Article Text

Abstract
Background Long-term prognosis of WHO grade II, isocitrate dehydrogenase (IDH)-mutated low-grade glioma (LGG) is poor due to high risks of recurrence and malignant transformation into high-grade glioma. Immunotherapy strategies are attractive given the relatively intact immune system of patients with LGG and the slow tumor growth rate. However, accumulation of the oncometabolite D-2-hydroxyglutarate (D-2HG) in IDH-mutated gliomas leads to suppression of inflammatory pathways in the tumor microenvironment, thereby contributing to the ‘cold’ tumor phenotype. Inhibiting D-2HG production presents an opportunity to generate a robust antitumor response following tumor antigen vaccination and immune checkpoint blockade.
Methods An IDH1R132H glioma model was created in syngeneic HLA-A2/HLA-DR1-transgenic mice, allowing us to evaluate the vaccination with the human leukocyte antigens (HLA)-DR1-restricted, IDH1R132H mutation-derived neoepitope. The effects of an orally available inhibitor of mutant IDH1 and IDH2, AG-881, were evaluated as monotherapy and in combination with the IDH1R132H peptide vaccination or anti-PD-1 immune checkpoint blockade.
Results The HLA-A2/HLA-DR1-syngeneic IDH1R132H cell line expressed the IDH1 mutant protein and formed D-2HG producing orthotopic gliomas in vivo. Treatment of tumor-bearing mice with AG-881 resulted in a reduction of D-2HG levels in IDH1R132H glioma cells (10 fold) and tumor-associated myeloid cells, which demonstrated high levels of intracellular D-2HG in the IDH1R132H gliomas. AG-881 monotherapy suppressed the progression of IDH1R132H gliomas in a CD4+ and CD8+ cell-dependent manner, enhanced proinflammatory IFNγ-related gene expression, and increased the number of CD4+ tumor-infiltrating T-cells. Prophylactic vaccination with the HLA-DR1-restricted IDH1R132H peptide or tumor-associated HLA-A2-restricted peptides did not enhance survival of tumor-bearing animals; however, vaccination with both HLA-A2-IDH1R132H and DR1-IDH1R132H peptides in combination with the IDH inhibitor significantly prolonged survival. Finally, tumor-bearing mice treated with both AG-881 and a PD-1 blocking antibody demonstrated improved survival when compared with either treatment alone.
Conclusion The development of effective IDH1R132H-targeting vaccine may be enhanced by integration with HLA class I-restricted cytotoxic T cell epitopes and AG-881. Our HLA-A2/HLA-DR1-syngeneic IDH1R132H glioma model should allow us to evaluate key translational questions related to the development of novel strategies for patients with IDH-mutant glioma.
- Brain Neoplasms
- Central Nervous System Neoplasms
- Drug Evaluation, Preclinical
- Vaccination
- Programmed Cell Death 1 Receptor
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Brain Neoplasms
- Central Nervous System Neoplasms
- Drug Evaluation, Preclinical
- Vaccination
- Programmed Cell Death 1 Receptor
WHAT IS ALREADY KNOWN ON THIS TOPIC
Low-grade gliomas are primary brain tumors with mutations in isocitrate dehydrogenase (IDH) and immunologically cold characteristics.
Despite their slow-growing nature, an overwhelming majority of patients eventually succumb to the disease, often with transformation into high-grade lesions. This delayed morbidity, however, presents a therapeutic window when immune therapies may prove to be beneficial.
WHAT THIS STUDY ADDS
Our studies demonstrate that inhibition of the mutant IDH1 enzyme in the HLA.A2/DR1 trangenic mouse model enhances inflammatory pathways as well as the expression of an immune checkpoint molecule PD-L1.
Inhibition of both mutant IDH1 and PD-1/PD-L1 signaling in vivo significantly improved survival. We showed that vaccination targeting both HLA.A2-restricted and HLA.DR1-restricted antigens, combined with IDH1R132H inhibition, significantly prolonged tumor-bearing animal survival.
Overall, our results suggest a need for developing strategies that would coordinately target both major histocompatibility complex class-I and class II antigens, tumor-specific metabolic molecules (eg, IDH mutation), and immune-checkpoint molecules.
Introduction
Diffuse gliomas represent the most common primary brain tumor in adult patients, and presence of a mutation in the Isocitrate Dehydrogenase 1/2 (IDH1/2) genes is now one of the required molecular determinations for the diagnosis of two of the three adult diffuse glioma subtypes—astrocytoma and oligodendroglioma.1 All IDH1/2 mutations result in the accumulation of the oncometabolite R-2-hydroxyglutarate (R-2HG), and the most common mutation IDH1R132H (arginine (R) to histidine (H) at position 132) accounts for over 80% of all IDH mutations in gliomas.2 Due to their infiltrative nature and high frequency of malignant transformation into more aggressive WHO grade 3 or 4 high-grade gliomas (HGG), the vast majority of patients with IDH-mutant low-grade gliomas (LGG) succumb to the disease within one to two decades of diagnosis.3 Although IDH-mutant LGG are described as immunologically cold,4 immunotherapeutic modalities, such as vaccines, may offer a safe and effective option for these patients due to their tumors’ slower growth rate (in contrast with HGG), which should allow sufficient time for multiple immunizations and, therefore, higher levels of antiglioma immunity. The immune system of patients with LGGs may not be as compromised as that of patients with HGG since patients with LGG demonstrated an excellent immunological response to vaccines in a previous clinical trial.5 Further studies are warranted to develop effective immunotherapy approaches for these patients by integrating our in-depth understanding of immunobiology and novel therapeutic approaches.
The impacts of the IDH1R132H mutation on tumor cell metabolism and epigenetic programming have been studied and profiled in great detail since the prevalence of the mutation in LGGs was uncovered.6 7 In addition to its oncogenic functions that are intrinsic to tumor cells, R-2HG also impacts the antitumor immune response.8 9 Our lab has shown that the presence of IDH1R132H and R-2HG accumulation can drive immune evasion through suppression of type-1 inflammatory genes resulting in a reduced recruitment of anti-tumor CD8+ T-cells.9 Studies by Bunse et al have demonstrated that R-2HG, taken up by T-cells in the tumor microenvironment (TME), impairs T-cell receptor signaling and altered cytokine production.10 Comparisons of human wildtype and IDH1R132H gliomas by Amankulor et al additionally showed decreased infiltration of immune cells within mutant tumors,8 while recent work by Friedrich et al revealed complex and dynamic changes in the activation of IDH1R132H glioma-associated myeloid cells leading to an immunosuppressive phenotype.11
Even though immunotherapies have emerged as a promising new anticancer treatment strategy in many tumor types, clinical trials of immunotherapies in gliomas have yet to result in improved patient outcomes.12 13 Given the high prevalence of IDH1R132H in LGGs and the immunosuppressive effect of R-2HG, IDH1R132H gliomas present an opportunity to combine a targeted treatment, via a small molecule inhibitor of mutant IDH1/2, with immunotherapy in the form of a therapeutic IDH1R132H peptide vaccine. Previous work by Schumacher et al demonstrated that the major histocompatibility complex (MHC) class II neoepitope resulting from the R132H mutation can be immunogenic in the context of human leukocyte antigens (HLA)-DR presentation.14
To establish a highly clinically relevant mouse model of IDH1R132H glioma, we describe the development of an orthotopic glioma model in an HLA-A*0201-double and HLA-DR*0101-double transgenic (dTG) mouse strain. We engineered this glioma model to carry the IDH1R132H mutation and thus produce 2-HG. Using this new model, we evaluated the therapeutic and immunological impacts of IDH1R132H inhibition alone and in combination with immunotherapeutic approaches. Our data indicate the ability of IDH1R132H inhibition to upregulate interferon-γ (IFNγ) signaling and delay tumor growth but without long-term survival benefit. We show that delay in tumor growth is dependent on both CD8+ and CD4+ T-cell responses and is boosted by prophylactic vaccinations targeting both HLA class I and class II glioma epitopes. Finally, we demonstrate the enhanced survival of tumor-bearing dTG mice treated with both an IDH1R132H inhibitor and an α-PD1 antibody when compared with either treatment alone, suggesting the utility of this model for future preclinical studies investigating IDH1R132H-targeting immune therapies.
