Article Text

Abstract
Background People living with HIV (PLWH) have increased risk of developing cancers after controlling traditional risk factors and viral suppression. This study explores whether T cells can serve as a marker of risk for cancer among HIV-infected virally suppressed patients.
Methods A nested case control study design was pursued with 17 cancer cases and 73 controls (PLWH without cancer)ouidentified among the US Military HIV Natural History Study cohort, and were matched for CD4 + count, duration of HIV infection, and viral suppression. Cells were obtained from PLWH on an average of 12 months prior to clinical cancer diagnosis. Expression of inhibitory receptors (PD-1, CD160, CD244, Lag-3, and TIGIT), and transcription factors (T-bet, Eomesodermin, TCF-1, and (TOX) was measured on CD8 +T cells from that early time point.
Results We found that cases have increased expression of PD-1 +CD160+CD244+ (‘triple positive’) on total and effector CD8 + compared with controls (p=0.02). Furthermore, CD8 +T cells that were both PD-1 +CD160+CD244+ and T-betdimEomeshi were significantly elevated in cases at time point before cancer detection, compared with controls without cancer (p=0.008). This was driven by the finding that transcriptional factor profile of cells was altered in cancers compared with controls. Triple-positive cells were noted to retain the ability for cytotoxicity and cytokine secretion mediated by expression of CD160 and PD-1, respectively. However, triple-positive cells demonstrated high expression of TOX-1, a transcription factor associated with T cell exhaustion.
Conclusion In conclusion, we have found a subset of dysfunctional CD8 +T cells, PD-1 +CD160+CD244+T-betdimEomeshi, that is elevated 12 months before cancer diagnosis, suggesting that peripheral T cell alterations may serve as a biomarker of increased cancer risk among PLWH.
- CD8-Positive T-Lymphocytes
- Tumor Biomarkers
- Translational Medical Research
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Patients Living with HIV (PLWH) are at increased risk for AIDS-defining cancers as well as some non-AIDS defining cancers, and have a worse prognosis.
PLWH have evidence of high levels of a subpopulation of CD8+ T cells with an activated/exhausted phenotype (PD-1+CD160+CD244+TbetdimEomeshi) 1 year prior to their cancer diagnosis, and which is significantly elevated compared with PLWH without cancer.
This CD8+ T cell subset is highly differentiated and activated, but harbors some functional impairment, with high levels of transcription factor thymocyte selection-associated HMG box.
The ability to identify a T cell subset in peripheral blood 12 months prior to cancer diagnosis, highlights the possibility of using peripheral blood biomarkers to identify patients at high risk for cancer.
Introduction
The introduction of combination antiretroviral therapy (cART) has led to a decrease in incidence of AIDS-defining cancers (ADCs) such as Kaposi sarcoma and non-Hodgkin’s lymphoma (NHL) in people living with HIV (PLWH).1 However, ADCs remain increased in incidence in PLWH compared with HIV-uninfected individuals, even in the setting of cART.2 In addition, some non-AIDS-defining cancers, including non-small cell lung cancer, Hodgkin lymphoma, hepatocellular carcinoma, anal and other human papilloma virus (HPV)-related cancers, are also increased in incidence among PLWH.
A higher prevalence of known risk factors for malignancy, such as smoking, alcohol use, and viral co-infections, contribute to this increased risk among PLWH, but large epidemiologic studies have consistently identified HIV infection as an independent risk factor.3 Untreated HIV infection is associated with CD4+ T cell depletion, chronic immune activation, and evidence of immune dysfunction.4 5 With cART, there is evidence of significant immunologic recovery with increased circulating CD4+ T cell counts, decreases in T cell activation, and improvement of other immunoregulatory markers including decreases in regulatory T cells and programmed death-1 (PD-1) expression.6–8 Despite this improvement, many immune parameters remain abnormal compared with those without infection, including depressed CD4:CD8 ratios, higher levels of chronic immune activation, higher rates of pro-inflammatory cytokines, decreased naïve T cell counts, and high proportions of terminally differentiated CD4 +and CD8+T cells.9–11
T cell exhaustion has also been described in the setting of HIV infection,12 13 including evidence of high-level expression of multiple inhibitory receptors, including PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), CD244 (2B4), lymphocyte-activation gene-3 (LAG-3), CD160 Ig-like glycoprotein (CD160), T cell immunoglobulin and ITIM domain (TIGIT) and T-cell immunoglobin and mucin domain containing-3 (Tim-3).6 14–20 Most significantly, even after ART-induced viral suppression, markers of T cell dysfunction remain elevated compared with uninfected individuals.6 21 Additionally, several transcription factors that play a crucial role in T cell exhaustion in HIV such as T-bet, Eomesodermin (Eomes), Forkhead Box O1 (FOXO1), Basic Leucine Zipper ATF, thymocyte selection-associated HMG box (TOX), and transcription factor T cell factor 1 (TCF-1),6 22–24 have additionally been associated with HIV pathogenesis. However, the presence of these altered circulating cellular markers of immune dysfunction have never been evaluated in the context of cancer risk because of the absence of appropriate longitudinal cohorts.
