Article Text

Abstract
Recent advances in understanding the roles of immune checkpoints in allowing tumors to circumvent the immune system have led to successful therapeutic strategies that have fundamentally changed oncology practice. Thus far, immunotherapies against only two checkpoint targets have been approved, CTLA-4 and PD-L1/PD-1. Antibody blockade of these targets enhances the function of antitumor T cells at least in part by relieving inhibition of the T cell costimulatory receptor CD28. These successes have stimulated considerable interest in identifying other pathways that may bte targeted alone or together with existing immunotherapies. One such immune checkpoint axis is comprised of members of the PVR/nectin family that includes the inhibitory receptor T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory domains (TIGIT). Interestingly, TIGIT acts to regulate the activity of a second costimulatory receptor CD226 that works in parallel to CD28. There are currently over two dozen TIGIT-directed blocking antibodies in various phases of clinical development, testament to the promise of modulating this pathway to enhance antitumor immune responses. In this review, we discuss the role of TIGIT as a checkpoint inhibitor, its interplay with the activating counter-receptor CD226, and its status as the next advance in cancer immunotherapy.
- Costimulatory and Inhibitory T-Cell Receptors
- Immunotherapy
- Lymphocytes, Tumor-Infiltrating
- Review
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Coinhibitory or immune checkpoint receptors such as CTLA-4 and PD-1 have important roles in limiting immune responses, providing a mechanism to prevent overactivation of T cells and consequent immune-mediated damage to the host.1 2 In cancer, where there is generally an abundance of antigen relative to the number or capacity of antitumor T cells, coinhibitory receptors can be highly expressed, reflective of an ‘exhausted’ T cell phenotype that further restricts antitumor activity.3 Certain coinhibitory receptors are also expressed by other cytotoxic lymphocytes such as natural killer (NK) cells, and presumably also restrict antitumor immunity.4 The importance of at least some coinhibitory receptors in cancer immunity has now been amply demonstrated by a host of preclinical and clinical studies showing that monoclonal antibodies (mAbs) that block the ability of CTLA-4 to sequester CD80/86 or inhibit PD-1 from binding its ligand PD-L1 exhibit robust activity.5–8 The precise mechanisms by which these antibodies act is still unclear, and may involve inhibiting the acquisition of an exhausted phenotype by tumor-infiltrating lymphocytes (TILs) or the expansion of antitumor T cells in lymphoid organs.
While survival benefits have been impressive, significant proportions of patients fail to respond to PD-1 or CTLA-4 blockade, either alone or in combination. This has provided the impetus for identification of other immune checkpoint pathways that may be amenable for therapeutic targeting to drive improved clinical responses, either in isolation or in combination with existing immunotherapies.9 10 One promising pathway is comprised of members of the PVR/nectin family that includes inhibitory receptor T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory (ITIM) domains (TIGIT). Indeed, mAbs against TIGIT are currently being evaluated in clinical trials, either as monotherapy or in combination with anti-PD-1/PD-L1 blockade.11 12 Here, we review the current understanding of TIGIT biology in the context of cancer and mechanisms of action for therapies targeting TIGIT, and provide an update on the status of clinical trials.
TIGIT, a member of the PVR/nectin family
Ligands and interactions
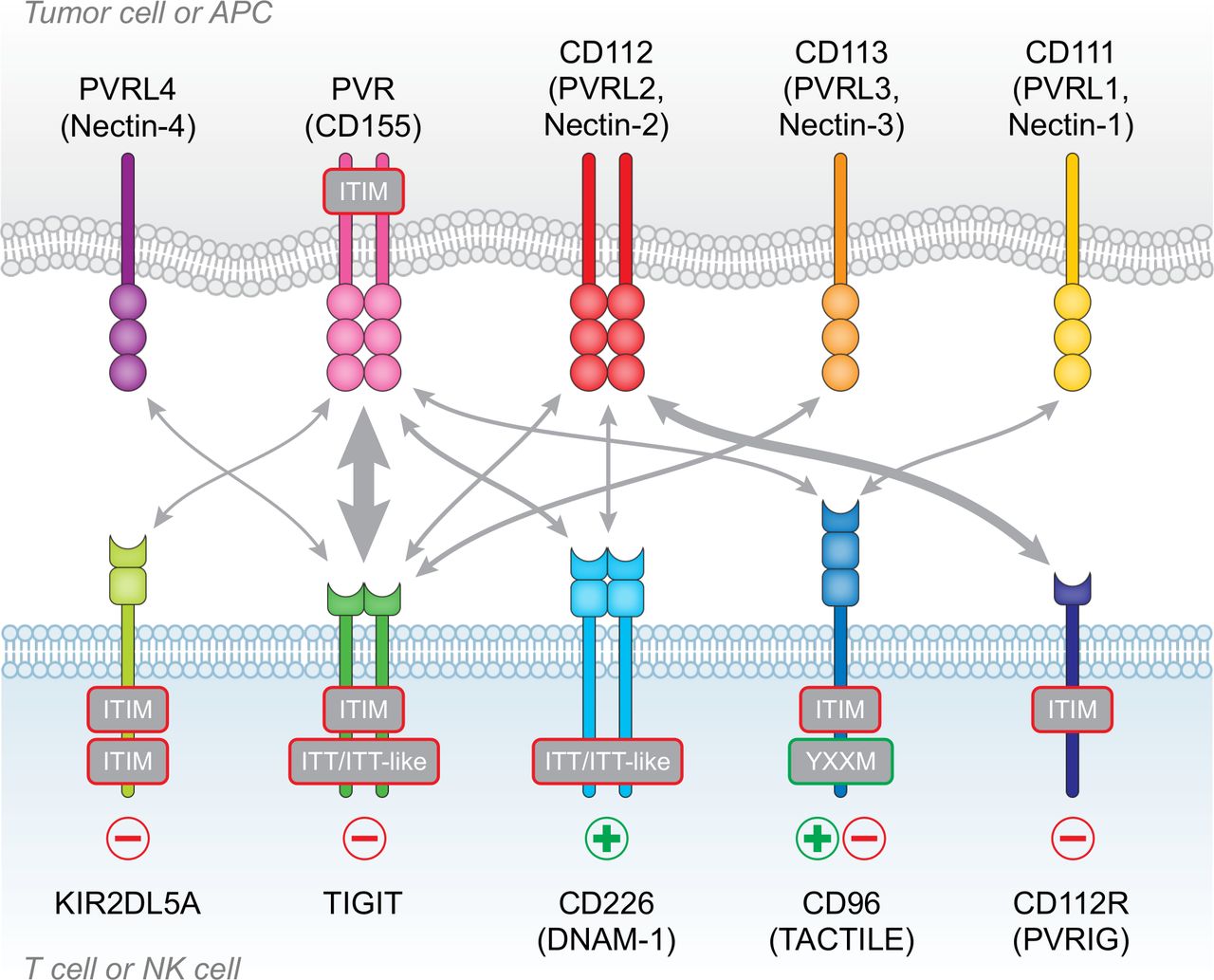
TIGIT (also known as WUCAM or Vstm313 14) was identified through a genomic search for genes expressed by immune cells and containing protein domain structures typically found in inhibitory receptors.15 TIGIT has one extracellular immunoglobulin variable-set (IgV) domain, a type I transmembrane domain, and an intracellular domain containing one ITIM and one immunoglobulin tyrosine tail (ITT)-like motif.15 16 The TIGIT IgV domain contains a conserved submotif, defined as (V/I)(S/T)Q, AX6G, and T(F/Y)P, found in PVR/nectin family members that includes TIGIT, CD226 (DNAM-1), CD96 (Tactile), CD112R (PVRIG), PVR (CD155), CD112 (PVRL2) and CD113 (PVRL3).17 This motif mediates a ‘lock and key’ trans-interaction with cis-homodimers of PVR. While the crystal structure of TIGIT complexed with PVR indicates that two TIGIT/PVR dimers can assemble into a heterotetramer with a core TIGIT/TIGIT cis-homodimer, the conserved ‘lock and key’ submotifs suggest that heterodimeric interactions between the various PVR/nectin family members may also occur.
The ligands for TIGIT are PVR, CD112 and CD113, with PVR serving as the principal ligand due to the high affinity interaction between TIGIT and PVR (figure 1). TIGIT binding affinities for CD112 and CD113 are much lower.15 CD226 and CD96 also bind to PVR, but with lower affinities; CD226 has the lowest affinity while CD96 binds with intermediate affinity.15 18 19 In addition, CD226 and CD112R can bind to CD112.20 21 PVRL4 may be the one ligand that is exclusive for TIGIT, as CD226, CD96 and CD112R do not appear to bind PVRL4.22 The shared ligands with differing affinities create a hierarchy of possible functional interactions, with the most important possibly driven by TIGIT-PVR engagement, due to the high affinity of this particular interaction. Importantly, TIGIT will ‘out-compete’ CD226 for their common ligand (PVR), suggesting already one mechanism whereby TIGIT may reduce T cell stimulation (ie, by blocking costimulation via CD226). Nevertheless, when considering the role of TIGIT in mediating cellular responses, it is prudent to take into account the complex regulation and integration of signals transduced by different receptors sharing common ligands.