Materials and methods
Reagents
The following reagents were purchased: DMEM (Corning, #10013CV), RPMI (Corning, #10040CV), GlutaMAX (Gibco, #35050038), sodium pyruvate (NA-Pyr, Gibco #11360070), MEM NEAA (Gibco, 11140076), Penicillin/Streptomycin (P/S, Gibco #15070063), HEPES (Gibco, #15630080), Premium Grade FBS (VWR Seradigm 97 068 Lot 340B17), 0.25% Trypsin (Corning, #25 053CI), poly-L-lysine (Sigma-Aldrich, #P4832), Incomplete Freund Adjuvant (IFA, Santa-Cruz Biotechnology #sc-24019). Poly-ICLC was generously provided by Oncovir Inc. All peptides were synthesized by A&A Labs LLC. In vivo treatment antibodies were purchased from BioXCell: anti-mouse CD8α clone YTS 169.4, anti-mouse CD4 clone GK1.5, anti-mouse PD-1 clone RMP1-14, rat IgG2a isotype control clone 2A3.
Generation of IDH1m HLA.A2/DR1 (dTG)-syngeneic tumor cell line
HLA-A*0201-double and HLA-DR*0101-double transgenic mice (endogenous MHC molecules have been knocked out) were obtained from the lab of Dr François Lemonnier (Pasteur Institute, Paris, France).15 Primary astrocytes were isolated from the cortices of neonatal pups as described by Schildge et al.16 The cells were transformed using the following plasmids pT2/C-Luc/PGK-SB1.3, pT/Caggs-NrasV12, pT2/shp53/GFP4/mPDGFβ, and pT3.5/Caggs-HsIDH1R132H. To isolate a uniform population, the resulting pool of transformed cells underwent single-cell cloning. Multiple clones were screened for the expression of IDH1m and GFP in vitro and their ability to reproducibly form tumors in vivo. Cells isolated ex vivo were used in the remainder of the studies (referred to as dTG-IDH1R132H, procedure outlined in figure 1A). For experiments evaluating responses to Class I tumor-associated antigens (TAAs), the dTG-IDH1R132H cells were transduced with a MMLV retroviral construct expressing the hgp100(209–217), hTYR(368–376), and h/mTRP2(180–188) peptides, and a blasticidin resistance gene. The plasmid was synthesized by VectorBuilder. The resulting dTG-IDH1R132H; TAA cells were selected using 10 µg/mL blasticidin (maintenance 5 µg/mL). Detailed description of protocols and plasmid backgrounds are available in online supplemental materials and methods.
Supplemental material

Generation and validation of the new dTG glioma model. (A) Schematic description of the genetic background of the dTG mouse strain and the workflow to generate the dTG glioma model. The transgenic mouse strain used in the current study lacks expression of mouse MHC class I (β2 m) and class II (H-2 IAβb) molecules. However, all nucleated cells (both immune cells and tumor cells) can express the human HLA-A*0201 and the human HLA-DR1*0101. (B) Immunofluorescent staining of DTG- IDH1R132H cells. Nuclei were visualized with DAPI (blue); intracellular IDH1R132H protein in yellow (bottom image); isotype control for primary antibody (top image; scale bar, 50 µm). (C) Cell surface expression of HLA-A2 (left panel) and HLA-DR1 (right) on IFNɣ treatment. DTG-R132H cells were cultured in the presence of 50 ng/mL IFNɣ (or solvent only media) for 72 hours and analyzed by FC. (D) Extracellular 2-HG produced by dTG-IDH1R132H cells in vitro. Within a 6-well plate, 1.5×105 normal human astrocytes (WT astrocytes) and 1.5×105 dTG-IDH1R132H cells were plated in 2 mL of media and cultured for 72 hours. Levels of 2-HG in the supernatant of each cell line were measured by LC-MS/MS. Data represent technical triplicates of each condition. (E) High levels of 2-HG produced by orthotopically inoculated dTG-IDH1R132H cells. Tumors were collected from mice injected with dTG-wt (n=5) or dTG-IDH1R132H (n=4) when tumor burden reached protocol endpoint. Single-cell suspensions were analyzed for the presence of 2-HG. dTG, double transgenic; FC, flow cytometry; HG, hydroxyglutarate; HLA, human leukocyte antigens; MHC, major histocompatibility complex.
Pharmacodynamics and pharmacokinetic analyses
The following analytes were measured by LC-MS/MS: 2-HG, AG-120, and AG-881. In vitro sources were snap frozen cell pellets (1.5×105 cells cultured for 72 hours). In vivo data were derived from snap frozen tissues: 20 µL of plasma and tumor-bearing hemispheres. Detailed descriptions of detection methods are available in online supplemental materials and methods.
Orthotopic tumor induction
All experiments used 10–13-week-old dTG mice. Animals were handled in the Animal Facility at UCSF, per an Institutional Animal Care and Use Committee-approved protocol. 1×105 cells were stereotactically injected through an entry site at the bregma 2 mm to the right of the sagittal suture and 3 mm below the surface of the skull of anesthetized mice using a stereotactic frame. Prior to all treatment, mice were randomized such that the initial tumor burden in the control and treatment groups were equivalent. Animals were kept on study until the specified days post-treatment or until they reached a protocol-specified humane endpoint.
Bioluminescent imaging (BLI)
Tumor progression was evaluated by luminescence emission on a Xenogen IVIS Spectrum 10 min after an i.p. injection of 150 mg/kg D-luciferin prepared according to the manufacturer’s directions (Gold Biotechnology, #LUCK-100). The average radiance signal was used to generate all tumor growth data.
IDH1m inhibition
For in vitro studies, stock solutions of AG-120 and AG-881 were resuspended in DMSO at 50 mM. For in vivo use, both inhibitor solutions were prepared daily in 0.5% Methyl Cellulose 400 (Fujifilm Wako Chemicals #133–17815)+0.2% Tween-80 (Sigma-Aldrich #9490). AG881 was administered QD at 50 mg/kg via oral gavage. AG120 was administered two times a day at 450 mg/kg via oral gavage.
Flow cytometry
Single-cell suspensions were stained with fluorescently labeled antibodies using concentrations recommended by the manufacturers. A detailed description of the flow cytometry (FC) procedure is provided in online supplemental materials and methods. A list of the antibodies used is available in online supplemental table 1.
