Article Text

Abstract
Background Oncolytic virotherapy (OV) represents a method to treat a variety of solid tumors by inducing antitumor immune responses. While this therapy has been extremely efficacious in preclinical models, translating these successes into human patients has proven challenging. One of the major reasons for these failures is the existence of immune-regulatory mechanisms, which dampen the efficacy of virally induced antitumor immunity. Unfortunately, the full extent of these immune-regulatory pathways remains unclear.
Methods To address this issue, we generated a doubly recombinant, oncolytic myxoma virus which expresses both a soluble fragment of programmed cell death protein 1 (PD1) and an interleukin 12 (IL-12) fusion protein (vPD1/IL-12 (virus-expressing PD1 and IL-12)). We then tested the molecular impact and therapeutic efficacy of this construct in multiple models of disseminated disease to identify novel pathways, which are associated with poor therapeutic outcomes.
Results Our results demonstrate that vPD1/IL-12 causes robust inflammation during therapy including inducing high levels of tumor necrosis factor (TNF). Surprisingly, although expression of TNF has generally been assumed to be beneficial to OV, the presence of this TNF appears to inhibit therapeutic efficacy by reducing intratumoral T-cell viability. Likely because of this, disruption of the TNF pathway, either through genetic knockout or antibody-based blockade, significantly enhances the overall outcomes of vPD1/IL-12-based therapy that allows for the generation of complete cures in normally non-responsive models.
Conclusions These data suggest that some aspects of OV-induced inflammation might represent a double-edged sword during therapy and that specific blockade of TNF might enhance the efficacy of these treatments.
- immunotherapy
- oncolytic virotherapy
- oncolytic viruses
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
One of the major goals of oncolytic therapy is the induction of high levels of inflammation within the tumor microenvironment. While this inflammation is known to assist in the generation of antitumor adaptive immunity, its full impact on the function of this immunity remains unclear.
What this study adds
Here we show that high levels of tumor necrosis factor induced during oncolytic therapy can directly inhibit the efficacy of treatment by causing the loss of intratumoral T-cell viability. These data suggest that the role of the inflammatory responses induced by oncolytic viruses might be complex and play both positive and negative roles during treatment and could eventually improve the clincal application of oncolytic therapy.
Introduction
Immune checkpoint blockade (ICB) is revolutionizing the way in which we view cancer treatment.1 2 Unfortunately, while the clinical outcomes of these therapies are impressive in some patients, the overall response rates to single agent treatment remain relatively low.3 One approach to improve the efficacy of ICB is through the use of tumor-localized, immune-enhancing treatments such as oncolytic virotherapy (OV).4 These treatments generate high levels of localized inflammation with the goal of remodeling the overall tumor microenvironment (TME) to be more supportive of antitumor immunity.5 Numerous oncolytic agents have been used for this approach in both preclinical models and human patients.6 7 Interestingly, the inflammatory responses induced are often quite similar, frequently being highlighted by the induction of classical antiviral cytokines such as interferons (IFNs) and tumor necrosis factor (TNF).8 However, despite an extensive body of oncolytic literature and the similarities of the overall responses, the functional impact of these cytokines remains somewhat unclear. In particular, while the inflammatory responses seen during OV are typically viewed as beneficial, they are also known to have detrimental effects in other contexts. For example, the induction of TNF during OV is generally viewed as a positive event due to TNF’s well-established role as an inflammatory mediator as well as its ability to trigger apoptosis in cancerous cells.9 However, TNF has also been shown to be immune suppressive during bacterial infections,10 graft-versus-host disease,11 and contact hypersensitivity,12 and its presence can actually aid in cancer progression by promoting cell proliferation and angiogenesis.13 It is therefore possible that certain aspects of the localized inflammation induced by OV might represent a two-edged sword producing both positive and negative effects during treatment.
Here we show that a novel recombinant myxoma virus (MYXV) expressing both a soluble programmed cell death protein 1 (PD1) inhibitor as well as an interleukin 12 (IL-12) fusion protein (vPD1/IL-12 (virus expressing PD1 and IL-12)) causes potent inflammation during treatment including robust induction of TNF. In contrast to being beneficial, however, the presence of TNF appears to blunt the efficacy of viral treatment by directly killing intratumoral T cells. Consistent with this hypothesis, antibody-based blockade of TNF during vPD1/IL-12 treatment increases the viability of intratumoral T cells enhancing overall therapeutic efficacy including allowing for complete disease eradication of normally non-responsive tumors. These results suggest that certain aspects of the inflammation induced during OV can make tumors less supportive of immunotherapy and that blockade of these factors can enhance overall efficacy.
Materials and methods
Cell lines and reagents
BSC40 (catalog no. BRL-2761) and B16/F10 (catalog no. CRL06475) cells were purchased from the American Type Culture Collection (Manassas, Virginia, USA). LLC-A9F1 cells (a previously described derivative of parental Lewis lung carcinoma (LLC) cells which express enhanced levels of murine H2-Kb)14 were a kind gift from Dr Mark Rubinstein at the Medical University of South Carolina. MC38B cells were obtained from Dr Aaron Ring at Yale University. BR5 cells were obtained from Dr Rita Serda at the University of New Mexico Health Sciences Center. All cell lines were cultured in DMEM+10% fetal bovine serum+1× penicillin-streptomycin-L-glutamine (Mediatech, Manassas, Virginia, USA) and passaged for less than 6 months prior to use. The following blocking or depleting antibodies were used in these studies: anti-TNF (clone XT3.11), anti-PD1 (clone RMP1-14), anti-CD4 (clone GK1.5), anti-CD8 (clone 53–6.7), anti-IFN-γ (clone XMG1.2) and were obtained from BioXcell (Lebanon, New Hampshire, USA).
Viruses and infections
All viral constructs are based on the Lausanne strain of MYXV which has been previously studied as an oncolytic agent.15 16 Doubly recombinant viral constructs were based on a previously described recombinant MYXV expressing the soluble ectodomain (aa 1–168) of PD1 (termed vPD1).17 These doubly recombinant constructs were created using a novel poxviral recombination plasmid made from the pBluescript plasmid backbone and separate regions homologous to the M152R and M154R viral open reading frames, which flanked a Tomato Red (TdTR) cassette driven by the consensus poxviral synthesis early/late promoter. Individual transgenes were subcloned into this vector and their expression driven by a second consensus poxviral synthesis early/late promoter. Secondary transgenes included in this study corresponded to the murine versions of: the soluble ectodomains of six known T-cell checkpoint proteins, including B and T lymphocyte A-associated (BTLA) aa 1–184, T-cell immunoreceptor with Ig and ITIM domains (TIGIT) aa 1–139, V-domain Ig suppressor of T-cell activation (VISTA) aa 1–189, cytotoxic T-lymphocyte-associated protein 4 (CTLA4) aa 1–162, T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3) aa 1–195, and lymphocyte activating 3 (LAG3) aa 1–441, or four known proinflammatory cytokines including—IL-2 (encoded as a previously described high-activity superkine,18 IL-12 (encoded as a p40-GGGS4-p35 fusion protein), IL-15 (encoded as an IL15-GGS5-IL15R fusion protein), or IL-18. Each construct was generated through homologous recombination of vPD1 and the corresponding poxviral recombination plasmid. Viruses were then clonally isolated through four or more rounds of sequential plaque purification for GFP+/TdTR+ foci, amplified in BSC40 cells, and purified using gradient centrifugation as previously described.19 vIL-12 was generated by combining the IL-12 recombination plasmid discussed above with virus expressing green fluorescent protein (vGFP). Viral titer was determined through serial dilution and infection of BSC40 cells.