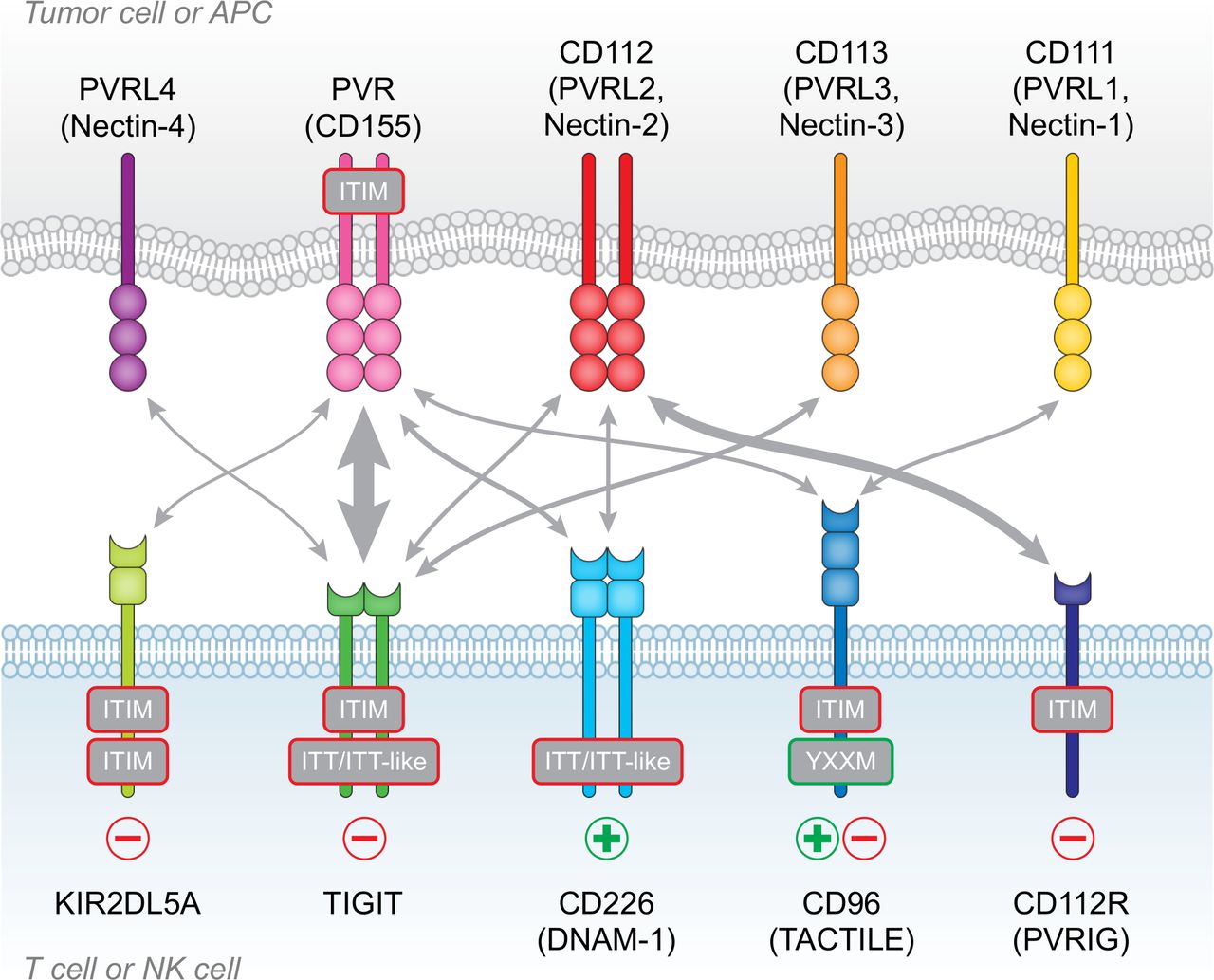
The PVR/nectin family and receptor/ligand interactions. TIGIT, CD226, CD96, CD112R and KIR2DL5A are expressed on T cells and NK cells, whereas the ligands PVR, CD111, CD112, CD113 and PVRL4 are expressed on antigen-presenting cells (APCs) or frequently overexpressed on tumor cells. T cell immunoreceptor with Ig and ITIM domains (TIGIT) contains both immunoreceptor tyrosine-based inhibitory (ITIM) and immunoglobulin tyrosine tail (ITT)-like domains and has inhibitory (-) function in T cells and NK cells. CD112R and KIR2DL5A also contain ITIM domains and deliver inhibitory signals (−). CD96 contains an ITIM domain, but human CD96 also contains an YXXM motif. While CD96 has inhibitory function in NK cells, it may act as either an inhibitory (−) or activating (+) receptor in T cells. CD226 is an activating receptor (+), despite containing an ITT-like domain. interactions between ligands and receptors are denoted by arrows, with arrow thickness proportional to relative affinities. The strongest interactions are between TIGIT and PVR, and CD112R and CD112.
Expression and function of TIGIT
TIGIT is expressed on CD4+ and CD8+ T cells, as well as innate lymphocytes including NK cells and γδ T cells. On conventional CD4+ and CD8+ T cells, TIGIT is not expressed in the naïve state but is induced shortly after activation and is most abundant on effector and memory T cells. TIGIT is also expressed on regulatory T cells (Tregs) and follicular helper CD4+ T (TFH) cells.13–15 23–26 Transcriptional control of TIGIT expression is not well understood, but Eomesodermin (EOMES) may positively regulate expression in CD8+ T cells as shown in patients with newly diagnosed acute myeloid leukemia (AML).27 BLIMP1, together with low Bach2 expression, may control TIGIT expression in TFH cells.28
CD8+ T cells
The expression pattern of TIGIT on CD8+ TILs supports TIGIT as an attractive target for modulation of antitumor responses. In cancer, sustained T cell receptor (TCR) stimulation can result in differentiation of CD8+ TILs to an exhausted state where the TILs become less responsive to stimulation, have reduced proliferative capacity and no longer exert full killing activity. Exhausted cells express high levels of PD-1 and other inhibitory molecules, including TIGIT.29 The nuclear receptor transcription factor NR4A (NUR77) is an important regulator of exhaustion, with ATAC-seq analysis showing that NR4A opens the chromatin of the gene encoding PD-1 and other markers up-regulated by exhaustion, and closes the chromatin of genes containing binding motifs for NF-κB and AP-1 that are repressed in exhausted cells.30 31 It remains to be determined whether TIGIT expression is also regulated by NR4A or other transcription factors involved in exhaustion such as TOX.32 33
Well prior to reaching a terminally exhausted state, naïve T cells that have been exposed to antigen and have received proper costimulatory signals progress to become either effector T (TEFF) cells or through stages of memory cell differentiation. Memory stem-like cells (TSCM) have high potential for self-renewal, and, together with central memory (TCM) cells, can respond rapidly to antigen, express effector molecules, and produce the effector pool as well as effector memory (TEM) and resident memory (TRM) populations.34–38 TIGIT and PD-1 expression may identify distinct populations of TSCM, with TSCM expressing these inhibitory receptors destined to give rise to dysfunctional, exhausted-like progeny CD8+ T cells. On the other hand, TSCM lacking expression of TIGIT and PD-1 may be committed to a more functional T effector or memory lineage.39 PD-1 is expressed at low/intermediate levels as activated naïve T cells differentiate, and PD-1 blockade may be exerting its effects on the pre-exhausted TILs (also called TPEX cells, for precursor exhausted T cells, or TMAP cells, for memory-like T cells associated with antigen persistence)37–39 in addition to potentially reinvigorating exhausted T cells.40–44 TIGIT expression has also been detected on TILs at varying stages of differentiation, often coexpressed with PD-1.45–47 Thus, therapies targeting TIGIT may have similar effects as those targeting PD-1/PD-L1, and could act at the T cell expansion step or at the level of exhausted T cells, or both.
NK cells
NK cells are thought to play important roles in tumor surveillance, particularly in controlling blood cancers and tumor metastasis. NK cell effector function against tumors is mediated through the integration of activating and inhibitory signals, some but not all of which are shared with CD8+ TILs.48 49 TIGIT, for example, inhibits NK cell cytotoxicity in both humans and mice, possibly contributing both to the maintenance of self-tolerance under conditions of acute inflammation and to the restrictive education of NK cells in a tumor setting.50–53 TIGIT is constitutively expressed by NK cells, although it is up-regulated in NK cells that are present in tumors.53 High TIGIT expression on intratumoral NK cells was found to be associated with NK cell exhaustion in a number of different mouse tumor models as well as in patients with colon cancer, and was also associated with tumor progression.53 In the mouse CT26 colon cancer, 4T1 mammary cancer and methylcholanthrene-induced fibrosarcoma models, treatment with anti-TIGIT mAb diminished NK cell exhaustion and impeded tumor growth, even in T cell-deficient mice. In patients with endometrial cancer, CD103+ NK cells resident in tumors had higher TIGIT expression than CD103- NK cells found in circulation, and in those patients with lymph node invasion, TIGIT expression on NK cells was significantly higher than in patients with no lymph node invasion.54
NK cells expressing high levels of TIGIT are subjected to suppressive mechanisms mediated through myeloid-derived suppressor cells (MDSCs), leading to blunted cytotoxicity against tumor target cells. Blockade of TIGIT led to resistance against MDSC suppression.55
Another TIGIT-mediated pathway for inhibition of NK cells comes from the observation that TIGIT may be expressed by tumor cells, and that engagement of PVR expressed on NK cells suppresses cytotoxicity and leads to enhanced tumor growth.56 Expression of TIGIT on both NK cells as well as T cells suggests that TIGIT-targeted immunotherapies have the potential to modulate both of these effector arms against tumors, unlike PD-1 and CTLA-4 which have limited expression in NK cells.