Supplemental material
Immunofluorescence
Cells were grown on poly-L-lysine coated coverslips to confluency and then fixed in 10% formalin at room temperature (RT) for 10 min and permeabilized for 10 min at RT in 0.1% Triton-X solution. Slides were then washed in 1× wash buffer (Dako, #S300685-2C) and incubated in 10% normal serum for 1 hour at RT. Primary antibodies were applied overnight in a moist chamber at 4°C. The next day, secondary antibodies were applied for 45 min at RT. Samples were dehydrated in graded ethanol (95%–100%) and mounted with DAPI (ProLong Gold antifade reagent with DAPI, Invitrogen #P36931). All staining steps were preceded by at least two 5 min incubations in 1X wash buffer. The antibodies used were: anti-IDH1m (1:200, Dianova, #DIA-H09), isotype control antimouse IgG2a (1:1000, Abcam, #ab18449), and goat antimouse IgG conjugated to Alexa Fluor 555 (1:500, Invitrogen, #A21425 2 mg/mL). Images were acquired using Zeiss Axio Imager 2 microscope (Zeiss, Oberkochen, Germany), using TissueFAXS scanning software (TissueGnostics GmbH, Vienna, Austria). Identical exposure time and threshold settings for each channel on all sections of similar experiments were used.
Nanostring
Tumor samples were either frozen in N2(l) immediately following dissection or dissociated enzymatically at 37°C (3.2 mg/mL Collagenase IV; 1 mg/mL DNAse I for 45 min) to generate a single-cell suspension. The latter was used to separate CD11b+ and CD11bneg fractions via the CD11b microbeads magnetic cell sorting kit (Miltenyi Biotec). mRNA was isolated from both whole tumor samples and isolated cell pellets using the RNeasy Plus Mini Kit (Qiagen, #74134). The Mouse (Mmv1)_Immunology codeset by Nanostring was used to measure the expression of immune-related genes. Data analyses used to generate the differential gene expression volcano plots are described in online supplemental material and methods.
Ex vivo T-cell suppression
CD3+ splenocytes from 6 to 10-week-old dTG naïve mice were collected using the Mouse CD3 T-cell Negative Selection Kit (BioLegend, #480031), labeled with 2 µM CFSE, washed, and added to all the wells of a round-bottom 96-well plate at 5×105 cells per well. T-cell activator beads (Gibco, #11 456D) were added at a 1:1 ratio to all wells except for negative control wells. Tumor-infiltrating CD11b+ cells were isolated as described above and resuspended at 8×106 cells/mL and serial dilutions were made starting at 4×105 cells per well. As a negative control 5×105 T-cells were cultured in complete RPMI only. As a positive control, 5×105 T-cells were cultured with activating beads only. Proliferation was measured by FC after 72 hours by evaluating the per cent of T-cells that had diluted CFSE.
Prophylactic vaccines and antibody treatments
Mice received two s.c. flank injections of 50 µL each of peptides emulsified in IFA and 20 µg poly-ICLC intramuscularly on days –21, –10, and –5 relative to the date of tumor inoculation. Emulsions contained equal volumes IFA and either PBS (control mice) or a combination of peptides hIDH1(R132H)(123–142), hgp100(209–217), hTYR(368–376), and h/mTRP2(180–188) each at the final concentration of 1 mg/mL. For PD-1 blocking and T-cell depletion studies, details on treatment timeline and antibody administration are described in the corresponding figures and online supplemental table 2.
Results
Generation and validation of the new dTG glioma model
To evaluate the HLA-DR-restricted IDH1R132H antigen-specific T-cell response, we generated a glioma cell line that is syngeneic to the dTG mouse strain (figure 1A). We obtained astrocytes from the cortical regions in the brains of neonatal dTG mice and transfected them ex vivo with SB vectors encoding oncogenes, NRas, PDGFβ, and a short-hairpin targeting p53, as well as a plasmid encoding the human IDH1R132H cDNA. Using serial dilutions, we established several clones of monolayer GFP+ cells (included in one of the SB vectors) that also displayed uniform IDH1R132H expression by immunofluorescence (figure 1B) and produced ~20 µg/mL of 2-HG in vitro (figure 1D). For in vivo studies, we chose two clones that robustly upregulated HLA-A2 and HLA-DR1 expression on IFNγ stimulation (figure 1C and online supplemental figure 1A). We harvested multiple tumors which emerged after orthotopic injections of the two clones and used the tissues to derive cell lines that grew reproducibly in vivo and produced high intratumoral concentrations of 2-HG (figure 1E and online supplemental figure 1B,C).
Supplemental material
Inhibition of IDH1R132H suppresses 2-HG production and tumor growth both in vitro and in vivo
To test the effect of IDH1R132H inhibition in our dTG model, we treated dTG-IDH1R132H cells in vitro with two mutant IDH inhibitors, AG-120 (ivosidenib; Servier Pharmaceuticals) and AG-881 (vorasidenib; Servier Pharmaceuticals). Both molecules have been shown to significantly inhibit the IDH1R132H function and are either already FDA-approved (AG-120)17 or currently in phase III clinical development (AG-881) for patients with mutant IDH tumors (ClinicalTrials.gov identifier NCT04164901). By measuring 2-HG levels produced by dTG-IDH1R132H cells, we determined that both inhibitors had nanomolar IC50 values (figure 2A). Next, we established the pharmacokinetic (figure 2B) and pharmacodynamic (figure 2C) properties of the two molecules in glioma-bearing dTG mice. Intratumoral concentration of AG-881 was highest 1 hour postadministration (~1800 ng/mL) and remained above 1000 ng/mL for 24 hours, while the concentration of AG-120 peaked 2 hours postadministration (~1500 ng/mL) and approached background levels by 24 hours (figure 2B). Four and 12 hours postadministration, both drugs successfully inhibited intratumoral 2-HG accumulation (figure 2C). Twenty-four hours after treatment, AG-881 yielded higher tumor exposure and more robust 2-HG suppression compared with AG-120. Due to enhanced brain penetration and target engagement, subsequent experiments used AG-881.

Inhibition of IDH1R132H suppresses 2-HG production and tumor growth in vivo. (A) Inhibition of IDH1R132H activity by AG-120 and AG-881 in vitro. Levels of 2-HG in the supernatant of dTG-IDH1R132H cells cultured in different concentrations of AG-881 (green) or AG-120 (purple) were examined by LC-MS/MS to determine the IC50 values for each inhibitor’s activity. (B) Levels of AG-881 (black) and AG-120 (pink) detected in the brain of dTG mice bearing dTG-IDH1R132H tumors. Beginning 7 days after the tumor inoculation, animals received oral administrations of vehicle or the corresponding inhibitor for 4 days. Samples were collected 1, 4, 12, and 24 hours after final drug administration and analyzed for the concentration of AG-881 and AG-120. (C) Both AG-881 and AG-120 suppressed intratumoral levels of 2-HG. Pharmacodynamics of AG-881 (black) and AG-120 (pink) in vivo were determined by analyzing the levels of 2-HG in samples from (B). (D,E) Tumor growth was significantly delayed by AG-881 treatment. Tumor-bearing mice were randomized to receive daily p.o. administration of AG-881 (n=5) or vehicle (n=4) beginning on day 7 post-tumor inoculation. (D) The tumor size, determined by BLI, and (E) weight of each mouse were measured every 3–4 days after treatment initiation and ended when control mice reached the protocol-specified endpoint due to tumor burden. BLI, bioluminescent imaging; dTG, double transgenic; HG, hydroxyglutarate.