T cell exhaustion is also well-described in the tumor microenvironment and likely a major mechanism by which tumor growth is enabled.25–27 Blockade of these immune checkpoints, including CTLA-4 and PD-1, has resulted in substantial improvement in clinical outcomes among a subset of patients harboring different cancer types, emphasizing the role of impaired T cell activity in controlling tumor progression.28 29 Given the prominent role of T cell activity in the antitumor response and rising numbers of cancers among people with HIV, we hypothesized that persistent T cell exhaustion due to chronic HIV infection, even after durable suppression with ART, may impair the ability of the immune system to perform effective tumor surveillance leading to increased cancer incidence. In order to address this question, we utilized The US Military HIV Natural History Study (NHS), a longitudinal cohort study and biorepository, to characterize the circulating immune phenotype, including markers of T cell exhaustion, activation, and function, among individuals virally-suppressed on cART who subsequently were diagnosed with cancer, compared with those who never developed cancer. By looking for markers with differential expression among those who subsequently develop cancer, we hope that a biomarker to identify patients at high risk may be developed.
Methods
Study design and subjects
The US Military HIV NHS is a well-characterized longitudinal cohort of Department of Defense beneficiaries with >5000 participants.30 To utilize a nested case–control design, the NHS repository was queried to identify PLWH with a diagnosis of lung cancer, HPV-associated cancers (defined as head and neck cancer, anal cancer, and cervical cancer), and NHL, with sustained viral suppression for at least 3 years on cART at the time of their cancer diagnosis. Within NHS, cancer diagnosis is coded based on chart review and abstraction. Peripheral blood mononuclear cells (PBMCs) were requested from a time point at least 6 months prior to cancer diagnosis. Controls were identified as PLWH without any cancer diagnosis, and identified utilizing the Greedy Algorithm requesting four controls for each case. Controls were matched to cases by the following criteria: CD4 count (±75), gender, age (±5 years), years of HIV suppression (±5 years). The controls selected for each case were those closest to the case in terms of the weighted sum of the absolute differences between the case and control matching factors.
Functional T cell assays were additionally performed on cryopreserved PBMC of 10 HIV-infected patients receiving cART and without a cancer diagnosis from Yale New Haven Hospital (YNHH) outpatient clinics (New Haven, Connecticut, USA). The current study was approved by Yale University School of Medicine and the Uniformed Services University (USU) institutional review boards, and informed consent was obtained.
PBMC staining and flow cytometry analyses
NHS samples: Cryopreserved PBMCs were thawed, rested and stained as previously described.9 Cells were stained with live/dead fixable red dead cell stain kit (Thermo Fisher Scientific, Massachusetts, USA), and then for 20 min at room temperature in the dark with fluorochrome-conjugated antibodies against surface markers CD3, CD8, PD-1, CD160, CD244, TIGIT, CD38, Human Leukocyte Antigen-DR (HLA-DR), LAG-3, CCR7, and CD45RA. For each panel of markers, we used fluorescence minus one controls for each marker of interest as the most rigorous method to standardize gating to determine positive versus negative events.31 32 After washing with buffer, cells were permeabilized with intracellular fixation and permeabilization buffers (Thermo Fisher Scientific, Massachusetts, USA) prior to being washed and stained for T-bet and Eomes antibodies for 1 hour at 4°C. Finally, the cells were fixed in phosphate buffered saline containing 1% paraformaldehyde prior to acquisition on the LSRFortessa flow cytometer (BD Biosciences). FlowJo software (BD BioSciences; California, USA) was used for analysis.
YNHH samples: Cryopreserved PBMCs from PLWH with no known cancer diagnosis were thawed and rested overnight . PBMCs were washed and stimulated with 1 μg/mL Staphylococcus endotoxin B (SEB; List Biological Laboratories, California, USA) in the presence of Brefeldin A (Thermo Fisher Scientific, Massachusetts, USA) at 37°C and 5% CO2 for 16 hours. PBMCs were stained with live/dead marker, and antibodies to CD3, CD8, PD-1, CD160, and CD244 antibodies for 20 min at room temperature. After intracellular fixation and permeabilization as above, cells were stained with antibodies against : granzyme B, perforin, CD107a, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α for 1 hour at 4°C, prior to fixation and acquisition on the flow cytometer. For expression of TOX and TCF-1, cryopreserved PBMC from PLWH were stained with CD3, CD8, PD-1, CD160, and CD244 antibodies followed by intracellular staining with TOX and TCF-1.