Regulatory T cells
TIGIT is considered a marker of Tregs as it is highly expressed on a subset of natural Tregs as well as activated Tregs.57 TIGIT may contribute to Treg stability as defined by maintenance of FOXP3 and suppression of IFN-γ.58 59 TIGIT has been reported to limit PI3K-AKT signaling, thereby inhibiting acquisition of a T helper type 1 (Th1) cell-like phenotype.60 Tregs expressing high levels of TIGIT are more suppressive than those expressing low levels, exhibiting superior suppression of Th1 and Th17 responses.24 Activation of Tregs through stimulation with agonist anti-TIGIT mAbs induced a suppressive program that included upregulation of IL-10 and fibrinogen-like protein 2.24 TIGIT stimulation increased suppressive function and normalized IFN-γ in Tregs from multiple sclerosis patients, while agonistic mAbs alleviated EAE in mice.61 In human melanoma patients, Tregs with high TIGIT expression are enriched in tumors and possess a stable, suppressive phenotype.62 The role for TIGIT in regulating the suppressive activity of Tregs in tumors was demonstrated in the mouse B16F10 melanoma model, whereby transfer of TIGIT-deficient Tregs together with wild-type CD4+ and CD8+ T effector cells into tumor-bearing Rag-/- mice resulted in reduced tumor growth similar to that observed in TIGIT knockout mice.63 Conceivably, Treg depletion by antibody-dependent cellular cytotoxicity (ADCC) via anti-TIGIT antibodies may contribute to antitumor efficacy.
Follicular helper T cells
TFH cells play an important role in establishing and maintaining the germinal cell (GC) reaction that leads to efficient generation of plasma B cells. TFH cells induce TIGIT expression while CD226 is concurrently down-regulated.25 It was proposed that initial priming by dendritic cells (DCs) in the T-cell zone is mediated by CD226 interactions with PVR. TFH cells then switch to TIGIT as they mature and reside in the GC, supporting B cell propagation and differentiation while TIGIT limits TFH expansion to provide rigorous B cell selection. Expression of TIGIT in TFH cells leads to the possibility that TIGIT may have a role in TFH-like cells found in tertiary lymphoid structures (TLS) that form in tumors, which structurally and functionally resemble GCs.64 65 TFH-like cells have been found in human malignancies such as breast cancer, where the presence of these cells was associated with better prognosis.66 67 As TLS generally shape the tumor microenvironment to one that is favorable for controlling tumor progression, and may also host the expansion of CD8+ T cell responses, it is possible that manipulation of TLS through anti-TIGIT treatment could have therapeutic benefit.
γδ T cells
γδ T cells comprise a small population of T cells compared with T cells using the αβ TCR. Unlike αβ T cells, γδ T cells are generally not restricted by major histocompatibility complex presentation of peptide antigens. In principle, this feature confers γδ T cells with the ability to exert antitumor activity through interaction with stress-induced molecules expressed by tumor cells, similar to NK cells.68 69 In humans, γδ T cells are primarily categorized into three subsets on the basis of TCRδ-chain usage, with Vδ1+, and likely Vδ3+ (or Vδ1-Vδ2-), being primarily resident in mucosal tissues, and Vδ2+ comprising the bulk of γδ T cells in blood circulation.68 Ex vivo expanded Vδ1+ T cells have been shown to possess broad cytotoxic potential but have not been well characterized in solid tumors.70 As tumors do grow in mucosal tissues rich with Vδ1+ cells, these cells may express checkpoint molecules that inhibit their antitumor function. Within the Vδ2+ population, those paired with the Vγ9 chain may be mediators of antitumor immunity through either direct cytotoxicity or production of IFN-γ and TNF-α.69
TIGIT expression has been reported on γδ T cells, particularly in the context of allogeneic hematopoietic stem cell transplantation (allo-HSCT) and AML. γδ T cells, together with NK cells, reconstitute early after allo-HSCT, and are key mediators of the graft-versus-leukemia effect of allo-HSCT. Vδ1+ T cells expressed higher levels of TIGIT than Vδ2+ T cells or NK cells, CD8+αβ T cells, or CD4+αβ T cells, and expression was increased by priming with IL-15.71 The Vδ1+ T cells expressing higher TIGIT were functionally inferior to Vδ2+ T cells expressing lower TIGIT, consistent with the possibility that TIGIT may be suppress effector function.71 In patients with de novo AML, a higher frequency of γδ T cells express TIGIT compared with γδ T cells in healthy individuals or patients in complete remission following chemotherapy.72 γδ T cells from AML were not characterized on the basis of TCRγ or TCRδ usage, and it remains to be determined if TIGIT expression affects γδ T cell responses in AML. An analysis of γδ T cells from cytomegalovirus (CMV) seronegative or seropositive donor blood demonstrated that TIGIT expression was higher on γδ T cells compared with αβ T cells or NK cells.73 Vδ2-γδ T cells, which respond against CMV and include Vδ1+γδ T cells, from CMV seropositive donors had higher TIGIT expression than those from CMV seronegative donors. Effector activity of Vδ2-γδ T cells induced by stimulation with anti-Vδ1 mAb was abrogated in the presence of soluble PVR, but was restored with the addition of anti-TIGIT mAb, indicating that TIGIT has a suppressive role in γδ T cells.
Mechanisms of action
Cell-Intrinsic direct signaling
The cytoplasmic domain of TIGIT contains one ITIM and one ITT-like motif, imparting TIGIT with the potential to directly transmit inhibitory signals. Much of what is known in regards to TIGIT signaling comes from experiments using NK cells. For human TIGIT signaling, the ITIM motif was essential as mutation or truncation of this motif resulted in loss of inhibition of NK-mediated cytotoxicity.50 For mouse TIGIT, an inhibitory signal could be transduced via either the ITIM or ITT-like motif alone, with mutation of both required to ablate inhibition of NK cell activity.51 Detailed analysis of TIGIT signaling in human NK cells revealed that following ligand binding, phosphorylation of the ITT-like motif may lead to recruitment of the cytosolic adaptor proteins growth factor receptor-bound protein 2 (Grb2) and β-arrestin 2.74 75 The Grb2 pathway interferes with NK cell cytotoxicity as Grb2 recruits SH2-containing inositol phosphatase-1 (SHIP-1), resulting in inhibition of phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase.74 β-arrestin 2 inhibits IFN-γ production through the recruitment of SHIP-1 resulting in inhibition of NF-κB activation.75 Coculturing human NK cells with MDSCs demonstrated that TIGIT signaling could directly inhibit NK cell cytotoxicity by reducing ERK1/2 and ZAP70/Syk phosphorylation.55
Elucidation of TIGIT inhibitory signaling pathways in T cells has been more elusive. TIGIT has been shown to inhibit T cells in a cell-intrinsic manner as agonistic TIGIT antibodies suppress T cell responses to stimulation with anti-CD3 and anti-CD28.14 23 76 Whole genome microarray analysis indicated that TIGIT down-regulated genes associated with TCR and CD28 signaling, T cell activation and cell cycle progression. Efforts to identify proteins recruited to the TIGIT cytoplasmic tail using human Jurkat T cells or cell-free reconstitution systems using large unilamellar vesicles have not been fruitful to date.47 While TIGIT contains a putative ITIM motif based on consensus sequence, ITIM motifs found in various immunoreceptors may have differential activities in recruiting SH2-containing phosphatases, with polarity and size of the amino acid residue following the ITIM phosphotyrosine (pY +1) playing a key role.77 As the TIGIT ITIM contains a bulky phenylalanine at pY +1 position, it may not be a suitable binding site for SH2-containing phosphatases such as SHP1. Furthermore, TIGIT does not contain an immunoreceptor tyrosine-based switch motif (ITSM). The ITSM is largely responsible for recruiting SHP2 in PD-1, rather than the ITIM, and this may explain why TIGIT does not recruit SHP2 (47, 77).
