms including direct cytotoxicity and induction of antitumor immunity. Unfortunately, the majority of studies examining OVT in the neoadjuvant setting have produced correlative, but not necessarily causative, data. Because of this, the exact mechanism(s) mediating the impact of OVT as a NAT remain unclear. For the most part, however, these studies seem to align with current oncolytic paradigms suggesting that the effects are mostly immune mediated and that T cells likely play a major role.
Direct cytotoxicity
Several published studies investigating OVT in the neoadjuvant setting have demonstrated direct killing of infected cells suggesting this as one mechanism through which these therapies may assist in tumor control postsurgery. HSV, armed with IL-12 or not, decreased the viability of Morris hepatoma cells,33 and a decrease in the number of viable cells was also found in mouse and human liver carcinoma and mouse neuroblastoma following HSVG47Δ treatment.26 Similarly, treatment with VSVd51 led to a necrotic phenotype in 4T1, MDA-MB-231 and BT-549 TNBC cells as demonstrated by light and electron microscopy, as well as increased high mobility group box protein 1 (HMGB1) and ATP in supernatants and increased cell surface calreticulin.53 Another study used H&E staining to show that NAT-Maraba caused an increase in necrotic tissue within TNBC patient-derived xenograft (PDX), luminal B breast cancer PDX, and 4T1 tumors.54 To our knowledge, however, none of these studies demonstrated a functional impact of this direct cell lysis on the efficacy of OVT in the neoadjuvant setting. Additionally, while this mechanism may be important in decreasing the size of the treated tumor to facilitate surgery or radiation, the beneficial effects of OVT as a NAT against disseminated disease will more likely lie in their ability to prime the immune system to target local recurrence and micrometastases.
T cells
A study using Maraba virus as a NAT found increased CD3+ T cells as well as T regulatory cells (Tregs) in virally treated tumors compared with untreated controls in a murine model of TNBC. This treatment resulted in decreased volume of primary and rechallenge tumors as well as improved survival. These effects were likely to be T-cell dependent, as they were not observed in nude mice. Additionally, the requirement for the second tumor to be of the same genotype as the first tumor was observed, demonstrating antigen specificity. The authors also showed increased interferon-γ (IFNγ) on restimulation of splenocytes which was abrogated by blocking antibodies against IFN-αR1 but not CXCR3.35 Interestingly, this work also demonstrated that media from Maraba-infected cells increased the migration of splenocytes in vitro and that this was dependent on CCL2, CCL5, and CXCL9–11 suggesting that infection altered the cellular secretome.35 Similarly, NAT treatment of pancreatic or lung cancer with VVΔTKΔN1L also caused an increase in the number of IFNγ+ splenocytes following ex vivo stimulation with either growth-arrested cells or mesothelin peptide. Additionally, treatment with VVΔTKΔN1L also increased the number of effector cells (CD44hiCD26Llo) as a per cent of CD45+ CD3+ CD8+ splenocytes in both models. A further increase in effector cells as well as CD8+ and CD4+ cells was observed following treatment with VVΔTKΔN1L-mIL12; however, this was only tested in the pancreatic model.25
In Yamada et al’s study using HSV-G47Δ as a NAT to RFA, viral treatment increased CD8+ but not CD4+ cells as a percent of CD45+ cells in contralateral untreated tumors. There was also an increase in T helper and Treg cells in cohorts treated with RFA plus NAT-G47Δ compared with control, but these differences were not significant compared with either monotherapy. Functionally, combination therapy significantly increased IFNγ in the serum 7 days after radiation as well as increased the number of IFNγ+ splenocytes observed following stimulation with Neuro2a or control Sal/N cells. Importantly, addition of anti-CD8 antibody completely abrogated the combination therapy-mediated decrease in the volume of untreated contralateral tumors directly implicating T cells as the effectors.26 In contrast, in a pancreatic mouse model the increase in survival observed following NAT-VVΔTKΔN1L was not lost following depletion of either CD8+ or CD4+ cells, although depletion of CD8+ cells did cause a slight increase in tumor volume.25 These results suggest that depletion of CD8+ T cells plays differing levels of importance in different tumor models.