The following antibodies used: anti-CD3-AlexaFluor700, anti-PD-BV42, anti-CD244-PerCP-Cy5.5, anti-CD107a-BV650, anti-granzymeB-APC/Fire750, anti-perforin-APC, and anti-CCR7-BV650 (BioLegend; California,USA); anti-CD8-V500, anti-IFN-γ-BV421, anti-HLADR-V500, CD25-PE, CD127-BV421, CCR4-PE-Cy7 and anti-CD38-APC (BD BioSciences; California,USA); anti-CD8-APC-eFluor780, anti-CD160-FITC, anti-LAG-3-PECy7, anti-TIGIT-PerCP-eFluor710, anti-CD45RA-APC-eFluor780, anti-T-bet-PE, anti-Eomes-eFluor660, FoxP3-APC and anti-TNF-α-Pe-Cy7,anti-TOX-APC- (Thermo Fisher Scientific, Massachusetts,USA) and anti-TCF-1Pe-Cy7 (Cell Signaling, Massachusetts,USA).
Statistical analyses
Categorical variables are summarized as frequencies and percentages, and continuous variables as median (IQR). Differences between cases and controls were evaluated using χ2 test or Fisher’s exact test for categorical variables, and Wilcoxon-rank-sum test for continuous variables. To account for the correlation on measurements taken from the same subject, the proportion of cell expression in PD-1+, CD160+, CD244 +or PD-1 +CD160+CD244+ were compared between T-betdimEomeshi and T-bethiEomesdimusing a linear mixed-effects model. Spearman correlation between the proportion of T-betdimEomeshi cells and PD-1+, CD160+, CD244+, or PD-1 +CD160+CD244+CD8+T cells was also computed. P values less than 0.05 were considered to be statistically significant. Statistical analyses were performed using R V.4.1.0.
Results
Cases were defined as individuals with HIV infection and with viral suppression at the time of cancer diagnosis, whereas controls were matched individuals with HIV infection who were never diagnosed with cancer (‘controls’). Twenty-three cases and 73 control subjects were identified for analysis. Seventeen cases had samples available prior to cancer diagnosis with median time between sample collection and subsequent cancer diagnosis among cases of 1.0 year (IQR, 0.93, 1.13). These cases and controls were well-matched (table 1) by age, gender, race, duration of HIV infection, CD4+ T cell count, CD4 nadir, CD8+ T cell count, absolute lymphocyte count, and years of viral suppression with no statistical difference between these variables (p>0.05 for each). The median age of cases and controls at time of sample collection was 48.0 (IQR: 43, 54) and 49.0 (IQR: 44, 55) years, respectively. At the time of cancer diagnosis, cases and controls had been infected with HIV for 14.0 (IQR: 7, 21) and 16.0 (IQR: 12, 20) years, respectively. All subjects were receiving cART and had HIV viral loads of <400 copies/mL. The median CD4 count among cases was 471 cells/mm3 and among controls was 448 cells/mm3. The CD4 T cell nadir among cases was 115 cells/mm3 and among controls was 210 cells/mm3. The median time between sample collection and subsequent cancer diagnosis among cases was 11 (IQR, 11–13) months.
Baseline characteristics of cancer cases and controls
CD8+T cells co-expressing PD-1, CD160, and CD244 are elevated in HIV-infected patients with cancer
Expression of immune inhibitory receptors PD-1, CD160, and CD244 was quantified using flow cytometry (figure 1A) from cancer cases compared with control subjects with no cancer diagnosis, and no difference was noted between the two groups when each of the receptors were measured individually. Distribution of CD8 +T cells based on expression pattern of all three receptors was measured. We found that mono-expression or dual-expression of PD-1, CD160, and/or CD244 among CD8+ T cells was comparable between cases and controls (figure 1C). However, the co-expression of all three inhibitory receptors (PD+CD160+CD244+, or ‘triple-positive cells’) was significantly increased in cases compared with controls (p=0.02) (figure 1C). In a subgroup analyses limiting the inclusion of cases to only those with samples available >12 months prior to cancer diagnosis (n=8), the increase in triple-positive CD8+ Tcells compared with controls without cancer remained significant despite the small number of samples (p=0.04, data not shown).

Expression of inhibitory receptors on CD8 +T cells: (A) Representative flow cytometry plots showing expression of inhibitory receptors PD-1, CD160 and CD244 on CD8 +T cells. (B) The percentage of cells expressing PD-1, CD160, and 2B4 among total CD8 +T cells in control (gray)and case (red) groups. (C) Proportion of cells among CD8 +T cells expressing combination of PD-1, CD160 and CD244 among total CD8 +T cells from cases and controls. Plot represents median with IQR. Non-parametric Wilcoxon rank-sum test was applied to compare between two groups. PD-1, programmed death-1.