Mouse models
All mice used in these studies were between 6 and 8 weeks of age. For the B16/F10, MC38 and LLC models, 1×106 cells from each cell line were injected subcutaneously into the flank(s) of syngeneic C57/Bl6 mice. Treatment was initiated when both tumors reached ~25 mm2. While some experiment-to-experiment variation was observed, this was typically around days 7–9 for B16/F10 and MC38 tumors and around days 14–20 for LLC tumors. Once tumors had reached 25 mm2, mice were randomly binned into the required groups and virally treated as indicated (contralaterally injected mice which displayed growth of only a single tumor were removed from the study prior to binning). Viral treatment typically consisted of three injections (given on days 0, +2, and +4). Each injection consisted of 1×107 focui forming units (FFU) of the indicated virus in 50 μL of sterile PBS and was delivered intratumorally into the larger of the two established tumors. Tumor area was then measured either every 2 days (for B16/F10 and MC38 tumors) or twice weekly (for LLC tumors) using digital calipers and is presented as tumor area (mm2) determined using the formula (area=length×width). For survival studies, animals were euthanized when the total area of their tumors combined to exceed 400 mm2. For the BR5 ovarian cancer model, 1×106 BR5-luc cells were injected into the intraperitoneal cavity of syngeneic FVB mice in 50 μL of sterile PBS. After 10 days, mice were injected with 200 μL luciferin and analyzed for apparent tumor burden by bioluminescence imaging. Imaged mice were then binned into their designated groups and either mock treated or treated with 1×107 FFU of the indicated virus in 50 μL of sterile PBS injected directly into the intraperitoneal cavity every 2 days for three treatments (given on days 0, +2, and +4). Bioluminescence analysis was performed every 7 days and the torso width and weight of each mouse were measured every other day using digital calipers. Animals were euthanized when their torso width exceeded 30 mm and body condition score worsened. Knockout mice used in these studies include: RAG-/- (B6.129S7-Rag1tm1Mom/J), TNFɑ-/- (B6;129S-Tnftm1Gkl/J), IFN-γ-/- (B6.129S7-Ifnγtm1Ts/J), IFN-γR1-/- (B6.129S7-Ifnγr1tm1Agt/J), and NOD/Scid/IL2Rγ-/- mice (NSG) (NOD.Cg-Prkdcscid/IL12rgtm1Wjl/SzJ).
Flow cytometry
Tumor-infiltrating lymphocyte phenotypes were analyzed using standard flow cytometry techniques. In short, excised tumors were mechanically disrupted into single-cell suspensions by grinding over a 40-µm nylon mesh filter into phosphate-buffered saline (PBS). Cells were then pelleted and immediately stained for flow cytometry using standard methodologies. Antibodies for these studies were obtained from BD Biosciences (San Jose, California, USA), Biolegend (San Diego, California, USA), and Invitrogen (Waltham, Massachusetts, USA) and included: Ly6c (clone HK1.4), CD45 (clone 30-F11), CD11b (clone M1/70), CD11c (clone HL3), Ly6G (clone 1A8), F4/80 (T45-2342), NK1.1 (clone PK136), I-Ab (clone M5/114.15.2), IL-17R (clone A7R34), CD3 (clone KT3.1.1), NKp46 (clone 29A1.4), CD49a (clone Ha31/8), CD49b (clone HMa2), CD25 (clone PC61), CD4 (clone GK1.5), B220 (clone RM2619), CD200RI (clone 0×110), and CD8a (clone 53–6.7).
Measurement of soluble cytokines
For analysis of intratumoral cytokines, excised tumors were weighed and then mechanically disrupted by grinding over a 40-µm nylon mesh filter into 3 mL of PBS. Resulting cell suspension was clarified through two rounds of centrifugation at 3000×g. Clarified supernatant was then separated into single-use aliquots for ELISA analysis. IFN-γ (catalog no. 551866) and TNF (catalog no. 558534) were measured using OPTEIA duo ELISA kits (BD Biosciences, Franklin Lakes, New Jersey, USA) per manufacturer’s recommendations. Cytokine concentrations were then normalized to initial tumor weights and data presented as cytokine per gram of tumor. For in vitro analysis of secreted cytokines, supernatant from infected cells was harvested and transgene expression was analyzed using the anti-PD1 DuoSet ELISA (catalog no. DY1086; R&D Systems, Minneapolis, Minnesota, USA) and the anti-IL-12 OPTEIA duo ELISA (catalog no. 555256) (BD Biosciences) per manufacturer’s recommendations.
In vitro viral characterization
All in vitro assays were carried about by adsorbing virus to cells for 60 min at room temperature and subsequently removing viral inoculation media and replacing with new complete growth media. To assay intracellular viral replication, 5×105 cells from each indicated cell line were infected with each virus at a multiplicity of infection (MOI) of 10. Seventy-two hours after infection, cells were harvested, and intracellular virus was released by repeated rounds of mechanical lysis involving alternating freeze/thaw cycles and sonication. Amount of infectious virus in each sample was then determined by serial dilution followed by quantitation of GFP+ infectious units using a standard viral foci forming assay on BSC40 cells. To assay the acute lytic capacity of each virus, each indicated cell line was infected with the indicated virus at MOIs ranging from 1.0 to 0.005. Forty-eight hours after infection, cellular viability was determined using the CellTiter 96 Non-Radioactive Cell Proliferation (MTT) assay (Promega, Madison, Wisconsin, USA) per manufacturer’s recommendations.
RNA-seq and bioinformatics
Bioinformatics analysis was conducted using the contralateral B16/F10 model. Syngeneic C57/Bl6 mice were implanted with contralateral B16/F10 tumors, binned, and treated as detailed above. Tumors were subsequently excised on day 6 following the initiation of treatment and disassociated into single cell suspensions over a 40-µM nylon mesh filter. Cells were then pelleted, total RNA extracted using an RNEasy kit (Qiagen, Hilden Germany), and RNA-seq performed by Novogene (Cambridge, UK) using an Illumina sequencer. Raw RNA-Seq data were downloaded in compressed FASTQ format, mapped against the Mus musculus reference cDNA sequence (GRCm38.p5; ENSEMBL release v98), quantified at transcript level with kallisto (v0.46.0), and summarized at the gene-level using BioMART’s gene-to-transcript mapping (accessed on January 22, 2019) via tximport (v1.15.6). Principal component analysis was conducted using finalized transcript counts. Disease Ontology Semantic and Enrichment analysis R package (v3.14) was used for gene set enrichment analyses defining differentially expressed genes as having a log2FC ≥2 and p value <0.05.