In human patients, Dummer et al saw increased CD8+ T-cell density after NAT with T-VEC.38 Critically, patients with an increase in IT CD8+ cell density displayed improved RFS (35% vs 10%) and 2-year OS rates (96% vs 70%) compared with patients who showed no change or decreased CD8+ cells although the authors asserted that the sample size was too small for true comparison and further validation was required. Similarly, in a phase 1 trial with T-VEC plus chemotherapy followed by surgery for non-metastatic stage II–III TNBC, Soliman et al saw a significant increase in T-cell clonality in resected tumors after combination treatment suggesting an ongoing clonal response. There was also a significant decrease in FoxP3+ Tregs, increased infiltration by lymphoid cells, a significant decrease in CD45RO+ memory T cells, and a trend toward increased CD8+ T cells. Unfortunately, no data matching immune response to clinical response were presented, and the small sample size begs for more patients to provide a more definitive pattern.40 Furthermore, in patients with bladder carcinoma, rF-GM-CSF or rF-TRICOM both demonstrated increased T-cell infiltration by positive immunohistochemistry (IHC) staining for CD3, CD4, CD8, and CD45RO compared with comparable cystectomy specimens used as untreated controls. Interestingly, there was no difference between tumor and non-tumor adjacent tissues. Taken together, these data fairly strongly supported the concept that viral NAT can increase the overall recruitment of T cells into remaining tumor beds even after the majority of the tumor is excised and that these T cells might influence the outcomes of surgery.
NK cells
An important role for NK cells in controlling tumor metastasis has been previously demonstrated.55 In this context, NK cells recovered from mice given viral-NAT displayed increased cytotoxicity in Yamada et al’s model of surgical stress26 and Ahmed et al saw increased NK cells (CD3-CD49b+) as a percentage of CD45+ cells in the blood of lung cancer bearing mice when treated with NAT-VVΔTKΔN1L.25 Additionally, the neoadjuvant use of JX-594 in human patients increased NK-mediated cytotoxicity postoperative compared with preoperative in a phase 2 clinical trial.34 Interestingly, despite the consistent increase in NK cell populations, the functional impact of these cells remains unclear. Ahmed et al saw that the delay in mortality resulting from NAT-OVT in their lung cancer model was fully dependent on NK cells.25 In contrast, the abrogating effect of ORFV and JX-594 on surgery-induced metastases was not dependent on NK cells (though a slight decrease in the effect is seen).34 More work is therefore needed to fully elucidate the functional impact of NK cells on OVT-NAT.
Innate immunity
While most of the effects of OVT have been attributed to adaptive immunity, a critical role for innate immunity has also been observed in some settings, including activation of the cellular cGAS-STING pathway on OVT-mediated sensitization to cisplatin.25 35 50 56 While only a limited number of studies have examined the role of innate immunity in OVT-NAT, treatment with VVΔTKΔN1L-mIL12 was shown to result in increased CD11b+GR1+ and decreased CD11b+F4/80+, as a percentage of CD45+ cells, in a murine LLC model. An increase in intratumoral IL-1α and IL-1β, IL-6, granulocyte colony stimulating factor (GCSF), regulated upon activation, normal T cell expressed and presumably secreted (RANTES), macrophage inflammatory protein 1a (MIP-1α) and keratinocyte chemoattractant (KC) was also observed in this setting. Additionally, ex vivo infection of dendritic cells and macrophages with VVΔTKΔN1L-mIL12 caused increased concentrations of KC, GCSF, MIP-1α, IL-1α and IL-1β, and IL-18 as well as increased expression of CD80, CD86, and major histocompatibility complex type I (MHCI).25 Similarly, the neoadjuvant use of Maraba virus in either 4T1 or EMT6 tumors increased expression of PD-L1 and a large panel of innate inflammatory mediators. Infected cells also demonstrated increased p-STAT, and p-IRF3 which was dependent on RIG-I expression as well as increased IL-6 expression which was dependent on MYD88.35 In patients with human bladder cancer, the NAT use of rF-GM-CSF or rF-TRICOM resulted in increased dendritic cell infiltration as demonstrated by IHC staining for factor XIIIa compared with comparable cystectomy specimens used as untreated controls.50 Unfortunately, none of these works examined the functional impact of these innate immune changes, suggesting that more work in this area is needed.