Having established an effective in vivo dose level of AG-881, we sought to determine the effect of IDH1R132H inhibition on tumor growth. We randomized dTG-IDH1R132H tumor-bearing mice 7 days post-tumor inoculation (dpi) to receive either vehicle control or AG-881 daily for 14 days. Since dTG-IDH1R132H cells encoded luciferase, we monitored the tumor size via BLI every 3–4 days (figure 2D). Inhibition of IDH1R132H in vivo resulted in significantly smaller tumors at days 17 and 20. In addition, AG-881-treated mice displayed fewer visible signs of disease progression (pain, neurological symptoms) and had lost significantly less weight than vehicle-treated ones (figure 2E) at both day 17 (p=0.04) and day 20 (p=0.03).
Inhibition of 2-HG production in vivo does not alter the tumor-infiltrating myeloid cell population, but results in increased recruitment of CD4+ T-cells
Since the composition of glioma-infiltrating leukocytes has been shown to affect clinical outcomes of patients with IDH1 mutant gliomas,18 we sought to profile the effects of IDH1R132H inhibition on the immune composition of dTG-IDH1R132H tumors in vivo. In dTG mice treated with AG-881 for 14 days as described in figure 2D, we confirmed that the AG-881 treatment regimen significantly reduced 2-HG levels in the tumor (figure 3A). Furthermore, as 2-HG can be secreted by tumor cells and myeloid cells constitute the large majority (~90%) of tumor-infiltrating immune cells (figure 3B), we measured 2-HG levels in CD11b+ cells isolated from AG-881-treated and vehicle-treated tumors. The 2-HG levels in control CD11b+ cells were nearly as high as those measured in the whole tumor samples and were significantly reduced in inhibitor-treated mice (figure 3C). However, the treatment did not affect the recruitment of CD11b+ cells to the TME (figure 3B). In-depth FC analyses of tumor myeloid cells (within the CD45+ compartment) revealed no significant changes in cells expressing activated cell-surface markers (“proinflammatory CD11b+ cells”, online supplemental figure 2A), abundance of tumor-associated macrophages (online supplemental figure 2B), monocytic myeloid-derived suppressor cells (M-MDSC, online supplemental figure 2C), polymorphonuclear MDSC (PMN-MDSC, online supplemental figure 2D), or brain resident microglia cells (online supplemental figure 2E). To evaluate whether reduction of 2-HG affected the immunosuppressive properties of tumor-derived myeloid cells, we tested the ability of CD11b+ cells from vehicle-treated and AG-881-treated tumors to suppress T-cell proliferation ex vivo. We cocultured CD3/CD28 activated T-cells from naïve dTG mice with tumor-derived CD11b+ cells at different ratios and measured T-cell proliferation via CFSE dilution (figure 3D). CD11b+ cells inhibited both CD8+ and CD4+ T-cell proliferation largely in a dose-dependent manner, but we observed no differences in the suppressive phenotype of myeloid cells derived from inhibitor-treated vs control-treated animals (black vs dark gray bars, figure 3D). While we did not detect changes in the phenotype or immunosuppressive function of myeloid cells in dTG tumors following 2-HG reduction, importantly, we observed recruitment of T-lymphocytes into tumors harvested from AG-881-treated animals (p=0.018 vs vehicle-treated), and those T-cells were predominantly CD4+ (p=0.006, figure 3E). The proportion of CD8+ T-cells was not significantly altered (p=0.25).
Supplemental material
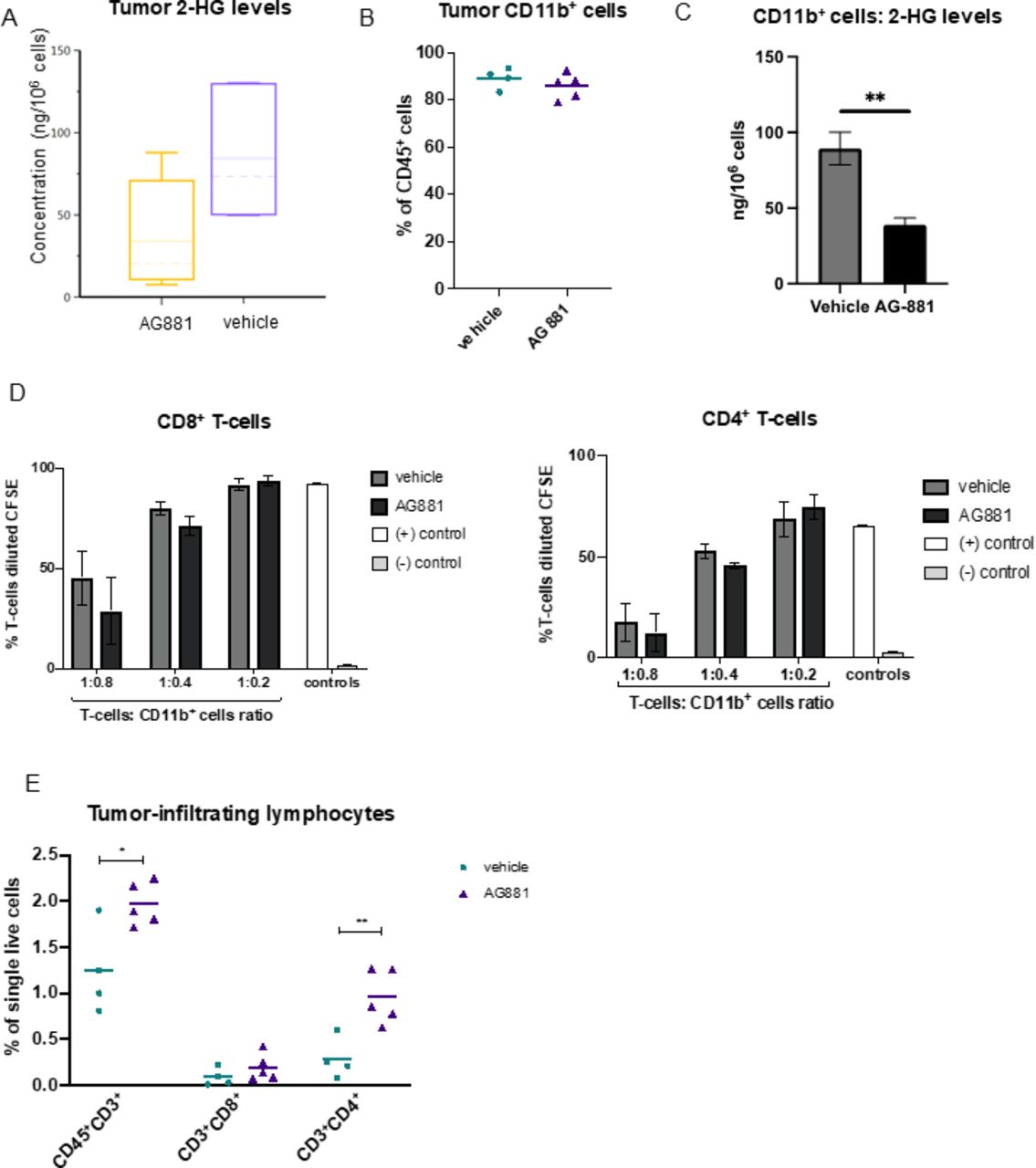
Inhibition of 2-HG production by AG-881 does not alter the tumor-infiltrating myeloid cell population but results in increased recruitment of CD4+ T-cells. (A) Mice bearing dTG-IDH1R132H tumors were treated daily with 10 mg/kg AG-881 or vehicle. Levels of 2-HG in the tumor-bearing hemisphere after 14 days of treatment were measured by LC-MS/MS. (B–D) A separate cohort of mice were treated as described in (A), and single-cell suspensions of the tumors were either analyzed by FC to determine the frequency of CD11b+ (B) cells among tumor-infiltrating leukocytes or used to isolate CD11b+ cells using a column separation protocol (C,D). 2-HG levels were significantly reduced in CD11b+ cells from AG-811-treated mice (C); however, their ability to suppress CD8+ ((D), left panel) and CD4+ ((D), right panel) T-cell proliferation ex vivo was unchanged. T-cell proliferation as a result of polyclonal CD3/CD28 activation was measured by dilution of CFSE 72 hours post-coculture initiation. (E) Single-cell suspensions generated in (B) were used to examine the frequency for CD8+ and CD4+ T-cells by FC. In 2-HG quantification and T-cell suppression studies n=3 mice per group. For FC analyses, n=4 in control group and n=5 in AG-881 groups. dTG, double transgenic; FC, flow cytometry; HG, hydroxyglutarate.