Regulation of CD226 activity
While TIGIT functions as an inhibitory receptor, it is often coexpressed with its activating costimulatory receptor, CD226 (also known as DNAM-1 and Nectin-2). CD226 has a broader cell expression pattern than TIGIT, being expressed not only on T cells and NK cells but also on NKT cells, B cells, monocytes/macrophages, DCs, megakaryocyte/platelet lineage and hematopoietic precursor cells, as well as endothelial cells and mast cells.78–81 Thus, CD226 has multifaceted roles in regulating activity of a wide variety of cells. In CD8+ T cells, CD226 is constitutively expressed by naïve cells and has important roles in various stages of T cell priming and activation. CD226 contributes to the formation of the immune synapse through its interactions with PVR expressed by antigen-presenting cells (APCs).82–85 As an adhesion molecule, CD226 participates in transendothelial migration of effector memory cells, allowing cells to leave blood circulation and enter sites of inflammation such as a tumor.86 In tumors, CD226 may facilitate functional interactions between effector cells and tumor cells many of which express PVR.87
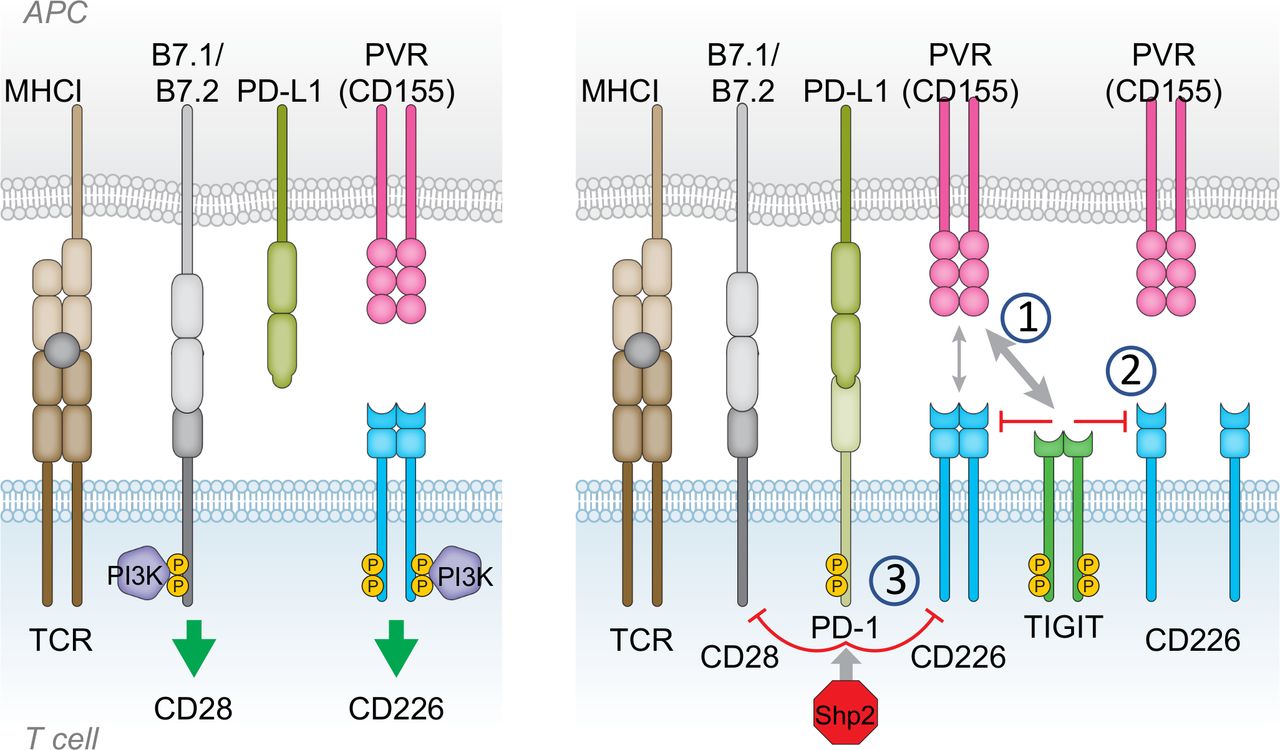
By virtue of TIGIT having higher affinity for the shared ligands PVR and CD112, TIGIT can outcompete CD226 for binding, thereby preferentially exerting its immunosuppressive effects over CD226 when both molecules are present on the same cell.16 Time-resolved fluorescence resonance energy transfer (TR-FRET) demonstrated that TIGIT could interact in cis with CD226 and disrupt CD226 homodimers reducing its ability to transmit activation signals.26
More recently, CD226 was found to be regulated not only by TIGIT but also by PD-1. CD226 was identified as a substrate for dephosphorylation by the PD-1-SHP2 complex, indicating that activation of the PD-1 pathway negatively regulates CD226 functional activity.47 88 Similarly, TIGIT also impedes CD226 activation, although through a different mechanism.47 Disruption of CD226 activation was mediated through interactions of the extracellular domains of TIGIT and CD226, reflecting the ability of TIGIT to out compete CD226 for binding to their shared ligands PVR and CD112. Indeed, the TIGIT cytoplasmic domain was completely dispensable for TIGIT-mediated inhibition of CD226 signal transduction47 (figure 2).

Mechanisms of T cell immunoreceptor with Ig and ITIM domains (TIGIT) inhibition of CD226. Left: CD28 and CD226 provide costimulatory signals to enable T cell activation on T cell receptor (TCR) engagement. Right: Following activation, inhibitory checkpoint receptors such as PD-1 and TIGIT are expressed by T cells, mediating suppression of activating signals. TIGIT and PD-1 disrupt CD226 and CD28 signaling through several mechanisms. (1) TIGIT has higher affinity than CD226 for shared ligands, allowing TIGIT to outcompete and displace CD226. (2) TIGIT disrupts CD226 homodimer formation, disabling capacity for CD226 activation. (3) Suppression of CD226 signaling by TIGIT and PD-1. CD226 phosphorylation is impaired or reduced in the presence of TIGIT extracellular domain or activated PD-1. Recruitment of Shp2 to activated PD-1 also impairs CD28 phosphorylation.
By interfering with CD226 activity, TIGIT can act to regulate a variety of aspects of T cell function that CD226 normally controls. For example, CD226 engagement with PVR results in phosphorylation of FOXO1, a widely expressed transcription factor but one with important roles in NK cells, CD8+ T cells and Tregs.89 Phosphorylated FOXO1 is translocated out of the nucleus and accumulates in the cytosol where it is targeted for degradation by ubiquitination. Since FOXO1 is a negative regulator of NK cell homing and effector functions,90 CD226 activation can promote NK cell activity. Similarly, CD226-mediated regulation of FOXO1 may also impact T cell function, including the activity of CD8+ tumor infiltrating T cells.89 A key target for repression by FOXO1 is T-BET, a critical transcription factor in both CD4+ and CD8+ T cells that controls a variety of effector mechanisms. EOMES and TCF7, transcription factors involved in the establishment of T cell memory, are also directly targeted by FOXO1.91 92 FOXO1 is also important for development and differentiation of Tregs as it regulates FOXP3.93 94 Deficiency or inactivation of FOXO1 in Tregs leads to enhanced IFN-γ production and abrogation of suppressive activity, whereas FOXO1-mediated transactivation of CTLA-4 manifests suppressive function.93 95 Thus, CD226-mediated suppression of FOXO1 may repress or functionally re-program Tregs, reducing their inhibitory potential (figure 3).

T cell immunoreceptor with Ig and ITIM domains (TIGIT) blockade may alleviate transcriptional programming mediated by Foxo1 in different immune cell populations. Left: TIGIT interferes with CD226 signaling, thereby inhibiting Akt phosphorylation and allowing Foxo1 to impair CD8+ T cell and NK cell function and enforce Treg suppressive function. Right: Blockade of TIGIT unleashes CD226 signaling, resulting in phosphorylation of Akt which subsequently phosphorylates Foxo1, leading to translocation of Foxo1 from the nucleus to the cytoplasm where Foxo1 is targeted for degradation. Reduction of Foxo1 transcriptional regulation may enhance CD8+ T cell and NK cell activity and impair suppressive activity of Tregs.
Competition between CD226 and TIGIT for binding to PVR may also have a regulatory role in Tregs.96 CD226 may not be involved in the development and function of Tregs under homeostatic conditions, but CD226 deficiency or blockade results in enhanced Treg-mediated suppression of conventional T cell activation.57 96 97 While CD226 deficiency maintained Foxp3 stability and Treg function, CD226 signaling did not have a direct effect; rather the absence of CD226 permitted TIGIT interaction with PVR that resulted in a signaling cascade that suppressed the TCR-AKT-mTORC1 pathway.96
CD226 is an important player in driving response to checkpoint immunotherapies
Single cell RNAseq analysis of CD8+ T cells isolated from tumors of non-small cell lung carcinoma (NSCLC) patients has revealed that the costimulatory activating receptors CD226 and CD28 are differentially expressed among distinct CD8+ T cell subsets, with TEFF and TRM subsets predominantly expressing CD226 and TEM cells favoring CD28.47 The principal CD8+ T cells that were double positive for CD28 and CD226 were those exhibiting a gene expression profile consistent with an activated or proliferative phenotype. Thus, the cellular distribution of CD226 in TILs, together with the convergence of PD-1 and TIGIT to regulate CD226 activity, strongly suggests that CD226 is a central player in regulation of antitumor CD8+ T cell responses. CD226 may even compensate for the absence of CD28 on important TIL subsets. Evidence for this comes from the recent identification of a family with a genetic defect in CD28.98 Affected individuals were generally healthy, with the exception of sensitivity to human papillomavirus infection, suggesting a compensatory relationship between the two costimulatory receptors.