Results
Coexpression of IL-12 and soluble PD1 during localized oncolytic treatment induces systemic antitumor responses
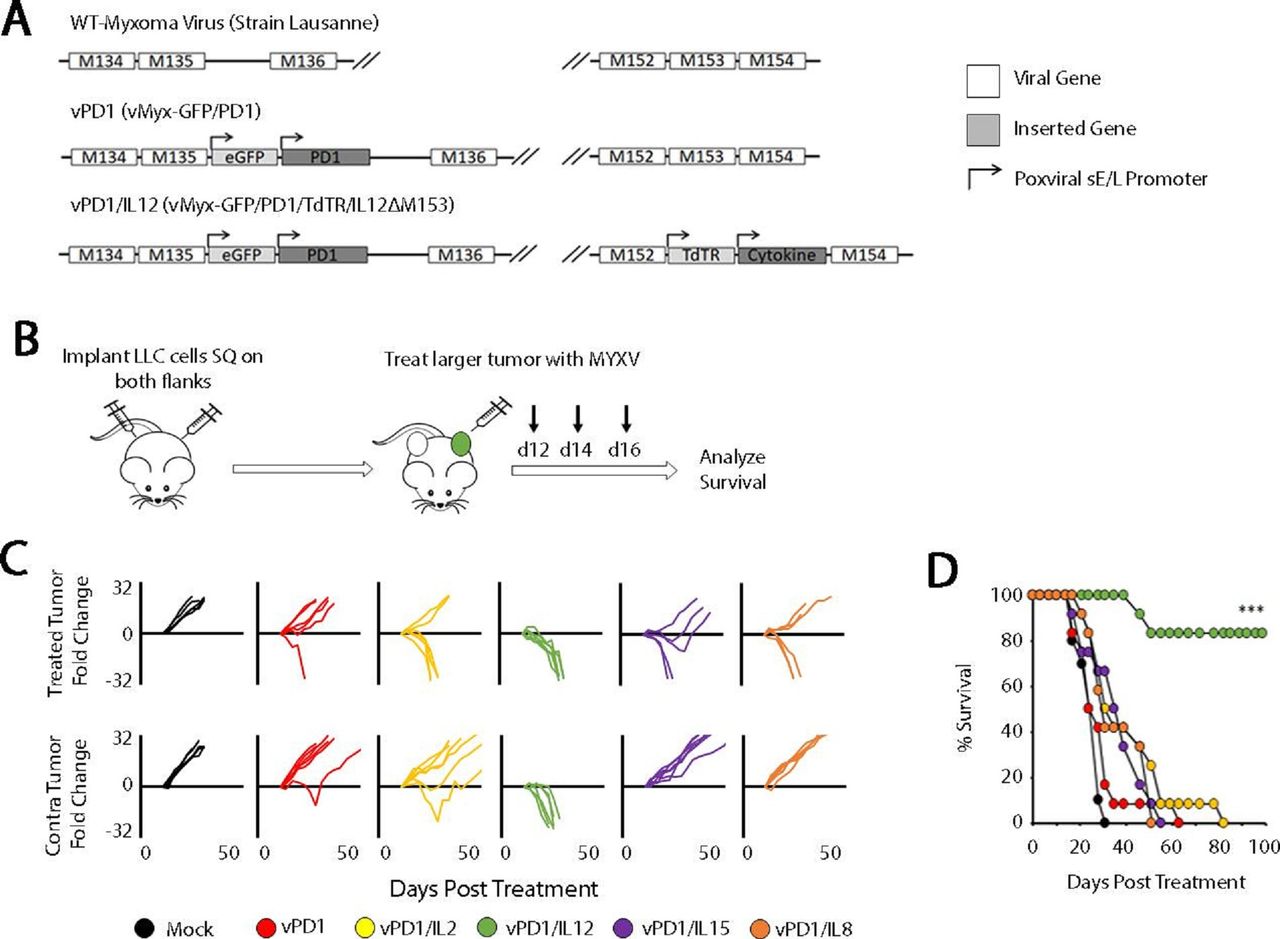
Our group has previously pioneered a novel method of incorporating ICB into OV through the use of a recombinant MYXV, which encodes a soluble fragment of the PD1 protein (vPD1).17 20 This virus is highly effective against directly injected tumors; however, the vast majority of patients with cancer present with metastatic disease and our previous work had suggested that this singly modified virus was ineffective in this context. To overcome this challenge, we hypothesized that a feasible path forward would be to create a next-generation recombinant virus expressing both soluble PD1 and a second therapeutic transgene. To test this hypothesis, we created a series of 10 viral constructs which expressed both soluble PD1 and either a proinflammatory cytokine (IL-2, IL-12, IL-15, or IL-18) or an additional soluble checkpoint inhibitor (LAG3, TIM3, VISTA, BTLA, CTLA4, or TIGIT) (figure 1A; online supplemental figure S1). To test whether these novel viruses would be more effective against disseminated disease, we then assessed their ability to regress non-injected lesions in a contralateral LLC tumor model (figure 1B). The results indicated that, of the viruses tested, only the virus coexpressing soluble PD1 and IL-12 (encoded as a p40-p35 fusion protein) displayed significantly improved efficacy over the vPD1 backbone. Impressively, treatment with this virus (vPD1/IL-12) was able to fully regress both injected and non-injected lesions in virtually all treated mice (complete phenotypic responses lasting >100 days in 10/12 animals) (figure 1C,D).