Enhanced survival, resulting from IDH1R132H inhibition, depends on activity of both CD4+ and CD8+ T-cells
To determine whether the antitumor response of IDH1R132H inhibition (figure 2D) was dependent on T-cell activity, we compared the survival of tumor-bearing mice treated with AG-881 in the absence of CD8+ or CD4+ cells. Three days after tumor cell inoculation, mice were randomized to receive a regimen of depleting antibodies targeting CD8 or CD4-expressing cells or an isotype control, in addition to receiving daily AG-881 treatment (figure 4A). Four days after the animals received a second round of antibodies, we examined samples of peripheral blood for the abundance of CD8+ or CD4+ T-cells. Even several days after administration of the depleting antibodies, levels of CD8+/CD4+ T-cells remained efficiently diminished in the circulation compared with levels in the isotype-treated group (online supplemental figure 3). As we observed in previous experiments, treatment of tumor-bearing mice with AG-881 alone significantly improved overall survival (figure 4B (MS=28 days, p=0.008 vs vehicle)). Depletion of either T-cell lineage, however, was equally sufficient to reverse the survival benefit conferred by IDH1R132H inhibition (figure 4B). These data indicate that even though we only detected changes in the CD4+ T-cell compartment of the TME following IDH1R132H inhibition (figure 3E), both T-cell types contributed significantly to the antitumor response in our model.
Supplemental material
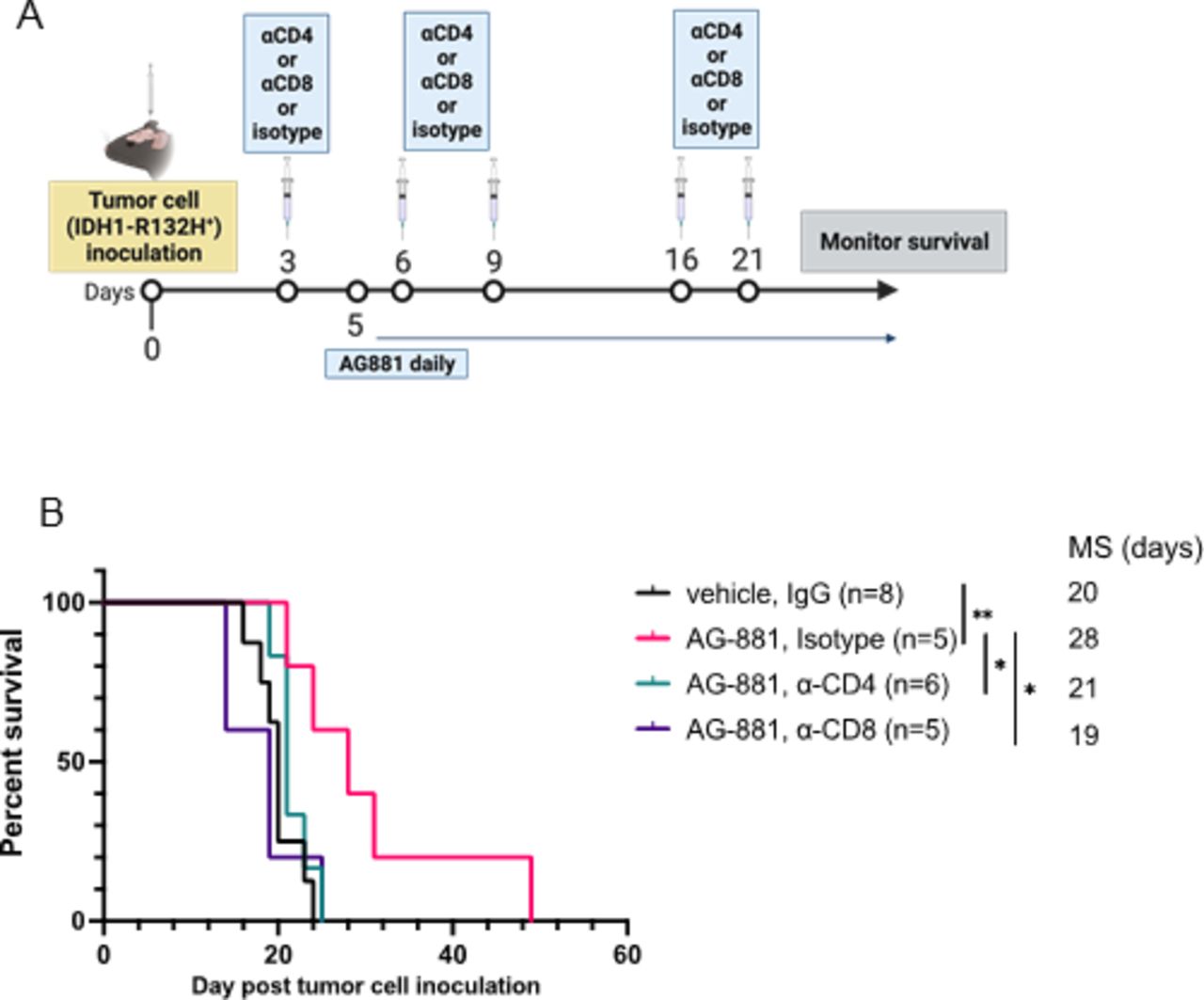
Enhanced survival outcomes, resulting from IDH1R132H inhibition, depend on activity of both CD4+ and CD8+ T-cells. (A) Schematic description of the T-cell depletion treatment regimen. (B) Kaplan-Meier survival curve. *P<0.05, **p<0.002; log-rank test.
Prophylactic IDH1R132H peptide vaccination extends survival only in combination with class I antigen vaccine and IDH1R132H inhibition
Previous studies using dTG mice demonstrated the ability of an IDH1R132H peptide vaccine to generate an effective immune response against subcutaneously implanted IDH1R132H-expressing sarcomas.14 To evaluate the efficacy of the peptide vaccine against our intracerebral tumor model, we first needed to confirm that the regimen induced a systemic anti-IDH1R132H response. We vaccinated 6-week-old dTG mice with two 100 µg doses of the synthetic IDH1R132H (p123-142) peptide (from here on referred to as R132Hp), emulsified in IFA, with poly-ICLC as an adjuvant. Ten days after the second dose, we harvested splenocytes and isolated T-cells. Ex vivo, we cocultured naïve syngeneic CD11b+ splenocytes pulsed with either wildtype IDH1 (p123-142) peptide or R132Hp together with T-cells from vaccinated mice for 72 hours. To assess T-cell activation, we measured the levels of IFNγ in the supernatant by ELISA (figure 5A). When CD11b+ cells were not pulsed with either peptide (media only control), we detected very low or no IFNγ. T-cells from all three tested mice produced significantly higher levels of IFNγ when stimulated with R132Hp-pulsed splenocytes than control wildtype-pulsed ones (figure 5A, green vs pink bars). In one of the three mice, we observed a significant increase of IFNγ production in the presence of the wildtype peptide compared with media only (Mouse 3), suggesting potential cross-reactivity of T-cells to wildtype IDH1.