CD8 +T cell subsets further identified as distinct subpopulations of naïve and memory cells by using surface receptors CD54RA and CCR7: naïve (CD45RA+CCR7+), central memory (CD45RA−CCR7+, or CM), effector memory (EM) (CD45RA–CCR7–, or EM), and terminally differentiated EM re-expressing CD45RA (CD45RA+CCR7−, or TEMRA) (figure 2A). There was no difference in the distribution of the four naive/memory subsets between cases and controls (SF1; p>0.05 for all subsets). However, among EM CD8 +T cells, the proportion of triple-positive cells (PD+CD160+CD244+) was significantly higher among cases compared with controls (p=0.04) (figure 2B) suggesting that cells with a more differentiated state have altered phenotypic characteristics among patients at risk for cancer.

Inhibitory receptor expression among memory cell subsets: (A) A flow cytometry plot showing the subsets of CD8+ T cells using cell surface markers central memory (CD45RA− CCR7+), naïve (CD45RA+CCR7+), TEMRA (CD45RA+CCR7−) and effector memory (CD45RA−CCR7−) subpopulations. (B) Proportion of PD-1 +CD160+CD244+ expression pattern among CD8 + memory cell subsets among cases and controls. Median and IQR presented . Non-parametric Wilcoxon rank-sum test was applied to compare between two groups. EM, effector memory; PD-1, programmed death-1.
Other T cell inhibitory receptors (TIGIT, Lag-3, and inducible T cell costimulator (ICOS)) and markers of immune activation (CD38 and HLA-DR) were also studied, but no difference was noted between cases and controls (SF2). CD4 +T cell nadir was not associated with frequency of CD8+ ‘triple positive cells’ (data not shown).
T-bet and Eomes expression on CD8+T cell subsets differentiate cases from controls
The transcription factors T-bet and Eomes play an important role in the regulation of exhaustion and memory fates of CD8 +T cells.33 T-betdimEomeshi CD8 +T cells have been associated with an EM phenotype in patients in acute and chronic infection, associated with expression of inhibitory receptors, and associated with poor viral control among PLWH.6 In the cancer tumor microenvironment, high T-bet expression on tumor-infiltrating lymphocytes is also associated with improved survival.34 35
We thus evaluated subsets of cells for expression of T-bet and Eomes (figure 3A). We found that cases demonstrated a significantly higher proportion of T-betdimEomeshi CD8+ T cells compared with controls (p=0.047; figure 3A). This was largely driven by differences between the two groups within the EM CD8 +T cell subset (p=0.009, figure 3B). We further evaluated whether subsets differentiated by inhibitory receptor expression have altered T-betdimEomeshi. Of note, because PD-1–CD160+CD244- and PD-1 +CD160+CD244– subsets on CD8 +T cells were minimally present among both patient groups (figure 1C), further characterization of these subsets was not pursued. Thus, six phenotypes of CD8 +T cells based on the expression of PD-1, CD160, and CD244 were studied. As expected, cells that do not express any inhibitory receptors (PD1–CD160–CD244–) had the lowest expression of T-betdimEomeshi cells (figure 3C). Cells that express all three inhibitory receptors (triple-positive, PD-1 +CD160+CD244+) had the highest proportion of cells that are T-betdimEomeshi, significantly higher than those cells that express zero, one or two inhibitory receptors (p<0.05 for all comparisons, figure 3C). This was true for both cases and controls, suggesting that triple-positive cells have an altered transcriptional state compared with other subsets. Of notable interest, among all PD-1 expressing subsets, whether single (PD-1 +CD160 CD244), double (PD-1 +CD160 CD244), or triple positive, there was a significantly higher proportion of T-betdimEomeshi cells among cases compared with controls (p=0.02, p=0.04, and p=0.02, respectively, SF3A), suggesting an altered transcriptional profile among CD8+ T cells from patients with cancer, even among cell subsets that express comparable levels of inhibitory receptors. This finding led to the discovery that the percentage of CD8+ T cells that are both triple positive and maintain a T-betdimEomeshi phenotype are significantly higher among cases compared with controls (p=0.008, figure 3D). The combination of these markers thus identifies a small, but highly differentiating, subset of CD8+ T cells among PLWH with viral suppression who are at risk of cancer. Consistent with this association, a positive correlation was observed between proportion of T-betdimEomeshi CD8+ T cells and expression levels of PD-1, CD160, CD244 individually or collectively (p<0.05 for all correlations; online supplemental figure SF3B).
Supplemental material

CD8 +Transcriptional factor expression: (A) Gating strategy to distinguish the T-betdimEomeshi and T-bethiEomesdim gated on CD8 +T cells. Proportion of cells with expression of T-betdimEomeshi and T-bethiEomesdim among total CD8 +T cells within controls (gray) and case (red) group. (B) The proportion of cells with expression of T-betdimEomeshi expression on subsets of CD8 +T cells in control and case group. (C) The proportion of cells with expression of T-betdimEomeshi within mono and co-expression of PD-1, CD160 and CD244 on CD8 +T cell subsets in case and control group. (D) Among total CD8+ T cells, proportion of cells with expression of T-betdimEomeshi and PD-1 +CD160–CD244–, PD-1 +CD160 CD244+ or PD-1 +CD160+CD244+. Bar represents median with IQR. Non-parametric Wilcoxon rank-sum test was applied to compare between two groups. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. CM, central memory; EM, effector memory; PD-1, programmed death-1; TEMRA, Terminally differentiated effector memory re-expressing CD45RA.