Supplemental material
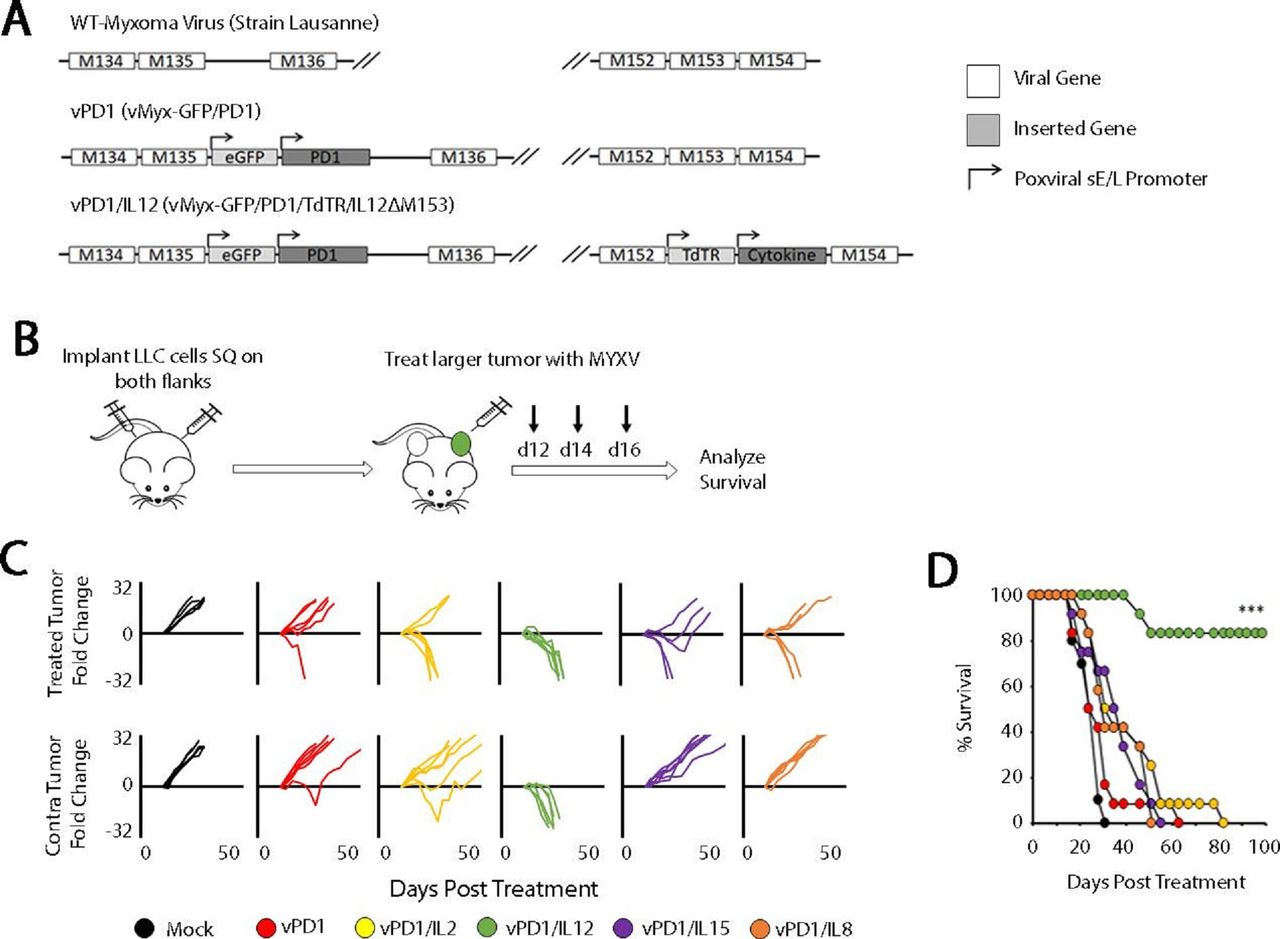
Localized oncolytic therapy involving IL-12 and soluble PD1 induces systemic antitumor responses. (A) Genomic schematic representation of the second-generation recombinant MYXVs used in these experiments. (B) Schematic representation of experimental design. Syngeneic C57/Bl6 mice were injected SQ on both the left and right flanks with LLC cells. Tumors were allowed to establish and then either mock treated or treated with three IT injections of the indicated virus (n=5 for all groups). (C) Progression of individual tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (D) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in B–D are from a single experiment, which is representative of two independent experiments. IL-12, interleukin 12; IT, intratumoral; LLC, Lewis lung carcinoma; MYXVs, myxoma viruses; PD1, programmed cell death protein 1; SQ, subcutaneously; vPD1, virus expressing PD1; vPD1/IL-12, virus expressing PD1 and IL-12.
vPD1/IL-12 displays unique therapeutic synergy in multiple tumor models
To advance on our initial finding, we next wished to understand both the breadth of vPD1/IL-12’s therapeutic efficacy as well as the relative contribution of its soluble PD1 and IL-12 transgenes. To accomplish this, we first generated a singly recombinant control virus expressing only an IL-12 fusion protein (vIL-12—online supplemental figure S2) and subsequently analyzed each virus for its in vitro replication properties (figure 2). As is seen for many immune-modulating oncolytic viruses, in vitro analysis indicated that vPD1/IL-12 displayed intracellular replication properties which are typical for MYXV in a variety of established tumor cell lines (figure 2A). Additionally, infection of multiple cell types across a wide range of multiplicities resulted in similar reductions in cellular viability (figure 2B) suggesting that vPD1/IL-12 possessed relatively normal direct lytic capacity and while it successfully secreted its intended transgenes (figure 2C,D), neither of these transgenes induced significant bystander killing. In contrast, in vivo efficacy studies indicated that localized treatment with vPD1/IL-12 was able to induce significant regression of both injected and non-injected lesions in a wide range of disseminated tumor models including: the previously detailed contralateral LLC lung cancer model, a contralateral B16/F10 melanoma model, a contralateral MC38 colon cancer model, and a peritoneally disseminated BR5 ovarian cancer model (figure 3). Critically, regression of non-injected tumors was not observed in any model following treatment with either singly recombinant control virus or in mice treated with an unarmed MYXV (vGFP) combined with an anti-PD1 antibody and recombinant IL-12 (online supplemental figure S3). Taken together, these data suggest that vPD1/IL-12 possesses a wide therapeutic breadth resulting from a unique form of combinatorial synergy, which is not readily duplicated through individual treatments.
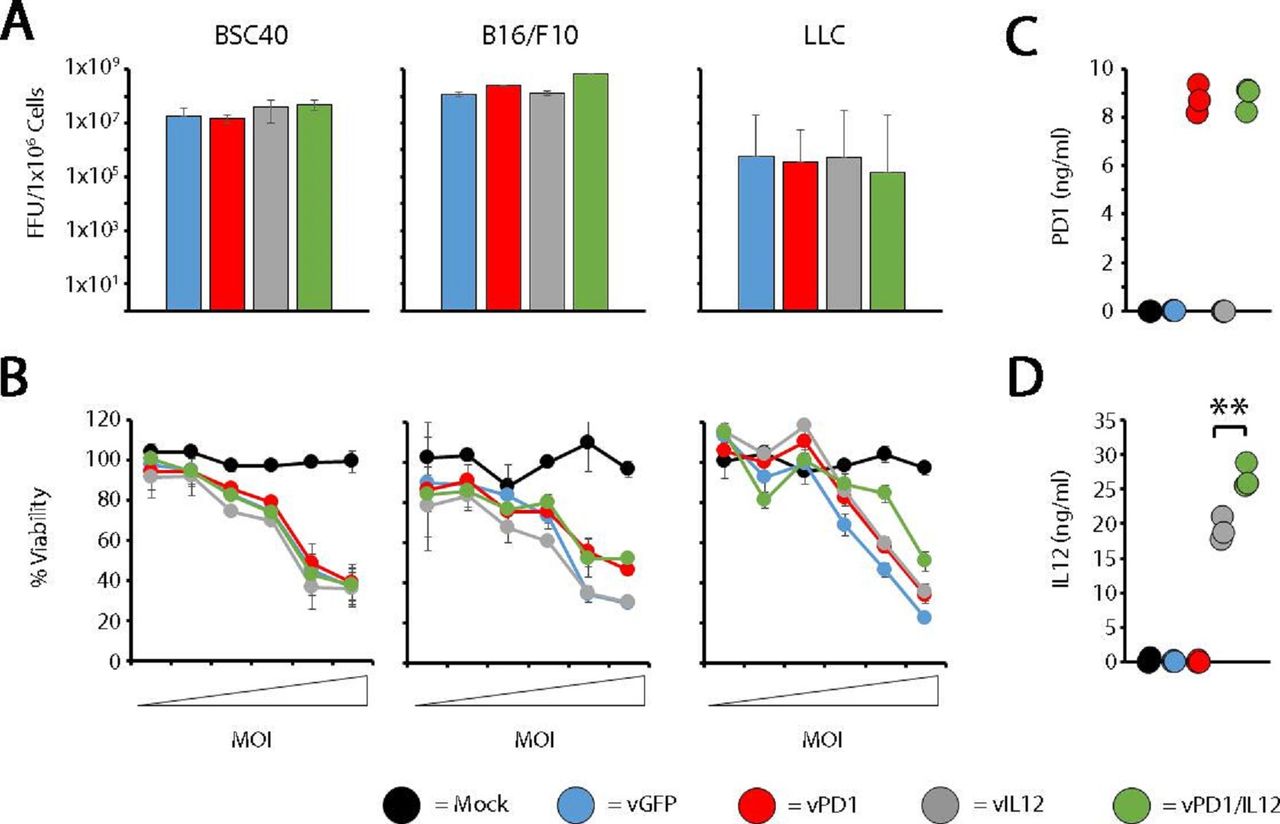
vPD1/IL-12 displays normal oncolytic potential in vitro. (A) The indicated cell types were infected with vGFP, vPD1, vIL-12, or vPD1/IL-12 at an MOI of 10. At 72 hours postinfection, cells were harvested and the number of infectious progeny virus was determined using standard foci forming assays. Data are representative of three independent experiments. (B) The indicated cell types were mock infected or infected with vGFP, vPD1, vIL-12, or vPD1/IL-12 at MOIs of 0.005, 0.01, 0.05, 0.1, 0.5, and 1.0. At 24 hours postinfection, cellular viability was determined using MTT assay. Data are shown as per cent viability compared with mock-infected control and represent the summation of two independent experiments. (C and D) BSC40 cells were infected with the indicated viruses at an MOI of 10. 72 hours postinfection, supernatant was harvested and analyzed for the presence of both soluble PD1 (C) and IL-12 (D) using ELISA. Data are representative of two independent experiments. Significance was determined using Student’s t-test (**p<0.01). FFU, foci forming units; LLC, Lewis lung carcinomav; vGFP, virus expressing green fluorescent protein; vIL-12, virus expressing interleukin 12; vPD1, virus expressing programmed cell death protein 1; vPD1/IL-12, virus expressing PD1 and IL-12.