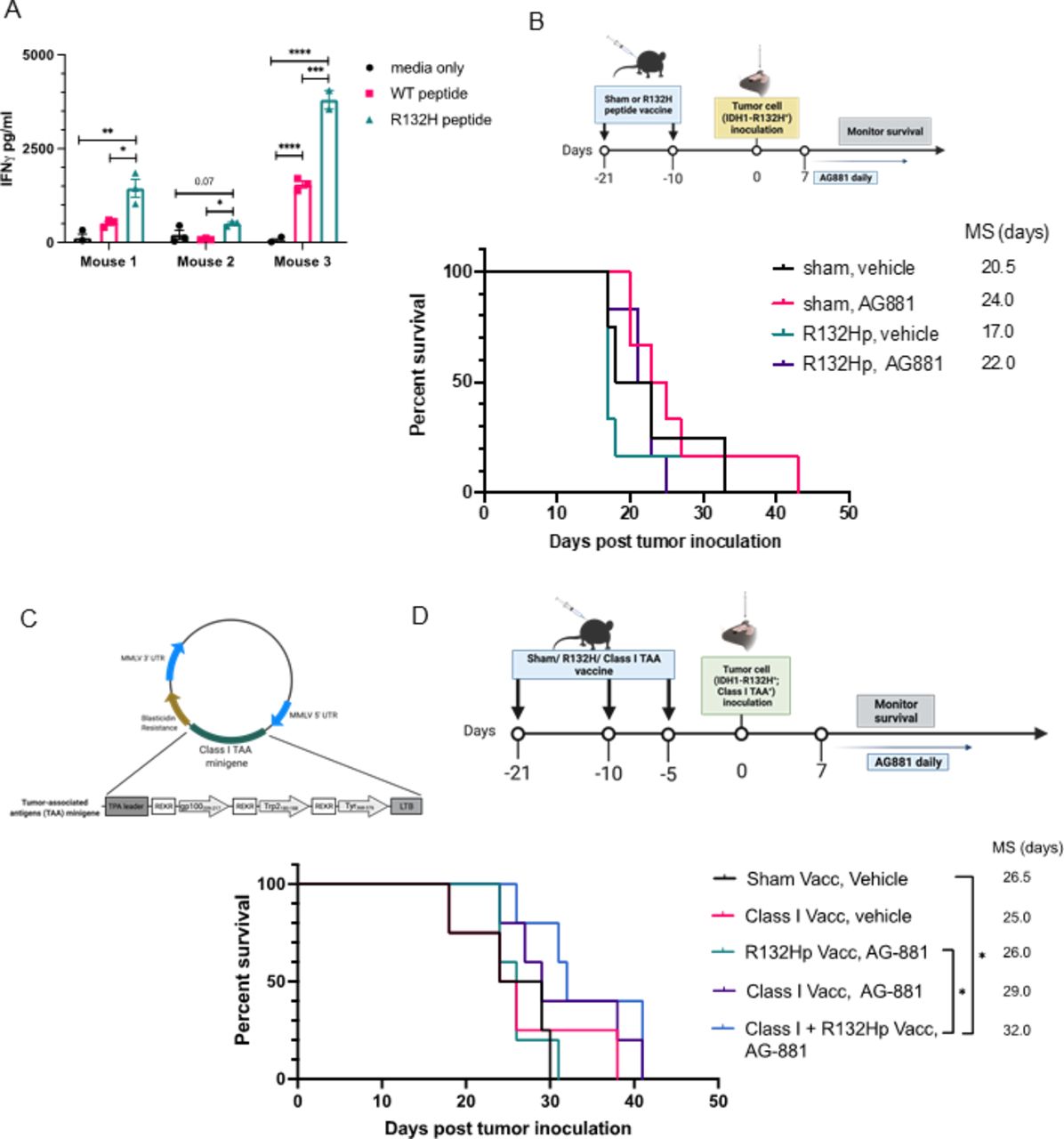
Prophylactic IDH1R132H peptide vaccine extends survival only when combined with class I peptide vaccine and IDH1R132H inhibition. (A) 9-week-old dTG mice received s.c. vaccinations with 100 µg of R132Hp emulsified in IFA and 20 µg intramuscular (i.m.) poly-ICLC on days 0 and 11. On day 21, mice were euthanized, and their spleens were harvested. IFNγ secretion by splenocytes stimulated ex vivo for 72 hours with peptide-pulsed syngeneic CD11b+ cells as indicated. Cells from three individual mice were plated in technical triplicates. Media only, no CD11b+ cells negative control. (B) Schematic representation of the treatment protocol for prophylactic vaccinations. 9–10 week-old dTG mice received s.c. vaccinations as described in (A) followed by an intracerebral challenge with dTG-IDH1R132H cells. After 7 days, tumor-bearing mice were randomized to receive daily oral administrations of 10 mg/kg AG-881 or vehicle control for up to 35 consecutive days. Kaplan-Meier curves demonstrate survival outcomes: Sham, Vehicle (n=4); Sham, AG-881 (n=6); R132Hp, Vehicle (n=6), R132Hp, AG-881 (n=6). (C) Diagram representing the retroviral vector used to transduce the dTG-IDH1R132H cells with HLA class I TAA epitopes derived from gp100, tyrosinase-related protein 2 (Trp2), and tyrosinase (Tyr). (D) Schematic representation of the treatment protocol. 7–10-week-old dTG mice received s.c. vaccinations with the class II R132Hp, peptides gp100209-217, Trp2180-188, Tyr368-376 (class I vacc), or PBS (Sham Vacc) emulsified in IFA and 20 µg i.m. poly-ICLC. Vaccines were boosted 10 and 15 days later. And 21 days after initial vaccine administration, all mice received an intracerebral challenge with the dTG-IDH1R132H; Class I TAA+ cells tumor cells (described in (C)). Seven days later, tumor-bearing mice started receiving daily oral administration of 10 mg/kg AG-881 or vehicle control for up to 35 days. Kaplan-Meier curves demonstrate survival outcomes: Sham, Vehicle (n=4); Class I Vacc, Vehicle (n=4); R132Hp, AG-−881 (n=5), Class I Vacc, AG881 (n=5), and Class I+R132 Hp, AG-881 (n=5). *P<0.05; long-rank test. dTG, double transgenic; HLA, human leukocyte antigens; IFA, incomplete Freund adjuvant; TAA, tumor-associated antigen.
Given our earlier data showing the importance of T-cells in the IDH1R132H inhibitor-mediated immune response (figure 4B), we asked whether prophylactic vaccination of dTG mice with the HLA-DR-presented R132Hp peptide could improve the survival of tumor-bearing mice (figure 5B). Surprisingly, the median survival of R132H-vaccinated animals (MS=17 days) was not different from that of sham vaccinated ones (MS=20.5 days, p=0.4). Treatment with AG-881 alone prolonged survival (MS=24 days), although the difference did not reach statistical significance due to the small cohort size. Combining R132Hp-vaccination with the IDH1R132H inhibitor resulted in MS of 22 days (vs MS=17 days in the vaccination-only cohort), which was comparable to the survival of the group treated only with AG-881, suggesting that there was no added benefit of the R132Hp vaccination.