Triple-positive cells retain cytotoxic and cytokine signature, but demonstrate some decrease in function compared with other subsets
To determine the function of CD8 +T cells expressing PD-1, CD160, and CD244, we stimulated cells from an independent cohort of PLWH (online supplemental table 1) with SEB, a polyclonal stimulus that engages the T cell receptor. Of the six subsets, the two subsets expressing CD160 expression had the highest levels of CD107a, granzyme B (GrB), and perforin on stimulation (figure 4A). The double-positive PD-1–CD160+CD244+ subset had significantly higher GrB compared with every other subset (p<0.05 for all comparisons), including triple positive (PD-1–CD160+CD244+) cells. Of note, subsets that expressed PD-1, in general, had lower GrB and perforin compared with those that did not express PD-1. In summary, on T cell receptor (TCR) engagement with antigen, expression of CD160 was associated with higher levels of GrB and perforin, whereas expression of PD-1 lowered GrB and perforin.

Impact of inhibitory receptors on CD8 +T cell Function: Cryopreserved PBMCs were from HIV-infected patients were stimulated with SEB for 16 hours cells and analyzed by multiparametric flow cytometry for intracellular accumulation cytotoxic molecules and cytokines. After acquisition of data on flow cytometer, cells were gated for live, CD3+CD8+T cells. Cells were further subdivided based on expression of PD-1, CD160, and CD244 into six distinct cellular subsets. Proportion of subsets with intracellular accumulation of (A). CD107a, granzyme, perforin (B) IFN-γ, and TNFα is shown. Of note, unstimulated cells were used as controls and percentage of granzyme, perforin, CD107 and cytokines were subtracted from SEB stimulated wells. Bar represents median with IQR. Non-parametric Wilcoxon rank-sum test was applied to compare between two groups. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IFN, interferon; PD-1, programmed death-1; SEB, Staphylococcus endotoxin B; TNF, tumor necrosis factor .
In contrast, the ability of CD8 +T cells to secrete cytokine was largely dependent on the expression of PD-1. The absence of PD-1 expression was associated with very minimal amounts of cytokine (IFN-γ and TNFα) production upon stimulation. Of note, PD-1+ single-positive cells generally produced more cytokines than double or triple-positive cells (figure 4B), suggesting that additional expression of CD244 and/or CD160 resulted in decreased cytokine production. Thus, the triple-positive cells appear to maintain some ability to secrete cytokines driven by PD-1 expression, but cytokine secretion was lower than the cells that express PD-1 alone (p=0.009 for TNFα and p=0.063 for IFN-γ).
Overall, triple positive (PD-1 +CD160+CD244+) CD8 +T cells demonstrated evidence of both cytotoxicity as measured by CD107a, GrB, and perforin production, as well as the ability to secrete cytokines upon polyclonal antigen stimulation. Cytotoxicity measures were facilitated by CD160 expression and blunted by PD-1 expression, whereas cytokine production was dependent on PD-1 expression but diminished on co-expression of CD160 and/or CD244.
Cells expressing multiple inhibitory receptors have a high level of TOX and low levels of TCF-1
TOX is one of the key drivers of T cell exhaustion in chronic infection and cancer23 24 36 37 whereas TCF-1 is highly expressed on naïve and central memory cells, playing a role in the generation of progenitor of CD8+ T cells.38 39 It has been previously shown that TOX expression is largely seen in memory and effector subsets and is low in naive CD8+ T cells. Expression of TOX has been associated with expression of T cell inhibitory receptors including PD-1, CD160, and Lag-3. Alternatively, TCF-1 expression is highest on naive and central memory T cells.40
TOX and TCF-1 expression was measured among T cell inhibitory receptors among HIV-infected patients (figure 5A). The percentage of TOX +TCF-1− cells was significantly higher on CD8 +T cells expressing PD-1, CD160 or CD244 (p=0.001 for each subset; figure 5A) compared with cells not expressing any of these receptors. Among CD8 +subsets with co-expression of multiple inhibitory receptors (double-positive cells or triple-positive cells) the proportion of TOX +TCF-1− cells increased while the proportion of TOX-TCF-1+ cells decreased (figure 5B). Among the TOX +TCF-1− subpopulation of cells, significantly higher percentages of cells expressing PD-1, CD160, and CD244 as single-positive cells, double-positive cells and triple-positive cells are present compared with TOX-TCF-1+ cells. Conversely, among the TOX-TCF-1+, the majority of cells are triple-negative cells (p<0.05; figure 5C).