vPD1/IL-12 displays distinct response patterns across a variety of tumor models. (A) Schematic representation of experimental design. Syngeneic C57/Bl6 mice were injected SQ on both the left and right flanks with LLC cells. Tumors were allowed to establish and then either mock treated or treated with three IT injections of vPD1, vIL-12, or vPD1/IL-12 (n=5–6 for all groups). (B) Progression of individual tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (C) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in A–C are from a single experiment, which is representative of two independent experiments. (D) Schematic representation of experimental design. Syngeneic C57/Bl6 mice were injected with B16/F10 cells on both flanks. Tumors were allowed to establish and then either mock treated or treated with three IT injections of vPD1, vIL-12, or vPD1/IL-12 (n=5 for all groups). (E) Progression of individual tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (F) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (**p<0.01). Data in D–F are from a single experiment, which is representative of two independent experiments. (G) Schematic representation of experimental design. Syngeneic FVB mice were injected with BR5-luc cells into the peritoneal cavity. Mice were sorted into their appropriate groups with equally representative tumor burden. Groups were then mock treated or treated with vPD1, vIL-12 or vPD1/IL-12 into the peritoneal cavity in 50 μL sterile PBS (n=5 for each group). (H) Bioluminescent signal indicative of tumor burden per group at 10 and 25 days after initial injection. (I) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (**p<0.01). Data in G–I are from a single experiment, which is representative of three independent experiments. (J) Schematic representation of experimental design. Syngeneic C57/Bl6 mice were injected with MC38 cells on both flanks. Tumors were allowed to establish and then either mock treated or treated with three IT injections of vPD1, vIL-12, or vPD1/IL-12 (n=5 for all groups). (K) Progression of individual tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (L) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in J–L are from a single experiment, which is representative of three independent experiments. IL-12, interleukin 12; IT, intratumoral; LLC, Lewis lung carcinoma; MYXV, myxoma virus; PBS, phosphate-buffered saline; PD1, programmed cell death protein 1; SQ, subcutaneously; vPD1, virus expressing PD1; vPD1/IL-12, virus expressing PD1 and IL-12.
vPD1/IL-12 displays distinct response patterns based on the functionality of antitumor T cells
Interestingly, our previous efficacy studies suggested that treatment with vPD1/IL-12 resulted in two distinct response patterns. In the LLC and BR5 models, treatment was associated with complete phenotypic elimination of all tumors and long-term survival. In contrast, in the B16/F10 and MC38 models, while treatment significantly improved overall survival, these improvements were non-curative and animals eventually succumbed to disease (figure 3). To better understand the cause of these response patterns, we next wanted to determine what cellular mechanisms mediated vPD1/IL-12’s efficacy in each type of model. To address this, we tested the efficacy of vPD1/IL-12 against each of our four tumor models in either immune-competent animals, animals lacking adaptive immunity (RAG-/- or T-cell-depleted), or completely immune-deficient animals (NSG). Consistent with most oncolytic paradigms, elimination of T cells severely restricts the efficacy of vPD1/IL-12 in our two curative models (LLC and BR5) including completely eliminating all long-term phenotypic regressions (figure 4B,C and online supplemental figure S4C). Additionally, while long-term survivors generated from immune-competent animals were able to reject subsequent tumor rechallenge, this ability was lost following antibody-based depletion of T cells (data not shown). In contrast, elimination of T cells from our non-curative models (B16/F10 and MC38) had only a minimal impact on vPD1/IL-12’s efficacy (figure 4D,E and online supplemental figure S4F). No delays in the growth of either injected or non-injected lesions were observed in any tumor model in completely immune-deficient NSG animals, suggesting that virtually none of vPD1/IL-12’s efficacy was due to direct viral lysis (online supplemental figure S5). Taken together, these data demonstrate that the ability of vPD1/IL-12 to generate completely curative responses is dependent on functional T-cell immunity and suggest that this immunity might be compromised in non-responsive models.