Current pre-clinical data suggest that both CD28 and CD226 play important roles in cancer immunity. CD28 is critical for the ability of CD8+ T cells to respond to PD-1 blockade, and cause tumor regression in mice.99 Similarly, there is a requirement for CD226 to achieve antitumor activity using PD-1/PD-L1-targeted therapies as well as anti-TIGIT mAb treatment.26 47 The absence of CD226 expression abrogates the therapeutic benefit of anti-CD137, anti-GITR and anti-CTLA-4 in various mouse tumor models.71 88 100 Clinically, high expression of CD226 was associated with better clinical outcome for NSCLC patients treated with the anti-PD-L1 mAb atezolizumab,47 and CD226 expression in CD8+ T cells was correlated with prolonged progression-free survival in melanoma patients treated with immune checkpoint blockade (ICB) therapies.71
Beyond the roles of TIGIT and PD-1 in regulating CD226 signaling, other factors may contribute to CD226 down regulation in CD8+ T cells, resulting in their dysfunction. The transcriptional regulator EOMES is upregulated in exhausted CD8+ T cells.101 An accessible intronic region of the CD226 gene contains an EOMES binding site, providing a means for EOMES to directly interact with regulatory elements to interfere with CD226 expression.100 Furthermore, interaction between CD226 and PVR can phosphorylate CD226 at a critical tyrosine residue (Y319), leading to the Cbl-b-mediated ubiquitination, internalization and degradation of CD226 in CD8+ T cells.71 Internalization and degradation of CD226 was also observed in NK cells following stimulation by membrane-bound PVR.102
Reverse signaling by PVR may modulate the immune microenvironment
PVR is constitutively expressed on epithelial cells and myeloid cells, but is upregulated in tumor-associated myeloid cells and is often over-expressed by tumor cells.103 High PVR expression in tumors has been associated with poor clinical response to anti-PD-1 immunotherapy.104 105 In metastatic melanoma, patients with high tumor PVR expression were less responsive to anti-PD-1 monotherapy or combination of anti-PD-1 with anti-CTLA-4.104 The dichotomy in response was even more pronounced when patients were further stratified on the basis of PD-L1 expression, with PD-L1negativePVRhigh patients faring the worst. Similarly, the combination of PVR and PD-L1 expression in non-small cell lung carcinoma (NSCLC) tumors may be predictive of response to PD-1 blockade, with patients expressing tumor PD-L1 but not PVR more likely to respond.105
That PVR is expressed on many normal cells in addition to tumor cells differentiates it from PD-L1, and suggests that PVR has a more generalized function in the balance of TIGIT-mediated suppression and direct activation of CD226. PD-L1 is typically expressed only after exposure to IFN-γ. However, in the context of TIGIT’s role as a coinhibitory receptor that works in concert with PD-1, it is important to account for PVR expression by DCs, which play a key role in triggering T cell-dependent tumor immunity and are a major site of regulation of T cell activation by PD-1.106 As such, PVR on DCs may play a key role along with PD-L1 in determining patient response to PD-1 and TIGIT blockade.
The intracellular domain of PVR contains an ITIM motif suggesting that interactions with TIGIT may result in modulation of myeloid cells or tumor cells themselves. However, ‘reverse signaling’ in myeloid or tumor cells following PVR binding to TIGIT has not been well characterized. Binding of a TIGIT-Fc fusion protein to PVR on human DCs was shown to enhance production of immunosuppressive IL-10 while reducing production of pro-inflammatory IL-12p40.15 Indeed, when added in the presence of DCs, TIGIT-Fc inhibited T cell responses while a blocking anti-TIGIT mAb enhanced responses in the same assay. TIGIT engagement of PVR induced phosphorylation of PVR and ERK, but did not induce DC maturation; PVR activation may instead contribute to a tolerogenic state in DCs. In addition to acting on DCs, TIGIT has been reported to inhibit macrophage activation and promote the skewing of peritoneal macrophages from a proinflammatory M1 phenotype to an immunosuppressive M2 macrophage profile.107 MDSCs are also present in the tumor microenvironment and play an important role in suppressing the antitumor immune response. Tumor MDSCs overexpress both PVR and PD-L1, indicating that their immunosuppressive properties may be enforced via reverse signaling mediated by both TIGIT/PVR and PD-1/PD-L1 pathways.108 Intriguingly, anti-PD-L1 treatment upregulated PVR expression on MDSCs, while anti-TIGIT upregulated PD-L1, suggesting that combination treatment may be needed to relieve MDSC suppressive activity.
TIGIT in cancer
TIGIT expression and association with clinical outcome
TIGIT is highly expressed on TILs in a wide variety of different human tumors.26 109 In a recent comprehensive analysis of immune checkpoint receptors in fresh biopsy samples from treatment-naïve cancer patients,~50% of PD-1+/CD8+ TILs coexpressed TIGIT, and at levels generally higher as compared with CD8+ T cells in the blood.110 Included in the cohort were breast cancer, kidney cancer, lung cancer, liver cancer, cervical cancer, esophageal cancer, gastric cancer and colorectal cancer. Other studies in melanoma, breast cancer, esophageal cancer, hepatocellular carcinoma (HCC), glioblastoma, NSCLC, AML and multiple myeloma (MM) reported similar TIGIT expression on TILs.111–121 Increased TIGIT expression has also been described for NK cells and Tregs that have infiltrated tumors.53 54 97
In a few studies, CD226 expression was included in the analysis, with increased TIGIT expression found to be accompanied by a decrease in CD226 expression. For example, in metastatic melanoma, the TIGIT/CD226 imbalance may correlate with decreased response of CD8+ T cells to TIGIT blockade.111 Down-regulated CD226 expression in CD8+ T cells in multiple myeloma (MM) was a characteristic of an exhausted phenotype.122 A similar pattern of TIGIT and CD226 expression was observed in CD8+ T cells from follicular lymphoma, although CD226 was frequently expressed in CD4+ T cells and TFH cells.123 A high TIGIT/CD226 ratio in tumor Tregs was correlated with a high frequency of Tregs in MM, and together these parameters were associated with poor clinical response to treatment with either anti-PD-1 or anti-CTLA-4.62
The correlation of high TIGIT expression with poor clinical outcome is consistent with the view that one of TIGIT’s activities is to establish an immunosuppressive tumor microenvironment. Patients with cutaneous melanoma who had high TIGIT expression on TILs had worse prognosis; PD-1 expression was not predictive of survival.118 In uveal melanoma, greater numbers of cells expressing TIGIT were found in patients that developed metastases than in patients that did not.112 In gastric cancer, TIGIT expression is up-regulated in cancerous tissues with high frequencies of infiltrating CD8+ T cells and high expression of TIGIT as well as PVR and CD112 are associated with poor prognosis.124 125 High TIGIT expression on CD8+ T cells, CD4+ T cells and Tregs has been detected in HCC and a TIGIT+PD1+CD8+ T cell profile was associated with accelerated disease progression and poor outcome.126 127
In addition to TIGIT expression on CD8+ T cells being a prognostic indicator in many cancer types, NK cell expression of TIGIT may also be predictive. Tumor resident CD103+ NK cells in endometrial tumors express high levels of TIGIT relative to non-resident NK cells, and these resident NK cells had an exhausted phenotype and were associated with advanced stages of disease and lymph node invasion.54 Similarly, in AML patients, TIGIT-expressing NK cells displayed a dysfunctional phenotype with reduced anti-leukemia activity; a high frequency of these TIGIT+ NK cells was correlated with poor prognosis risk.128
Therapeutic targeting of TIGIT in cancer
Preclinical mouse tumor models
The use of mice genetically engineered for TIGIT deficiency or anti-TIGIT mAbs has provided valuable insight into the mechanisms underlying TIGIT’s potential as a target for cancer immunotherapy. Studies in a variety of tumor models have shown that absence or blockade of TIGIT reduced tumor growth and increased survival. Tigit-/- mice have reduced growth of B16F10 melanoma tumors when implanted subcutaneously. Control of tumor growth in the absence of TIGIT was dependent on CD8+ T cells, not NK cells; interestingly, TIGIT expression on Tregs also had an important role in regulating CD8+ T cell antitumor activity.63 However, when B16F10 melanoma cells were inoculated intravenously in an experimental lung metastasis model, Tigit-/- mice had similar tumor burden as wild-type mice,129 130 although a separate study did report that TIGIT deficiency in NK cells alone was sufficient to confer protection by preventing NK cell exhaustion.53 The reasons for this discrepancy are not clear, but may reflect alterations in the balance between innate and adaptive tumor immunity in the presence or absence of TIGIT on NK vs CD8+ T cells. In multiple myeloma models using cell lines derived from Vk*MYC mice, Tigit-/- mice had reduced tumor burden and improved survival, with CD8+ T cells critical for controlling disease.120
The studies using Tigit-/- mice suggest that anti-TIGIT mAbs may be effective when administered as a single agent. Indeed, delivery of anti-TIGIT mAbs in a preventative setting, i.e. with treatment initiated prior to tumor becoming established, indicates that tumor growth can in fact be delayed with anti-TIGIT mAb alone.47 53 This has been demonstrated with subcutaneous tumors such as CT26 colon carcinoma, E0771 breast adenocarcinoma and methylcholanthrene-induced fibrosarcoma, and the B16F10 and 4T1 mammary carcinoma experimental metastasis models. Anti-TIGIT mAb treatment reduced tumor growth in a model of head and neck squamous cell carcinoma (HNSCC) through enhancement of CD8+ T cell responses and abrogation of the immunosuppressive capacity of Tregs and MDSCs.108 131 Anti-TIGIT single agent activity has also been observed in the Vk12653 multiple myeloma model, either following transplant of tumor cells or in the relapse setting following hematopoietic stem cell transplant.120 122
However, anti-TIGIT mAb alone may be not be sufficient to drive a clinical response in a therapeutic setting, where treatment is initiated in mice with established tumors.26 61 Here, combination of anti-TIGIT mAb with other immune checkpoint blockade therapies is generally required to promote antitumor activity. Dual blockade of both TIGIT and the PD-1/PD-L1 pathway resulted in nearly 100% complete responses in various tumor models including CT26, MC38, EMT6 mammary carcinoma and GL261 glioblastoma.26 61 132 In the CT26 model, coblockade of TIGIT and PD-L1 was required for tumor elimination and enhanced CD8+ T cell IFN-γ production in draining lymph nodes, while either antibody alone was without effect.26 Enhanced effector function of CD8+ T cells, as well as CD4+ T cells, was also observed with coblockade as compared with either monotherapy alone in the MC38 model.61 These findings are consistent with a mechanistic synergy between the PD1 and TIGIT pathways.47
As discussed above, both PD-1 and TIGIT repress CD226 phosphorylation and thus limit CD226 functional activity.47 88 Since the mechanisms by which TIGIT and PD-1 target CD226 for dephosphorylation are non-redundant, it is understandable why combining anti-TIGIT with PD-1/PD-L1 blockade is required to fully activate CD226. CD226 is necessary for activity of dual TIGIT/PD-1 combination therapy as CD226 deficiency or anti-CD226 mAb blockade abolishes the therapeutic benefit.26 47 The contribution of CD226 to activity of anti-TIGIT combination with anti-PD-1/PD-L1 indicates that considerations should be given to CD226 expression and functional availability.