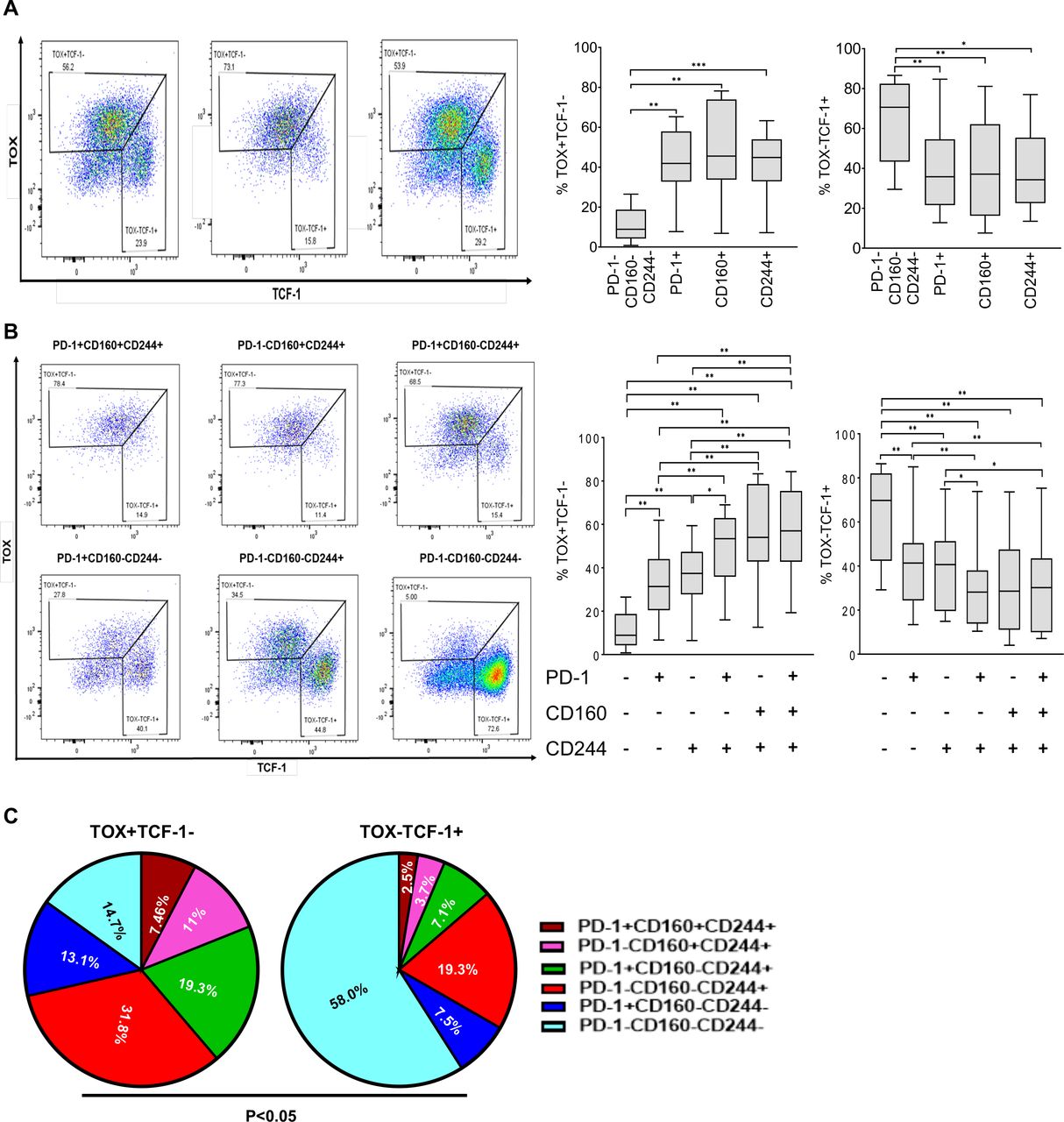
TOX and TCT-1 expression on T cell inhibitory cell subsets among patients with HIV infection: (A). Flow plot showing the expression of TOX +and TCF-1– on PD-1, CD160 and CD244 expressing subsets. Expression of cells that were TOX +TCF-1– and TOX-TCF-1+ among cell subsets shown (B). Flow plot showing TOX and TCF expression on CD8 +T co-expressing PD-1, CD160 and CD244. Proportion of cells expressing TOX +TCF-1– and TOX-TCF-1+ among each of the six CD8 +T subsets shown. (C). The proportion of cells co-expressing single and co-expression of PD-1, CD160 and CD244 within TOX +TCF-1– and TOX-TCF-1+ subsets. Bar represents median with IQR. Non-parametric Wilcoxon rank-sum test was applied to compare between two groups. *p<0.05, **p<0.005, ***p<0.0005. PD-1, programmed death-1; TCF-1, transcription factor T cell factor 1; TOX, thymocyte selection-associated HMG box.