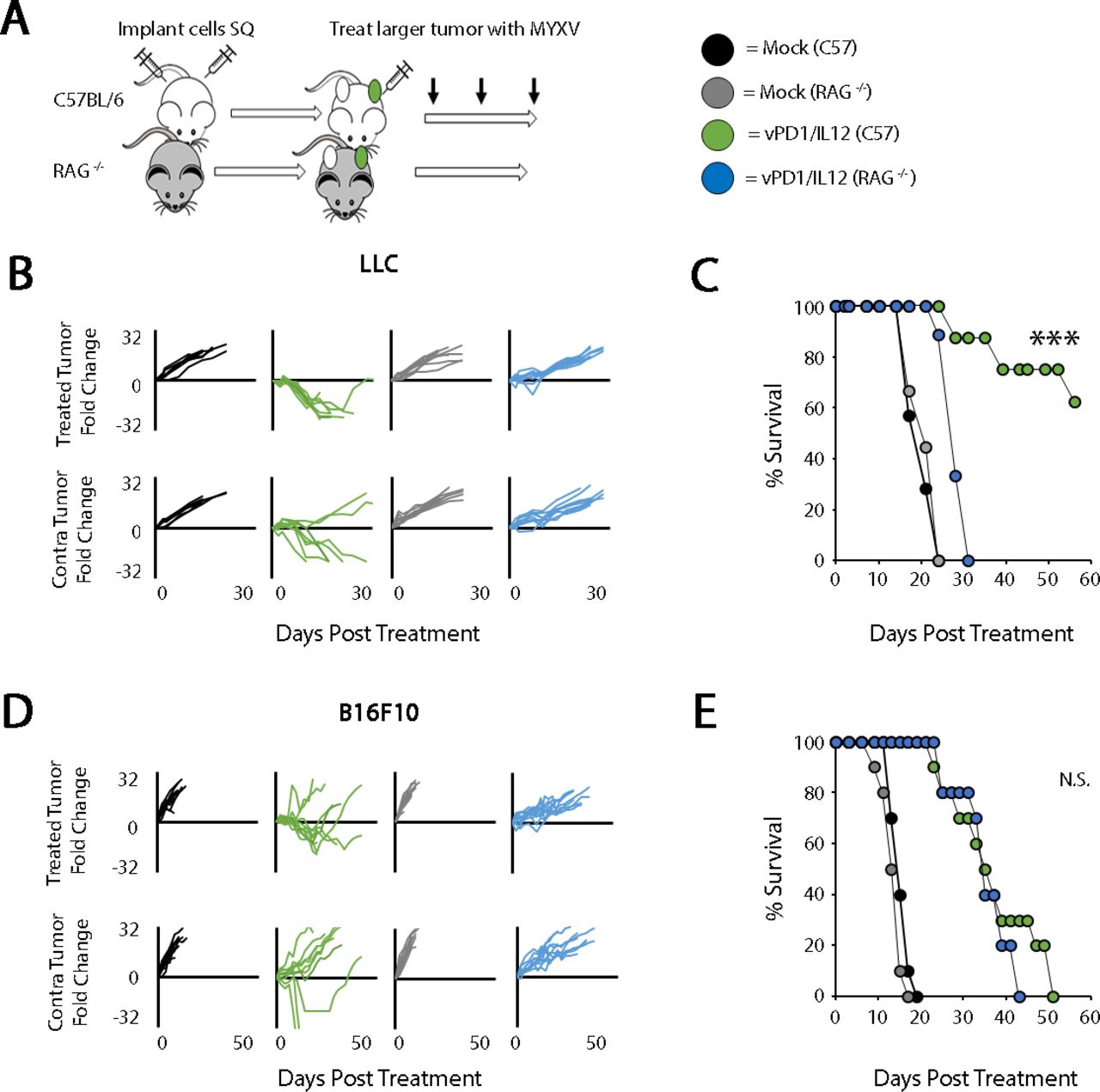
Response to vPD1/IL-12 treatment correlates with the impact of antitumor T cells. (A) Schematic representation of experimental design. C57/Bl6 or RAG-/- mice were injected SQ on both the left and right flanks with LLC or B16/F10 cells. Tumors were allowed to establish and either mock treated or treated with three IT injections of vPD1/IL-12 (n=5–9 for all groups). (B) Progression of individual LLC tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (C) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in B and C are from a single experiment, which is representative of two independent experiments. (D) Progression of individual B16/F10 tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (E) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis. Data in D and E are from a single experiment, which is representative of two independent experiments. IL-12, interleukin 12; IT, intratumoral; LLC, Lewis lung carcinoma; MYXV, myxoma virus; N.S., not significant; PD1, programmed cell death protein 1; SQ, subcutaneously; vPD1, virus expressing PD1; vPD1/IL-12, virus expressing PD1 and IL-12.
Genetic ablation of TNF enhances the efficacy of vPD1/IL-12 treatment
To determine what might be inhibiting the development of functional antitumor T-cell responses in our non-curative models, we performed transcriptomic profiling of B16/F10 tumors treated with either our singly recombinant control viruses (vPD1 or vIL-12) or vPD1/IL-12 6 days post-treatment (a time point just before tumor outcomes begin to diverge). Consistent with its profoundly enhanced therapeutic efficacy, this analysis indicated that tumors treated with vPD1/IL-12 displayed highly distinct RNA profiles compared with tumors treated with our other viruses (figure 5A). This distinct profile was highlighted by significantly increased cytokine production including increased levels of RNA coding for IFN-γ, IFN-β, and TNF (figure 5B,C). Consistent with this enhanced RNA expression, tumors treated with vPD1/IL-12 also displayed increased levels of IFN-γ and TNF protein (figure 5D) as well as gene signatures consistent with the presence of both these cytokines (figure 5E).
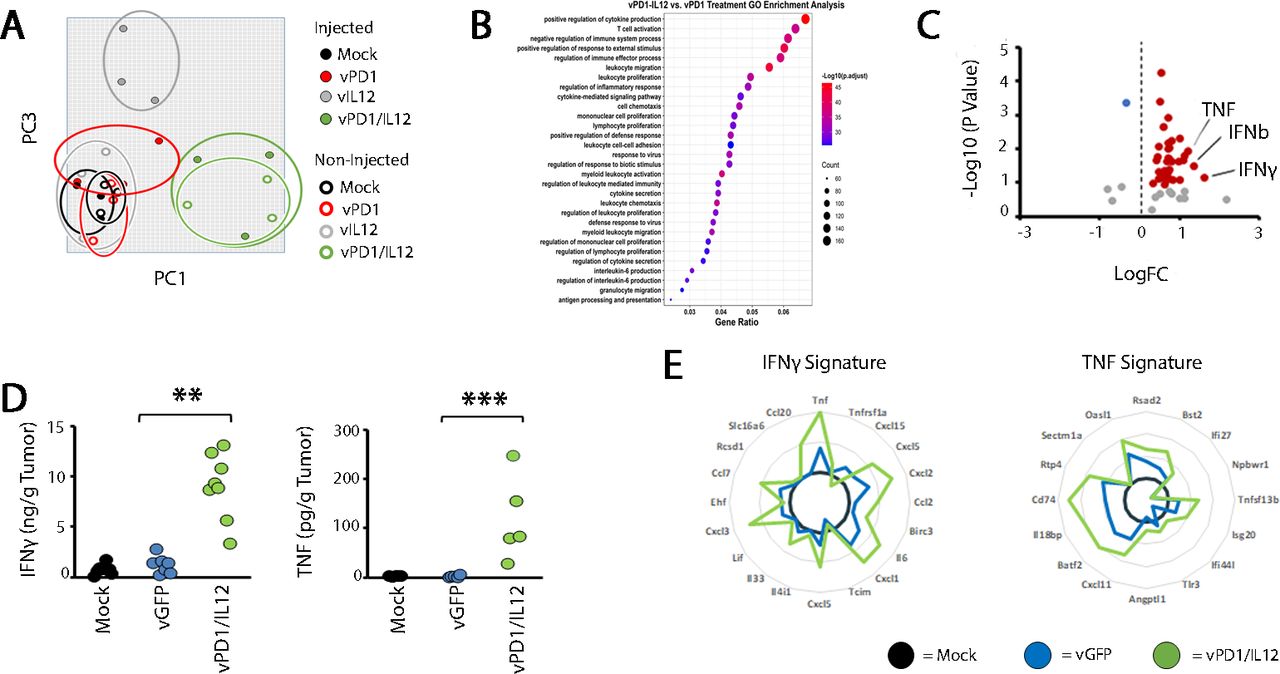
Localized vPD1/IL-12 treatment induces high levels of TNF. Syngeneic C57/Bl6 mice were injected SQ on both the left and right flanks with B16/F10 cells. Tumors were allowed to establish and then either mock treated or treated with three IT injections of vPD1, vIL-12, or vPD1/IL-12 (n=3 for all groups). Six days after the initiation of treatment, total RNA was extracted from each tumor and analyzed using RNA-seq. (A) Unsupervised principle component analysis of gene expression from the indicated cohorts. (B) Gene Ontology pathway analysis of all genes which were differentially expressed between vPD1/IL-12 treated mice and all other cohorts. (C) Volcano plot showing the expression of all cytokines and interleukins. Data shown compare tumors treated with vPD1/IL-12 to tumors treated with control virus. (D) ELISA data showing expression of IFN and TNF protein in the supernatants of tumors 8 days after being treated as indicated. (E) Analysis of RNA-seq data (from A) demonstrating the presence of previously identified gene response signatures to IFN and TNF. FC, fold change; IFN, interferon; IL-12, interleukin 12; IT, intratumoral; MYXV, myxoma virus; PD1, programmed cell death protein 1; SQ, subcutaneously; TNF, tumor necrosis factor; vGFP, virus expressing green fluorescent protein; vPD1, virus expressing PD1; vPD1/IL-12, virus expressing PD1 and IL-12.