Conclusions
Numerous studies have shown that OVT applied in the neoadjuvant setting can improve therapeutic outcomes and that multiple viral platforms can be effective in this context. Additionally, several lines of study, including “armed” recombinant viruses and the combination of viral-NAT with ICI appear poised to further improve this approach’s clinical potential. Unfortunately, due to the relatively recent interest in using oncolytic viruses as NATs, numerous questions remain unanswered. First, additional functional studies dissecting the relative causative versus correlative effects of OVT would be helpful in defining the effectors mediating this form of treatment. In this context, clinical NAT-OVT studies not only provide potential improvements to patient outcomes but also create an opportunity to explore the fundamental mechanisms involved in OVT. Excised tumor tissue can be examined for viral replication, expression of transgenes, tumor perfusion, immune infiltration, and potential impact of the cellular matrix and the longitudinal nature of NAT provide the opportunity to correlate these factors with clinical outcomes. Additionally, the potential use of NAT-OVT early in treatment avoids the potential confounding effects of treating patients who have already undergone immune-suppressive treatments, including chemotherapy or radiation. Second, in addition to the work reviewed here, numerous studies have also examined the direct injection of oncolytic viruses into the surgical cavities immediately post-resection (perioperative OVT),57 58 including the study by Markert et al detailed above, in which OVT was administered both as a NAT and perioperatively.43 While these studies suggest that this approach is also clinically viable, the mechanism(s) and therapeutic potential of OVT in the two settings might be quite different due to the limited amount of malignant tissue remaining postresection. For example, it is quite possible that delaying surgery to provide a course of NAT-OVT could have negative consequences. However, if OVT is administered after tumor resection, there are theoretically no tumor cells for the virus to infect, replicate in, or use as a source of antigens for presentation. Unfortunately, while there is a body of literature comparing the use of other immunotherapies in the perioperative versus neoadjuvant settings,20 59 60 basically no such literature exists for the use of oncolytic viruses. The field therefore needs to address the potential benefits or downfalls of perioperative versus NAT with OVT. Third, as is largely true for the oncolytic field in general, how the complex tumor microenvironment impacts overall therapeutic efficacy remains largely unknown. Because of this, it is difficult to rationally design the next generation of improved treatments. As a single potential example, tumors are known to contain highly heterogeneous populations of malignant cells. However, how this heterogeneity impacts the outcomes of OVT in either the context of primary treatment or NAT remains unclear. Studies defining the mechanisms behind therapeutic failure of oncolytic NAT are therefore urgently needed. Finally, it is worth noting that not all of the effects of NAT-OVT are likely to be positive to surgical outcomes. For example, stressed or dying cells have been shown to release HMGB1 in response to viral infection. This release, in turn, can cause localized edema, which is a potentially fatal complication during surgical resection.61 62 While clinical trials involving NAT-OVT have generally proven extremely safe, some care must therefore still be exercised when using this form of therapy. Nevertheless, the use of OVT in the neoadjuvant setting appears attractive and the concept should continue to be explored.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Contributors EB and RJT conceived of the manuscript; RJT researched and wrote the manuscript; EB provided edits and rewrites.
Funding This work was supported by grants to Dr Eric Bartee from NIH-NCI (R01-CA194090), NIH-NIAID (R21-AI142387), the ACS (RSG-17-047-01-MPC), and intramural funding from both the Medical University of South Carolina and the University of New Mexico Health Sciences Center, and to Dr Raquela Thomas (NIH-K12-2GM088021).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.