Previous studies by our group demonstrated that vaccine therapy using MHC-class I glioma-associated peptides significantly improved the efficacy of IDH1R132H inhibition in a syngeneic C57BL/6 glioma model.9 Here, we sought to determine the contribution of the class I-based immune response to the enhanced survival provided by IDH1R132H inhibition in our new dTG model. For this purpose, we generated a retroviral plasmid encoding three well-known HLA-A2-restricted tumor-associated antigens (TAAs)—gp100 (p209-217), TRP2 (p180-188), and TYR (p368-376).19 20 We transduced the dTG-IDH1R132H cell line with the vector and used the resulting dTG-IDH1R132H-TAA+ cells in the following experiment (figure 5C). We treated mice with prophylactic vaccines of either IFA-emulsified solvent control (sham vacc, n=4), R132Hp alone (n=5), class I TAAs alone (n=9), or combination of all four peptides (class I+R132 Hp vacc; n=5). All groups received orthotopic injections of the IDH1R132H-TAA+ tumor cells 5 days after the last vaccine dose. Seven days post-tumor inoculation, mice were randomized to receive daily gavage of either vehicle or AG-881 for up to 35 days and monitored for survival (figure 5D). Vaccination with class I TAAs alone did not affect median survival (MS=25). Although the addition of AG-881 to the class I vaccine resulted in a trend toward longer survival (MS=29 days), the difference from the control cohort was not significant (vs sham vacc, veh MS=26.5 days, p=0.26). As we observed previously, vaccination with R132Hp in combination with AG-881 resulted in median survival equal to that of the control group (MS=26). However, when mice were vaccinated with all four peptides and subsequently treated with AG-881, survival was significantly prolonged (MS=32 days vs control p=0.02). Using tumor samples collected when each animal reached the endpoint, we attempted to test the ability of tumor-infiltrating lymphocytes (TILs) to produce IFNγ ex vivo in response to the antigens used in our prophylactic vaccines. We obtained preliminary ELISpot data showing enriched antigen-specific reactivity to R132Hp in the group vaccinated with all four peptides (online supplemental figure 4A–C). Unfortunately, due to limited sample availability, we were not able to statistically confirm these observations. Nevertheless, these data suggest that, in the setting of IDH1R132H inhibition, immune activation involving both class I and class II epitopes may be required for the activation of an antitumor T-cell response that is sufficient to prolong the survival. In addition, we attempted to address whether R132H-specific CD4+ T-cells could recognize R132Hp which is endogenously presented by the dTG-IDH1R132H cell line; however, this proved technically challenging. Nonetheless, evaluation of HLA-DR expression by FC demonstrated that class II-expressing tumor cells constitute less than 30% of all live cells within the TME (online supplemental figures 4D and 5), suggesting that tumor cells may not be effectively presenting the IDH1R132H-derived peptide themselves.
Supplemental material
Supplemental material
Inhibition of IDH1R132H function in vivo results in an enhanced IFNɣ signature response and significantly extends survival when combined with PD-1 blockade
To further understand the global immune response in dTG tumor-bearing mice treated with AG-881, we examined the changes in gene expression in response to IDH1R132H inhibition. We isolated RNA from vehicle-treated or AG-881-treated tumors (n=3 per group) and analyzed gene expression using the nanoString nCounter platform with the Mouse Immunology v2 panel. Differential expression analysis revealed that interferon response factor 1 (Irf1), as well as Stat1, Cxcl9, Cxcl11, Socs1, and Pd-l1, were significantly upregulated in the AG-881-treated group compared with the control (figure 6A). In addition, many of the antigen presentation machinery-related genes, such as Cd74, Tap1, Psmb9, and Ciita, were also found to be upregulated by the AG881-treatment. Gene set enrichment analysis and KEGG pathway analysis both confirmed that IFNγ response, antigen-processing and presentation machinery, along with multiple proinflammatory signatures, were significantly enriched after IDH1 R132H inhibition compared with the control (figure 6B).21 As Pd-l1 is known to be upregulated in response to IFNγ signaling and PD-L1+ myeloid populations are enriched in gliomas,22 we used FC to evaluate the in vivo protein expression of HLA-DR and PD-L1 on the surface of immune cells in the TME and observed that both molecules were significantly upregulated in response to AG-881 treatment (figure 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of IDH1R132H function in vivo results in an enhanced IFNɣ signature response and therapeutic efficacy of PD-1 blockade. (A) Mice bearing dTG-IDH1R132H tumors were treated with vehicle (n=3) or 10 mg/kg AG-881 (n=3) for 14 days. mRNA from tumor samples was collected and analyzed using the Mouse Immunology v2 panel (Nanostring) for significant changes in gene expression resulting from IDH1R132H inhibition. (B) The top 5 (out of 136 gene sets) significantly upregulated pathways following AG-881 treatment. (C) CD45+ single-cell suspensions derived from the dTG-IDH1R132H tumors of mice treated as in (A) were analyzed by FC for the expression of HLA-DR and PD-L1. *P<0.05, **p<0.002; Student t-test. (D) Schematic representation of the combined AG-881 and PD-1 blockade treatment protocol and Kaplan-Meier survival curves. Vehicle control and isotype antibody (Veh, IgG; n=8), vehicle and PD-1 blocking antibody (Veh, α-PD-1, n=9), AG-881 and isotype antibody (AG-881, IgG; n=9), AG-881 and PD1 blocking antibody (AG-881, α-PD-1, n=8). *p<0.05; long-rank test. dTG, double transgenic; HLA, human leukocyte antigens; NES, normalized enrichment score; padj, adjusted p values.
Based on these data, we tested whether the antitumor effect of IDH1R132H inhibition could be improved by blocking the PD1/PD-L1 interaction. Five days after inoculation with dTG-IDH1R132H tumors, mice were randomized to receive vehicle and IgG control antibody, AG-881 treatment alone, αPD-1 blocking antibody, or a combination of the inhibitor and the blocking antibody (figure 6D). The survival analysis revealed no difference in the MS of control-treated mice (n=8) and those that received immune checkpoint inhibition alone (n=9; MS=20 vs 19 days). AG-881 therapy alone also did not significantly extend the median survival in this cohort (n=8; MS=22 days, p=0.2 vs vehicle), likely due to a cluster of early deaths in the treated group. Finally, treatment with both AG-881 and α-PD1 antibody resulted in enhanced survival (n=8; MS=24.5 days, p=0.01 vs vehicle), suggesting that AG-881-mediated upregulation of IFNγ molecules and immune checkpoint signals was necessary to observe a therapeutic benefit of PD-1 blockade.
Discussion
Multiple recent studies of both human gliomas and mouse preclinical models have demonstrated major differences in the immune composition and response to immunotherapies in mutant IDH tumors compared with non-mutated gliomas.8 10 11 These findings clearly highlight the need for a more in-depth investigation of how IDH1/2 mutations impact the glioma immune environment using the most clinically useful models. Using the dTG mouse strain, we aimed to develop a clinically relevant glioma model that would allow us to examine the effects of IDH1R132H inhibition and presentation of the R132Hp epitope by HLA-DR. As multiple clinical trials are evaluating the therapeutic efficacy of R132Hp vaccination in patients with glioma (reviewed in Platten, Bunse, and Wick, 2021),23 our mouse model can be a useful tool to extend these efforts in a translational bedside-to-bench approach.