To understand the functional impact of the inflammatory responses induced by vPD1/IL-12, we next tested how disruption of either the IFN-γ or TNF pathways would influence the efficacy of vPD1/IL-12. Interestingly, despite its well-documented antitumor properties,21 disruption of the IFN-γ pathway (either through the use of mice genetically lacking IFN-γ or IFN-γR1, treatment with IFN-γ-blocking antibodies, or disruption of STAT1 within tumor cells) did not reduce the therapeutic efficacy of vPD1/IL-12 against B16/F10 tumors (online supplemental figure S6). In striking contrast, disruption of the TNF pathway (using mice genetically lacking TNF) allowed for vPD1/IL-12 therapy to completely eradicate both treated and non-treated tumors from 100% of animals in both the B16/F10 and MC38 models (figure 6). As was initially observed in the LLC and BR5 models, these curative phenotypes were now T-cell dependent and generated T-cell dependent memory in long-term survivors (online supplemental figure S7). These data suggest that the presence of TNF during vPD1/IL-12 treatment inhibits either the development or functionality of antitumor T-cell responses preventing curative therapy.
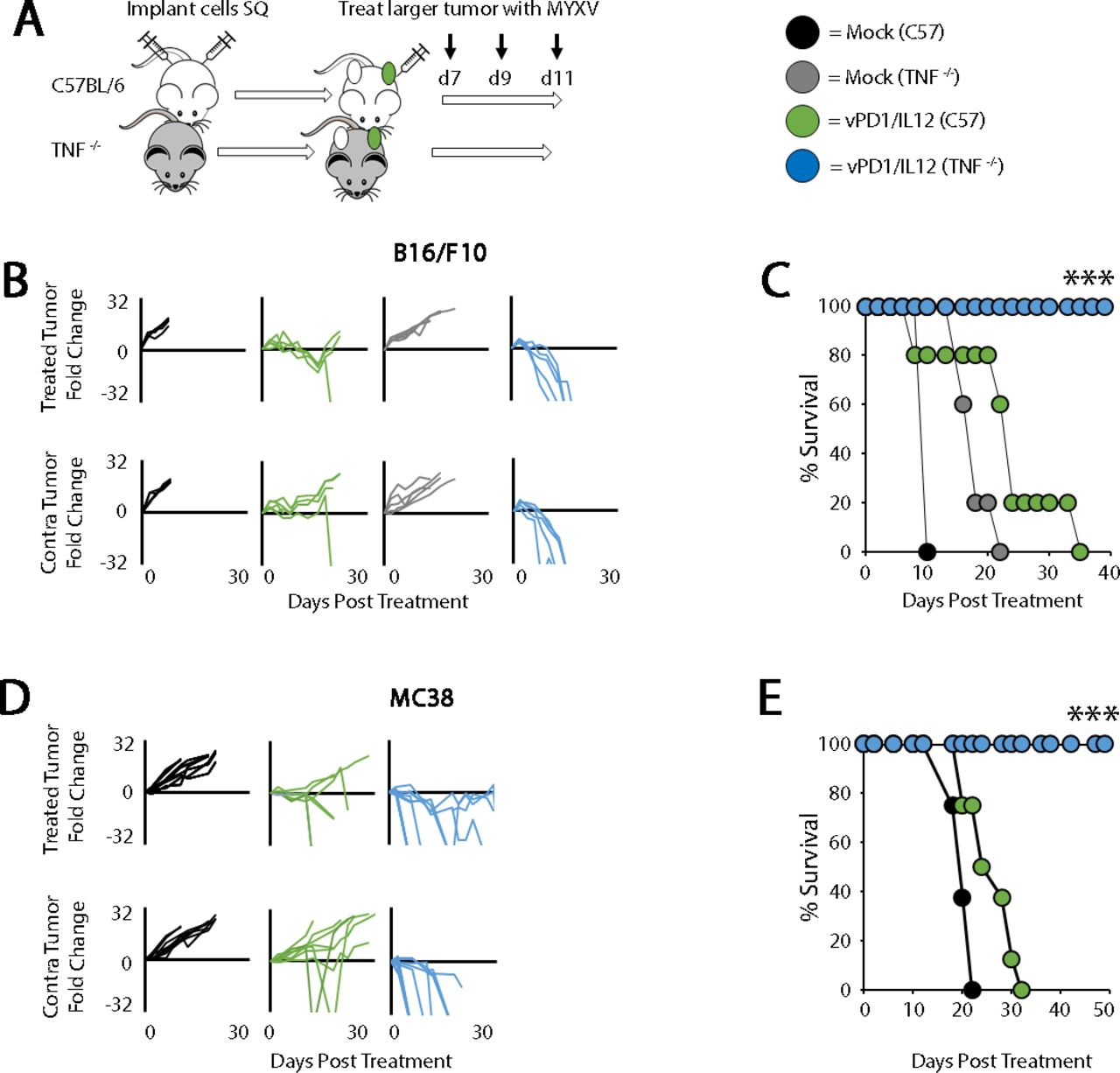
Genetic loss of TNF improves the efficacy of vPD1/IL-12. (A) Schematic representation of experimental design. C57/Bl6 or TNF-/- mice were injected contralaterally with B16/F10 or MC38 cells. Tumors were allowed to establish and then either mock treated or treated with three IT injections of vPD1/IL-12 (n=5–9 for all groups). (B) Progression of individual B16/F10 tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (C) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in B and C are from a single experiment, which is representative of two independent experiments. (D) Progression of individual MC38 tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (E) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (***p<0.001). Data in D and E are from a single experiment, which is representative of two independent experiments. IT, intratumoral; MYXV, myxoma virus; SQ, subcutaneously; TNF, tumor necrosis factor; vPD1/IL-12, virus expressing programmed cell death protein 1 and interleukin 12.