Beyond dual TIGIT/PD-1 blockade, other immunotherapies have demonstrated promise as a combination partner with anti-TIGIT. These are evident within the PVR-nectin family itself. CD96 blockade improves antitumor responses in Tigit-/- mice.133 Treatment with anti-CD96 mAb in Tigit-/- mice resulted in greater reduction of lung metastases in the orthotopic B16F10 and E0771 tumor models. The activity of anti-CD96 mAb was dependent on NK cells and also CD226, as depletion of NK cells or coadminstration of blocking anti-CD226 mAb abrogated the effects of anti-CD96 mAb. While CD96 has been well characterized as an inhibitory receptor for NK cells, its role in CD8+ T cells is less clear, as CD96 has recently been shown to possess costimulatory activity that enhances CD8+ T cell activation and effector responses.134 PVRIG may compete with TIGIT and CD226 for binding to CD112, and it is hypothesized that blockade of PVRIG/CD112 in addition to TIGIT/PVR blockade may allow CD226 to engage its ligands unencumbered. PVRIG is expressed on TILs and intratumoral NK cells. In vitro experiments have demonstrated that a combination of anti-PVRIG and anti-TIGIT mAbs enhanced human TIL and NK cell effector function.135–137 Combination of anti-PVRIG with anti-PD-L1 reduced tumor growth as did anti-PD-L1 treatment in PVRIG-/- mice,137 138 indicating that combination of anti-PVRIG with anti-TIGIT may be another strategy worth investigating.
Human clinical trials
To date, there are over a dozen clinical trials targeting TIGIT. The majority of the anti-TIGIT clinical candidates are of the IgG1 isotype well known to interact with high affinity Fcγ receptors (FcγR). Others are IgG1’s bearing Fc domain mutations that enhance or abolish FcγR binding, or are of the IgG4 isotype, which exhibits limited ability to interact with FcγR.139 Over 20 additional anti-TIGIT mAbs or variants such as bispecifics are in preclinical development, with nearly two dozen companies entered in this highly competitive space (figure 4).

Anti-TIGIT monoclonal antibody therapies in clinical development. Molecule, clinical development phase, and Fc activity are indicated. Landscape as of November 2021. TIGIT, T cell immunoreceptor with Ig and ITIM domains.
Several anti-TIGIT mAbs have been tested as single-agents in Phase Ia clinical trials and in combination with anti-PD-L1 or anti-PD-1 antibodies in phase Ib or phase 2 clinical trials. Tiragolumab is a fully humanized IgG1 anti-TIGIT mAb with an effector IgG1 backbone that was tested as a single agent in Phase Ia and in combination with the anti-PD-L1 antibody atezolizumab in Phase Ib.140 Tiragolumab was well-tolerated as a single-agent and in combination with atezolizumab, with a safety profile similar to that of a checkpoint inhibitor; no new safety signals were detected with tiragolumab. No objective responses occurred in Phase Ia, but the combination of tiragolumab and atezolizumab produced objective responses in phase Ib, particularly in patients with tumors not previously treated with prior cancer immunotherapy or with tumors having positive PD-L1 expression.
Given the preliminary findings in phase I, the combination of tiragolumab and atezolizumab was then tested in a randomized, double-blinded, placebo-controlled phase II study (CITYSCAPE) in patients with newly-diagnosed metastatic PD-L1-positive non-small cell lung cancer.141 Patients were randomized to receive tiragolumab plus atezolizumab or placebo plus atezolizumab and were treated to disease progression and/or loss of clinical benefit. The addition of tiragolumab to atezolizumab improved overall response rate, progression-free survival, and overall survival, especially in patients with tumors having high PD-L1 expression, compared with placebo plus atezolizumab. The combination of tiragolumab and atezolizumab was well-tolerated, and no new safety signals were detected. Tiragolumab plus atezolizumab is currently being studied in multiple solid tumors in Phase III clinical trials.
Vibostolimab is another humanized anti-TIGIT mAb with an effector IgG1 backbone. In a phase I study, vibostolimab was evaluated as monotherapy or in combination with pembrolizumab in patients with metastatic solid tumors or in patients with advanced or metastatic NSCLC that were either anti-PD-1/PD-L1 treatment naïve or refractory.142 Vibostolimab was generally well tolerated across all dose levels and demonstrated a manageable safety profile. While sample sizes are small, efficacy evaluations demonstrated antitumor activity, particularly in anti-PD-1/PD-L1 treatment naïve patients and in both PD-L1 status positive and low/negative patients; limited responses were observed in the anti-PD-1/PD-L1 refractory population. Vibostolimab is being further evaluated as monotherapy or combination therapy in patients with NSCLC or other select advanced solid tumors.
Etigilimab is yet another anti-TIGIT mAb using the IgG1 framework. In a Phase I study, patients with advanced or metastatic solid tumors were treated with etigilimab monotherapy or in combination with anti-PD-1 antibody nivolumab.143 Etigilimab had an acceptable safety profile. Preliminary signals of clinical activity were observed when etigilimab was combined with nivolumab, but limited conclusions regarding efficacy can be made due to small sample sizes in this early phase trial.
Other studies are ongoing, but have not yet been reported out. Assuming initial demonstrations of efficacy are confirmed, one immediate outstanding question is whether an intact Fc domain is required for the activity of anti-TIGIT mAbs. Another question is centered around identification of biomarkers. Thus far, PD-L1 expression has been the most useful and utilized tool. As analyses of biomarker data from clinical trials become more mature, additional biomarkers such as serum immune cell-derived proteins may be revealed.
Conclusions and future directions
Based on the expression patterns of TIGIT, CD226, and the shared ligand PVR, the mechanisms of action of TIGIT blockade encompass many facets of the antitumor immune response. Adding the potential contribution of an intact Fc domain may further expand the potential activity of anti-TIGIT mAbs. Another layer of complexity in targeting TIGIT is the mechanistic convergence of TIGIT with the PD-1 inhibitory pathway, necessitating dual blockade to fully unleash activation of the full repertoire of tumor-reactive CD8+ T cells (figure 5). Yet there is more to the story, as considerations beyond TIGIT blockade and combination with anti-PD-(L)1 mAbs should be taken into account to unlock the full potential of TIGIT-targeted therapies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
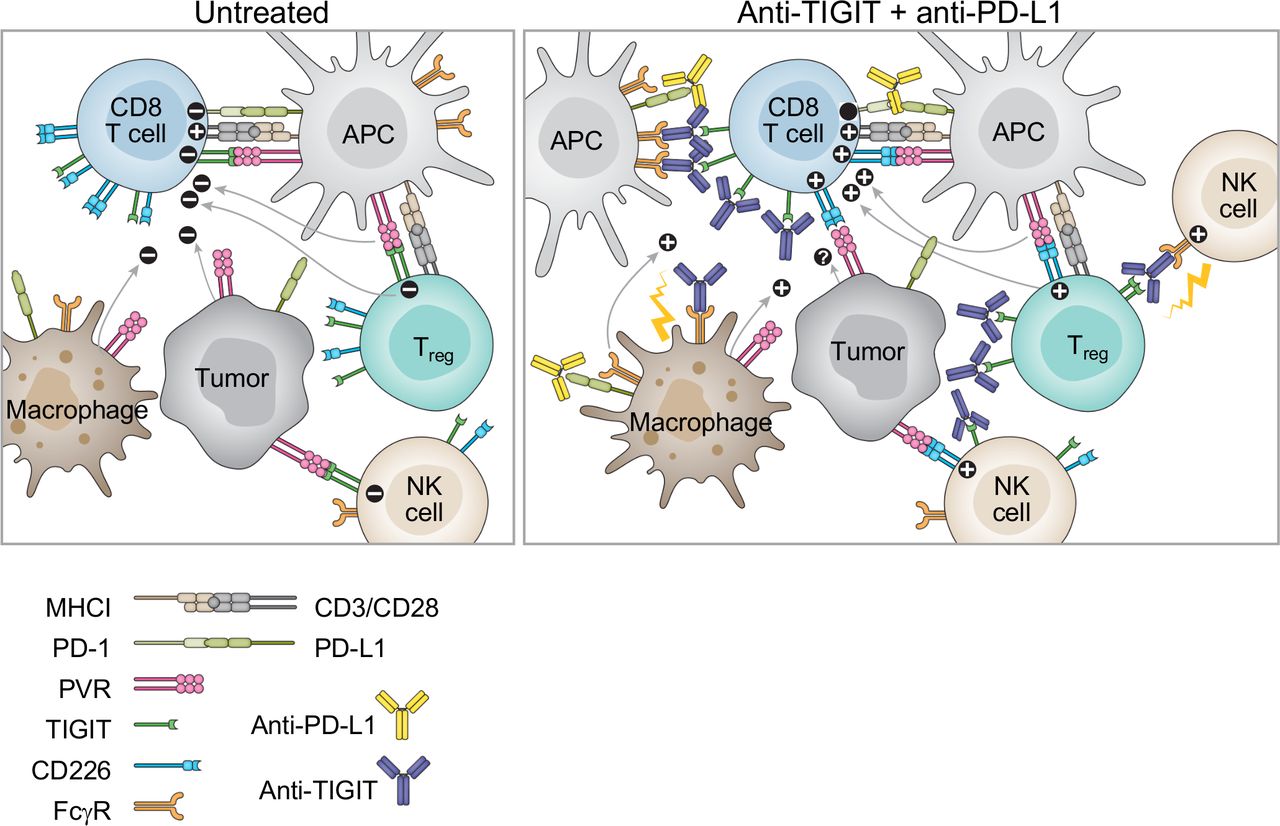
Effects of dual blockade of T cell immunoreceptor with Ig and ITIM domains (TIGIT) and PD-(L)1 on antitumor immune responses. Left: Tumor-reactive CD8+ T cells encounter various immunosuppressive mechanisms (denoted by ‘-’) mediated by TIGIT and PD-1. In addition to inhibiting CD8+ T cells, TIGIT also inhibits NK cell activity. TIGIT signaling supports Treg suppressive function. Additionally, myeloid cells such as antigen-presenting cells contribute to a suppressive tumor microenvironment through release of cytokines such as IL-10 and TGF-β following PVR engagement of TIGIT. Tumor cells also express PVR and may contribute to immune suppression. Right: Anti-TIGIT combined with anti-PD-L1 reverses immunosuppression and modulates the tumor microenvironment to support antitumor responses (denoted by ‘+’). Dual blockade of TIGIT and PD-(L)1 allows costimulatory activation by CD28 and CD226 for fully competent CD8+ T cell effector activity. Release of CD226 from TIGIT restraint enhances NK cell activity. Treg suppression is impaired through blockade of TIGIT, and CD226 may promote a proinflammatory phenotype. Anti-TIGIT mAbs with an effector competent Fc capable of engaging Fcγ receptors (FcγR) allow additional mechanisms such as ADCC mediated by NK cells and ADCP mediated by macrophages that may lead to depletion of cells expressing high levels of TIGIT such as Tregs. In addition, Fc-FcγR interaction may modulate myeloid cells to relieve suppression and promote inflammatory conditions. The effects of CD226 engagement of PVR on tumor cells remains to be determined (denoted by ‘?’). NK, natural killer.