Although most neoantigen-targeting immunotherapy clinical trials are focused on class I epitopes, the majority of tumor-specific immunogenic peptides may be presented by class II MHC and recognized by CD4+ T-cells.24 Furthermore, neoantigen-specific CD4+ T-cells can play critical roles in providing proper priming and costimulation of cognate CD8+ cytotoxic T-cells.25 Platten et al demonstrated the feasibility of eliciting a class II IDH1R132H-specific peptide vaccine response and its potential to influence disease progression in a first-in-human phase I trial published earlier in 2021.26 Therefore, it was not entirely surprising that depletion of either CD4 or CD8+ T-cell subset in our model was sufficient to reverse the therapeutic effect of IDH1R132H inhibition (figure 4B). Further in-depth studies are needed, however, to delineate the reasons why we observed no therapeutic benefit of the class II neoantigen-specific peptide vaccine (figure 5B).
Although this model enabled us to learn about how the R132H mutation in IDH1 impacts the glioma microenvironment, our experiments evaluating AG-881 in combination with either the peptide vaccines or PD-1 blockade did not result in the long-term survival of mice. While this raises a concern in our future clinical developments of these approaches, some critical differences with human IDH mutant glioma remain (eg, our model lacks a mutation in ATRX that is predominantly found in human astrocytoma). As such, direct extrapolation of the observed survival effect to clinical settings should be done cautiously as the drivers of clinical outcomes may differ. Nonetheless, in our preclinical studies which used a limited number of tumor-bearing mice (as this model is not commercially available and breeding a high number of experimental animals was not always feasible), we observed statistical differences in the median survival of mice treated with AG-881/anti-PD1 combination (p<0.05). These data are in line with our findings that AG-881 therapy increases IFNγ signaling and PD-L1 expression, which together with increased TIL infiltration could prime the activity of anti-PD1 therapeutics. Together, our results may provide some rationale for further evaluation of the proposed mechanism of action for AG-881 in combination with anti-PD1 for the treatment of patients with mutant IDH1 glioma.
Myeloid cells within the glioma TME have been shown to exist on a continuum of activation states depending on their location within the tumor and also the stage and mutational landscape of the tumor.27 Recent work by Friedrich et al demonstrated that glioma-associated myeloid (GAM) and lymphoid cells derived from IDH-mutant tumors can uptake R-2-HG and display multiple immunosuppressive features which were alleviated by suppressed production of R-2HG or inhibition of its downstream metabolic signaling.10 11 However, most analyses of human IDH1 mutant GAM are performed using samples from patients who have been previously treated with standard-of-care (SOC) therapy with radiation and temozolomide-based chemotherapy, which likely affects the cells’ phenotype. A study by Poon et al compared the inflammatory states of GAMs in IDH1-mutant and IDH-wildtype samples from previously untreated patients with glioma and showed that GAMs within IDH-mutant tumors displayed more proinflammatory phenotypes and this was associated with longer overall patient survival.28 These seemingly contradictory findings highlight the importance of carefully considering the differences among the samples being evaluated when comparing study outcomes. In our current study, we were not able to detect changes in myeloid cell phenotypes following AG-881 treatment. However, we observed significant upregulation of IFNγ response-related genes and antigen-processing and presentation machinery (figure 6A–C) suggesting that IDH1R132H inhibition led to a global shift towards a more proinflammatory immune responses within the TME. One of the mutant IDH1/2 inhibitors tested in our studies (AG-120) was recently also used by Wu and colleagues to examine the effects of mutant IDH1 inhibition in a preclinical model of cholangiocarcinoma.29 Notably, changes in the tumor immune microenvironment following AG-120 treatment described by the authors included induction of IFNγ response signature and the need for an intact immune system, particularly effector cytotoxic CD8+ Tcells.29 These findings are especially clinically relevant since the recently reported early data from the phase I trial of ivosidenib and vorasidenib in patients with IDH1 mutant glioma demonstrated that optimal suppression of 2-HG was associated with increased tumor infiltration by CD3+ cells and higher expression levels of type I IFN and antigen-presentation genes.30 31 These combined preclinical and clinical data suggest that upregulation of IFNγ transcriptional signatures is a common mechanism of action which precedes T-cell antitumor immune responses in solid tumors treated with mutant IDH1 inhibitors.
Our observation that pharmacologic inhibition of IDH1R132H leads to a more productive endogenous antitumor response was supported by another recently published study by Kadiyala et al.32 In a C57BL/6 murine model of astrocytoma, the authors showed that inhibition of R-2HG accumulation in combination with SOC therapy with radiation and temozolomide-based chemotherapy led to an increase in dendritic cell and anti-tumor specific T-cell trafficking to the TME coupled with a decrease in the accumulation of MDSCs, regulatory T-cells, and immunosuppressive macrophages.32 In addition, similarly to our findings, IDH1R132H inhibition resulted in higher levels of PD-L1 expression, prompting the authors to determine the effects of combined IDH1R132H inhibition, SOC, and anti-PD-L1 checkpoint blockade.32 They further established that long-term survivors of the combination treatment were resistant to tumor cell rechallenge in the contralateral hemisphere, strongly suggesting the development of anti-glioma immune memory.32 We were not able to test the memory as we did not observe long-term survivors when we combined AG-881 and checkpoint inhibition in our model (figure 6D). Multiple factors likely contributed to the different outcomes, such as the different models used (both in terms of mouse strains and also tumor cell mutational landscape) and the absence of SOC treatment in our study. Interestingly, Wu et al also demonstrated that, in cholangiocarcinomas, treatment of tumor-bearing animals with a checkpoint inhibitor (α-CTLA4) synergized with inhibition of IDH1R132C by AG-120 and lead to durable responses which were not observed in either treatment alone.29 Despite their differences, mounting evidence by all three studies indicate the need for combining immune checkpoint and IDH1R132H inhibition in order to more fully exploit the benefits of immune reprogramming stemming from suppressed R-2HG accumulation.
Viewed together, a pattern emerges from the findings of these several studies. Specifically, the data indicate that inhibiting mutant IDH1 in gliomas creates an enhanced immunostimulatory TME which could provide a critical therapeutic window for immunotherapies that engage both class I and II responses and alleviate immunosuppression via immune checkpoint inhibition. Our new HLA-A2/HLA-DR-syngeneic mouse model presents a unique opportunity to study these clinically relevant approaches in-depth with the goal of ultimately prolonging glioma patient survival.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
All members of the Okada laboratory; UCSF Laboratory for Cell Analysis Core Facilities: Flow Cytometry Core, Genome Analysis Core, and Preclinical Therapeutics Core. All experimental diagrams were created with BioRender.com.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors HO conceptualized the study. PC, AY, TC, MM, ALM, TN, KMD, and DD conducted the mouse-based experiments and generated the data. RN, SR, CH, AET, MLH, ML, and BN provided AG-881 and contributed to the design of mouse experiments using AG-881. These members also contributed to the PK assays of AG-881. All authors contributed to the writing and reviewing of the manuscript.
Funding Loglio Foundation from Dabbiere Family, NIH/NINDS R35 NS105068, NIH/NCI R01CA222965.
Competing interests CH, RN, SR, AET, MLH, and BN are employees of Agios (currently) Servier Pharmaceuticals, Boston, MA.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.