Discussion
A prominent feature of exhausted T cells is their expression of various combinations of inhibitory receptors. During acute infections, these receptors are typically upregulated on CD8+ T cells and play an important role in both promoting antiviral responses and attenuating pathologic inflammatory responses. The sustained high expression of inhibitory receptors in chronic viral infections constrains T cell function and compromises viral control, as evidenced by a beneficial antiviral effect of blocking these inhibitory receptors in both animal models14 41 42 and human studies.43 44 In chronic HIV infection, not only is T cell dysfunction noted in virus-specific cells but also has been reported in the global T cell population, persisting even after effective viral suppression on ART. However, the heterogeneity of T cell subsets with various combinations of inhibitory receptors is becoming increasingly evident and is illustrative of the complexity of the adaptive immune response in both tumors and infection.
With the knowledge that T cell exhaustion is evident in those with chronic viral infections and poor antitumor immune responses adversely impacts cancer outcomes, we hypothesized that PLWH with evidence of T cell exhaustion are at increased risk for cancer. Supporting this hypothesis, we show here that among PLWH who have durably suppressed viremia, those who are subsequently diagnosed with cancer harbor higher levels of a distinct population of CD8 +T cells with co-expression of three inhibitory receptors (PD-1 +CD244+CD160+) and a signature transcriptional factor profile (TbetdimEomeshigh). This population of cells exhibits evidence of both activation and suboptimal antigen-specific cytokine secretion, as noted in functional assays. This finding suggests that there is evidence in the peripheral blood of altered immune profiles that may identify those at risk of invasive cancer, potentially allowing for the development of predictive biomarkers among high-risk patients.
Prior studies have established that inhibitory receptors are elevated among PLWH, particularly among those harboring uncontrolled HIV replication, suggesting that chronic antigenic exposure may drive the presence of these cells. Peretz et al, reported that CD8+ T cells co-expressing PD-1 and CD160 (PD-1 +CD160+) represent an advanced exhausted T cell population and were more prevalent among viremic HIV-infected patients compared with elite controllers, individuals with excellent viral control in the absence of therapy.19 Another study revealed that mono-expression and co-expression of PD-1, CD160, and CD244 on CD8 +T cells was elevated in untreated HIV-infected patients compared with both HIV-infected patients with long-term viral suppression on ART and healthy uninfected individuals.6 Expression of these inhibitory receptors was associated with a transcriptional profile skewed towards activation and exhaustion. In the current study, we explore the clinical impact of this cellular subset was specifically explored, where we report for the first time that PLWH who progressed to a cancer diagnosis harbor higher proportions of CD8 +T cells co-expressing PD-1 +CD160+CD244+ 1 year prior to cancer diagnosis, compared with patients without cancer. Thus, the current study demonstrates that this cellular subset remains relevant among patients with durable viral suppression, and may serve as a marker for those at risk for future cancer diagnosis among PLWH. The finding that the frequency of this subset of CD8 +T cells is not associated with CD4 +T cell nadir is also of interest as it suggests that depth of immunosuppression based on CD4 loss may not drive ongoing abnormalities of CD8 T cell function.
T-bet and Eomes regulate differentiation of CD8 +T cells following an encounter with a foreign antigen. Previous studies have reported that CD8 +T cells expressing low levels of T-bet and high level of Eomes express multiple inhibitory receptors, display a transitional differentiation phenotype, and are associated with poor T cell function and increased immune activation in untreated HIV-infected patients.6 Consistent with these findings, we also found that T-betdimEomeshi cells were more likely to express PD-1, CD160, and 2B4. Similarly, we noted that these triple-positive cells (PD-1 +CD160+CD244+) were more likely to have a T-betdimEomeshi phenotype compared with cells not expressing these receptors. Surprisingly, we found that even within this small and well-defined triple-positive T cell subset, patients who developed cancer (cases) had a higher level of T-betdimEomeshi compared with controls. In fact, the combination of inhibitory receptor expression and a T-betdimEomeshi profile distinguishes future patients with cancer from controls. These findings are supportive of the hypothesis that some patients may have evidence of significant global T cell dysfunction, putting them at increased risk for cancer development. An alternative explanation of these findings is that an undiagnosed tumor is already presence and the circulating cells simply are reflective of changes occurring in the tumor microenvironment. However, this alternative hypothesis appears less likely given that a subgroup of patients all analyzed at time points >12 months prior to cancer diagnosis still harbor increased levels of triple-positive cells. Future time course experiments should be explored to help ascertain how many months (or years) prior to cancer diagnosis these cells may be prevalent at increased levels. Regardless, these findings in the peripheral blood remain important as they suggest that the peripheral blood may serve as a means by which to discriminate patients with HIV infection who are at increased risk for future cancer diagnosis. Further confirmatory studies should be pursued to evaluate their use as a predictive biomarker for cancer risk. Additionally, the question of whether this unique T cell phenotype may also serves as a biomarker for HIV-uninfected population is an important question that should be addressed if appropriate long-term cohorts of the general population are available.