Beyond CTLA-4 and PD-1/PD-L1
Based on our current understanding of its mechanism of action, the TIGIT/CD226-PVR axis provides an opportunity to synergize with or extend existing therapies targeting CTLA-4 and PD-1/PD-L1. It is perhaps not a coincidence that both of these pathways directly or indirectly involve the regulation of the CD28 costimulatory receptor on T cells. Although TIGIT does not regulate CD28 but rather CD226, the recent appreciation that both CD226 and CD28 are clients of PD-1 provides an attractive hypothesis for understanding why PD-1/PD-L1 blockade might combine with TIGIT blockade. Optimal costimulation in T cells expressing both PD-1 and TIGIT, when exposed to their respective ligands, will only be achieved by simultaneously blocking both coinhibitory receptors: TIGIT blockade is required to allow CD226 to engage PVR, and PD-1 blockade is required to prevent CD226 dephosphorylation in addition to its better known functions of blocking dephosphorylation of CD28 and (at least under some conditions) dephosphorylation of the CD3ζ subunit of the TCR. As CD226 is emerging as a major determinant of T cell function, optimal activation or re-activation of CD226 would appear to be an important adjunct to current treatments, especially PD-1/PD-L1 blockade given the convergence between the PD-1 and CD226 pathways.
The step, or steps, of T cell function that are regulated by TIGIT and PD-1 are a topic of intense current interest. PD-1 blockade is most often thought to relieve the exhausted phenotype typically associated with TILs, reactivating at least some effector activity. TIGIT blockade may work in parallel to achieve the same outcome. However, recent evidence has emphasized a role for PD-1 earlier in T cell activation, acting to expand a stem-like compartment that serves as a cellular resource to drive antigen-specific T cell responses. These TSCM cells are likely instructed by antigen-presenting DCs either in the tumor or lymphoid organs. Since we and others have found that TIGIT may coexist with PD-1 on these early cells, again there would be the opportunity for two coinhibitory receptors to act in concert. Although TIGIT’s ligand, PVR, is widely distributed, it is also expressed by DCs. Whether, as is the case for PD-L1, PVR on DCs is critical for its ability to regulate its receptor is unknown. It will be of great interest to determine if TIGIT and PD-1 blockade increase TCR clonality, as might be expected if DCs were in fact a relevant site for the function of both inhibitory pathways.
The significance of TIGIT function, and indeed the function of PD-1 and CTLA-4, must not be viewed solely through the lens of effector T cells. For example, while Tregs express all three coinhibitory receptors, CTLA-4 and TIGIT are expressed at high levels and both may enable Treg depletion if Fc domain-intact antibodies are used, although clear evidence for this possibility in clinical studies has yet to be found. TIGIT may also, independently, cause Treg reprogramming.
Unlike CTLA-4 and PD-1, TIGIT it also widely expressed by NK cells.144–147 The PVR-TIGIT interaction, via the TIGIT’s ITIM or ITT domains, may help drive NK cell inactivation, so TIGIT blockade might be expected to enhance NK antitumor activity. Similar considerations may also be relevant for TIGIT signaling on Tregs.
In the case of NK cells, the state of activation or ‘education’ may be important not only for direct tumor cell killing, but also for establishing the immunological phenotype of a tumor. Recent evidence suggests that NK cells may secrete chemokines that can recruit DCs or CD8+ T cells to the tumor microenvironment,148 thereby priming the landscape for adaptive responses. Thus, TIGIT blockade may combine with PD-1 blockade for reasons that are orthogonal to the reactivation of costimulatory receptor signaling in the effector T cell compartment.
Beyond PVR
The PVR-nectin family of receptors and ligands is continually expanding as interest in this family spurs efforts to identify new potential targets. Recently, Nectin4 (PVRL4) has been described as a ligand that exclusively interacts with TIGIT and not CD226, CD96 or PVRIG.149 Nectin4 is expressed abundantly during fetal development, but expression is restricted to primarily placenta and testis in human adult tissues.150 However, Nectin4 is expressed in many cancers, including breast, bladder, lung and pancreas, and expression is associated with poor prognosis.149 151 Nectin4 engagement of TIGIT on NK cells reduces cytotoxicity, but does not completely abrogate NK cell activity. Blocking anti-Nectin4 mAb attenuated growth of MDA-MB-453 human breast carcinoma cells, which express Nectin4 as well as other PVR-nectin family ligands, implanted into SCID-beige mice together with human NK cells.149
While CD111 (PVRL1) has not been demonstrated to be a ligand for TIGIT, it may still play a role in regulating TIGIT-expressing CD8+ T cell responses against tumors.152 In a mouse model of hepatocellular carcinoma, PVRL1 was reported to stabilize PVR expression on the cell surface, allowing for productive TIGIT interaction with PVR to mediate inhibition of CD8+ T cells and promote tumor growth. Further illustrating the complex network of PVR-nectin family interactions and how this may directly or indirectly modulate TIGIT activity, PVRL3 has been shown to mediate clathrin-dependent endocytosis of PVR upon cell-cell contact.153 As PVRL3 has higher affinity for PVRL1 than PVR, PVRL3 will preferentially bind to PVRL1, thereby disrupting the ability of PVRL3 to reduce PVR surface expression.152 153
Ligands for TIGIT may also come from sources other than tumor cells. The anaerobic Gram-negative bacterium Fusobacterium nucleatum is found in the tumor microenvironment, where it can bind to tumor cells.154 Interaction of the Fap2 protein of F. nucleatum with human TIGIT inhibited NK cell and TIL activity, but Fap2-mediated TIGIT inhibition was dependent on the ability of the bacteria to cause hemagglutination. CD226 did not interact with F. nucleatum, and the binding site for TIGIT-F. nucleatum interaction appeared to be distinct from the TIGIT-PVR site. As anti-TIGIT mAbs have been selected primarily on the basis of their ability to block interaction with PVR, consideration should be given to other ligands such as Fap2 that may engage TIGIT even in the presence of anti-TIGIT mAb.
Beyond the Fab
Selection of anti-TIGIT mAbs has focused predominantly on the variable antigen-binding fragment (Fab) as this region that provides target-binding specificity and determines whether the Ab will block interaction with PVR and other ligands. However, the constant region may also play an important role, as the fragment crystallizable (Fc) region can allow engagement with FcγRs to enable effector functions such as ADCC, complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), induction of cytokines/chemokines, and endocytosis of opsonized targets.155 Thus, the Fc portion of the anti-TIGIT mAb may be an important determinant of mechanism(s) of action.
Different IgG isotypes have distinct binding profiles to various FcγRs, which themselves have differing properties. For anti-TIGIT mAbs, IgG1 or IgG4 Fc formats are used. IgG1 has the highest affinity to all FcγRs relative to other isotypes, but affinities are variable for each specific FcγR. IgG1 can mediate effector functions including ADCC, ADCP and CDC. Mutations in IgG1 can abrogate effector activity, rendering it FcγR null, or enhance effector activity. By comparison, IgG4 has high affinity for FcγRI, but low affinity for FcγRIIa, FcγRIIb and FcγRIIIa, and therefore, can mediated ADCP but do not or have low potential to induce ADCC or CDC. FcγRI is expressed on monocytes/macrophages, DCs and activated neutrophils, and one its primary functions is to induce ADCP by myeloid cells when interacting with IgG1 or IgG4. FcγRIIa and FcγRIIIa are expressed on monocytes/macrophages, DCs, NK cells and platelets. FcγRIIa mediates ADCP when engaged with IgG1, whereas FcγRIIIa interaction with IgG1 can lead to ADCC by NK cells and macrophages.
The contribution of Fc-FcγR coengagement for functional activity of anti-TIGIT mAbs was investigated using a clone (10A7) that exhibited cross-reactivity for both mouse and human TIGIT.156 In mice, the IgG2a isotype is equivalent to human IgG1. Growth of CT26 tumors was controlled using a wild-type, fully effector competent IgG2a format, but mutations in the Fc to attenuate FcγR binding had no effect on tumor progression. Notably, while wild-type IgG2a has the capacity to mediate ADCC and ADCP, depletion of intratumoral Tregs was not observed, demonstrating that an effector competent Fc does not necessarily result in depletion as a mechanism of the Ab. When the anti-TIGIT Ab was engineered to the human IgG1 format or effector null variant, T cell responses were attenuated with the mutant variant and activity of the wild-type Ab was inhibited by blockade of FcγRIIIA.156 Studies using a different clone (T4), also with cross-reactivity for mouse and human, showed that an Fc-enhanced Ab improved on the therapeutic effect of wild-type Fc Ab, whereas activity was lost with an effector null mutant.157 While a reduction in intratumoral Tregs was observed, suggesting ADCC-mediated depletion, Tregs were still present. As FOXP3 was used as a marker for Tregs, it is possible that the reduction in Tregs may be attributable to destabilization of FOXP3 expression through blockade of TIGIT, and that enhanced engagement with FcγRs enforces the reprogramming of Tregs. A non-Treg depleting, effector competent Fc-dependent, mechanism was recently suggested. Engagement of FcγRs on myeloid cells by anti-TIGIT mAb (clone 18G10) with the IgG2a isotype resulted in enhanced antigen presentation as well as increased cytokine and chemokine production, thereby promoting a state of persistent immune activation supportive of robust antitumor activity.158
The possibility of Fc-mediated depletion activity cannot be ruled out, as ADCC or ADCP mechanisms may be applicable only to those cells expressing high surface levels of TIGIT. Studies using a human anti-TIGIT mAb (clone EOS-448) showed that a human IgG1 formatted Ab, but not IgG2 or IgG4, had cytotoxic potential that reduced Tregs in PBMCs from cancer patients, with less impact on CD8+ T cells or non-Treg CD4+ T cells. As Tregs had the highest TIGIT expression, there was a correlation between TIGIT expression level with susceptibility to depletion.73 Notably, however, the degree of Ab-mediated cytolysis of anti-TIGIT mAb was not as strong as rituximab, a well-characterized CD19 depleting Ab. The potential for depletion of cells expressing TIGIT extends beyond Tregs and may include tumor cells themselves. Tumor CD4+ T cells from patients with Sézary syndrome had higher expression of TIGIT than normal circulating CD4+ T cells, and the malignant CD4+ T cells were more susceptible to ADCC, indicating that direct cytotoxicity on TIGIT-expressing tumor cells may provide yet another mechanism for FcγR-engaging anti-TIGIT mAbs.73
Myeloid cells expressing FcγR may be potentially modulated on engaging Fc effector competent Abs, as the intracellular domains of FcγRs contain either ITIM or ITAM domains with inhibitory or activating activity, respectively. Another possibility consequent of Fc-FcγR interaction is sequestration of TIGIT, providing yet another mechanism contributing to the ability of CD226 to engage PVR. While it is evident that anti-TIGIT may have modulatory effects on DCs, macrophages, and MDSCs, as discussed earlier, it remains to be determined if these effects are dependent on Fc-FcγR or merely blockade of TIGIT-PVR interaction.
Engagement of various FcγRs expressed on different cells can produce a diverse array of functional consequences that can impact the tumor microenvironment and antitumor responses. As clinical trials investigating anti-TIGIT mAbs with differing Fc backbones read out their data, the importance of Fc in driving clinical responses, and whether Fc is a differentiator for the clinical candidates, will become clear.139
Beyond combination with other checkpoints
The cancer immunity cycle describes generation of immunity to cancer as a cyclic, self-propagating and reiterative process.159 160 Therapeutics targeting various steps of the cancer immunity cycle have revolutionized the treatment of cancer.161 The cycle is initiated by cancer cell death and release of tumor-specific neoantigens, and this process can be enhanced with chemotherapy, radiation therapy, photodynamic therapy, viral therapy and other tumor-directed therapies. NK cells play an important role at this stage. Antigen-presenting cells take up tumor antigens and present them to immune cells. Strategies to improve antigen presentation include vaccine approaches, IFN-γ, GM-CSF, TLR agonists, STING agonists and anti-CD73. T cells are primed and activated and start to undergo differentiation. Here, Ab blockade of inhibitory receptors such as CTLA-4 and PD-1, or agonism of costimulatory receptors such as CD27, GITR, 4-1BB, and OX40, can allow optimal T cell responses to be generated, and cytokines such as IL-2, IL-12 and IL-15 may be beneficial. T cells then traffic to tumors. At this step, chimeric antigen receptor (CAR)-T cell therapy or other adoptive cell transfer modalities increase the numbers of T cells that can respond against tumors, and bispecific T cell engagers direct T cells specifically to tumor cells. Once attracted to the tumor site, T cells must infiltrate the tumor. Antiangiogenesis factors and modulators of the tumor microenvironment can allow T cells to penetrate tumors. Finally, T cells recognize and kill cancer cells, and this is where many immune checkpoint inhibitors play a major role. Throughout the cancer immunity cycle, immunosuppressive pressure is applied by Tregs and the tumor microenvironment.
TIGIT may directly impact multiple steps of the cancer immunity cycle, thereby differentiating it from other immune checkpoints possessing perhaps more narrow functions. However, given the promise of immune checkpoint inhibitors such as anti-PD-1/anti-PD-L1, anti-CTLA-4, and now anti-TIGIT, there is intense interest in other immune checkpoints such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), V-domain immunoglobulin suppressor of T cell activation (VISTA), B7 homology 3 protein (B7-H3), inducible T cell costimulatory (ICOS), and B and T lymphocyte attenuator (BTLA).162 Clinical trials are underway for all of these targets, with LAG-3 gaining the most attention in terms of clinical candidates and numbers of trials. While early data has shown that targeting these immune checkpoints is generally well tolerated and promising early results have been reported, mature clinical data are still awaiting. Of note, clinical trials investigating these novel agents are predominantly designed to test combinations, with anti-PD-(L)1 being the partner of choice.
It remains to be seen if targeting these various immune checkpoints will result in a clear winner, the more likely scenario being that each may provide some degree of benefit based on its specific biology. Targeting TIGIT, with its role in not only mediating antitumor responses of multiple lymphocyte populations including TSCM, memory and effector T cells and NK cells, but also myeloid cells through PVR, may thus have an advantage. Rather than seeking additional immune checkpoint inhibitors, perhaps it is more prudent to evaluate other therapeutic strategies that leverage and build on the promise of TIGIT blockade or PD-1/PD-L1 coblockade. For instance, combination of anti-TIGIT with Abs targeting costimulatory receptors including 4-1BB, OX-40 or GITR demonstrated additive or synergistic effects in various mouse tumor models.73
While cytokines such as IL-15 expand lymphocytes, including those that are tumor-reactive, activity of these cells may be limited by TIGIT expression; however, combination of anti-TIGIT with IL-15 promoted control of metastatic tumors in mouse melanoma models.102 Cell therapy is another means of increasing numbers of tumor-reactive T cells in a patient.145 Adoptive T cell therapy with TILs may benefit from TIGIT blockade, as TIGIT, often together with PD-1, is expressed on TILs.26 CAR- T cells may face the same challenges posed by TIGIT expression, and anti-TIGIT mAb is one way to overcome this T cell inhibitory signal.163 Combination of TIGIT/PD-1 coblockade with CD40 agonism was demonstrated to overcome immune evasion mediated by the TIGIT/PVR axis and elicit robust antitumor responses in a mouse pancreatic adenocarcinoma model,164 validating DC-activating strategies as potential partners and opening the door to testing anti-TIGIT plus anti-PD-(L)1 with cancer vaccines.145
As the antitumor immune response must battle continually evolving immune-resistant mechanisms, therapeutics that can impact multiple steps of the cancer immunity cycle may elicit more potent clinical responses than those specifically targeting a particular step. While combinatorial approaches have become standard, the right combinations are needed. TIGIT is a target that is expressed by key immune cell players in the cancer immunity cycle, and the TIGIT/CD226-PVR axis has a role in nearly every step. TIGIT in cancer immunotherapy is still relatively in its infancy, but there is vast potential for anti-TIGIT as a combination partner for therapies beyond anti-PD-1/PD-L1. Thus, TIGIT-based immunotherapies hold tremendous promise in the fight against cancer.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Contributors EYC and IM wrote the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests EYC and IM are employees of Genentech, a member of the Roche group, which develops and markets drugs for profit.
Provenance and peer review Not commissioned; externally peer reviewed.