The mechanism by which this population of triple positive (PD-1 +CD160+CD244+) impacts T cell function and antigen-specific responses requires closer evaluation, particularly given the potential for therapeutic application. Triple-positive cells have significant stores of GrB and perforin and an ability to degranulate in the presence of antigen-specific TCR-mediated polyclonal stimulation, as evidenced by increased CD107a expression. Co-expression of CD160 and CD244 appears to be a driving factor in maintaining this cytotoxic profile as subsets not expressing CD160 have low levels of perforin stores. Interestingly, though triple-positive cells have high levels of granzyme and perforin, it is lower than those cells that express CD160 and CD244, but not PD-1 (ie, PD-1–CD160+CD244+). Thus, the addition of PD-1 expression appears to blunt the cytotoxic potential of CD8 T cells (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Characterization of CD8+T cell subsets expressing PD-1, CD160, and CD244 Heatmap representation of features of CD8+T cells expressing inhibitory receptors PD-1, CD160, and CD244. Green represents lowest expression for each marker (TOX, TCF-1, CD107a, granzyme B, perforin, IFN-γ, TNF-α) and red represents the highest level. IFN, interferon; PD-1, programmed death-1; TCF-1, transcription factor T cell factor 1; TNF, tumor necrosis factor; TOX, thymocyte selection-associated HMG box.
Despite the negative association of PD-1 on GrB/perforin production, we find that PD-1 is essential for cytokine production. Cells expressing PD-1 as a single inhibitory receptor exhibited the highest level of IFN-γ and TNFα compared with other subsets. However, co-expression of additional inhibitory receptors (CD244 or both CD244/CD160) decreased cytokine secretion among PD-1 expressing cells. The expression of inhibitory receptor CD244 has been shown to be increased in chronic infection and correlates with effector or EM subtypes,45 46 with CD244 blockade increasing virus-specific cytotoxic granule degranulation (CD107a and perforin) and cytokine production (IFN-γ and TNFα) further supporting its role in T cell exhaustion.47 48
TOX and TCF-1 are transcriptional regulators of lymphocyte development and maturation.40 49–52 Under the homeostatic condition, naïve and early differentiated memory CD8 +T cells primarily expressed TCF-1 whereas EM CD8 +T cells primarily expressed TOX.53 54 We found a higher percentage of TOX +TCF-1- and a lower percentage of TOX-TCF-1+ on CD8+T cells expressing one or more inhibitory receptors compared with CD8 +T cells expressing none, with those subsets expressing two or three receptors having the highest proportion of TOX +TCF-1– cells. CD244 expression was most highly associated with the TOX expression suggesting a dominant role of CD244 in driving cells toward an exhausted phenotype.
This study utilized a cohort of PLWH, followed longitudinally for greater than three decades with stored biologic specimens and well-annotated clinical data. The USU HIV NHS cohort is unique in that it has documented time of HIV seroconversion and a high rate of antiretroviral uptake, thus representing a population with preserved CD4 +T cell counts and high rates of viral suppression. Thus, the NHS allowed for a unique ability to probe for precancer changes in immune cells without the confounding impact of high levels of HIV replication e. Given durable viral suppression on ART among all participants as well as high CD4 T cell count among both cases and controls, risk of other opportunistic infections is extremely low. The largest limitations of the current study is its small sample size. Despite utilization of a large longitudinal cohort, only 17 patients with cancer were available for study. Similar studies should be pursued with other large observational cohorts. We limited the study to cancers (HPV-associated cancers, lung cancer, lymphoma) where it was likely that antitumor immunity is important for disease progression. Whether these markers are seen in other cancer types should additionally be studied. Lastly, the USU HIV NHS is a study of military personnel and beneficiaries, and participants studied are predominantly man. Additional cohorts with more balanced sex and gender distribution must be studied to further generalize findings. Lastly, given the fact that this is a small retrospective study, no multiple testing adjustment was performed in order to avoid missing important findings. Despite these limitations, this study demonstrates for the first time, using longitudinally well-preserved biologic specimens, evidence that alterations in cellular immune subsets may be used to determine individuals at high risk for cancer. Larger controlled prospective studies will be needed to determine whether these markers may be used as predictive biomarkers.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants but Yale University Human Investigations Committee, HIC Protocol #: 1401013195, exempted this study. Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @BrindaEmu
Contributors The authors confirm contribution to the paper as follows: Study conception and design: DT, BE, AG, BA, SMK, JO, RCM, KK, CMS, JMB; Data collection: OC, DT, CB; Analysis and interpretation of results: OC, DT, KW, YD, XW, XC, KAS, BE, AG. Draft manuscript preparation: OC, BE. All authors reviewed the results and approved the final version of the manuscript. BE will serve as the guarantor.
Funding This study was carried out with the funding from National Institute of Health, R01CA206483.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.