Blockade of TNF during vPD1/IL-12 treatment enhances therapeutic efficacy by improving intratumoral T-cell viability
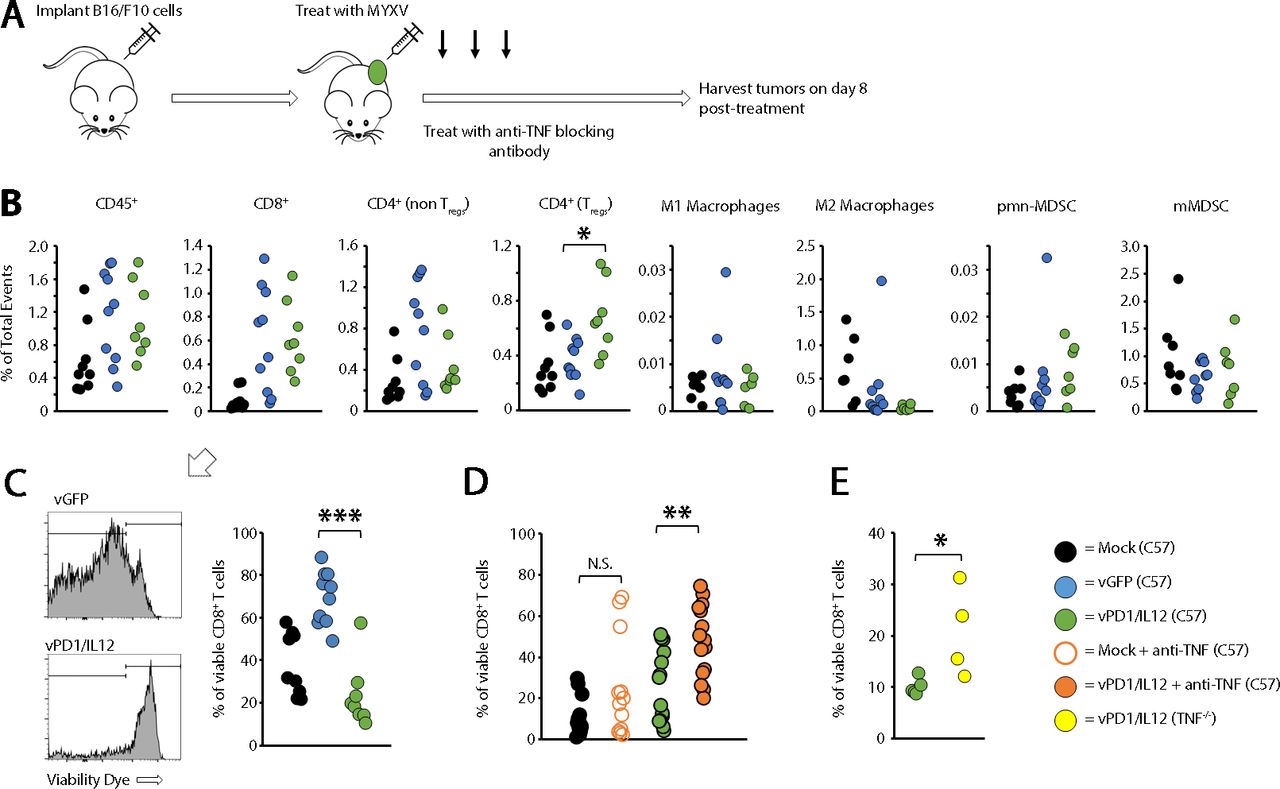
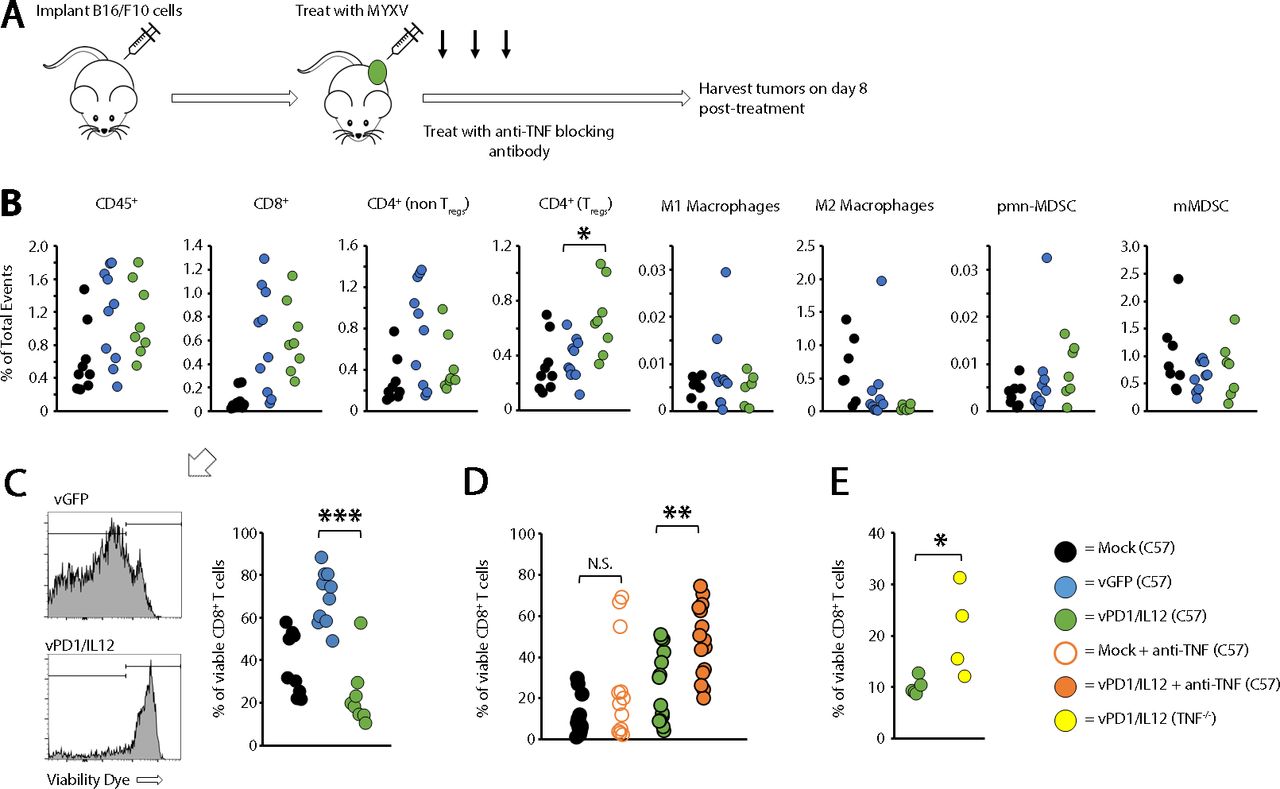
TNF has been implicated in immune development,22 23 raising the possibility that the increased efficacy seen in our previous studies was due to pre-existing immune differences between wild type (WT) and TNF-/- mice. Initial studies, however, suggested that the overall immune phenotypes in both the spleens and tumors of these mice were similar (online supplemental figure S8). We therefore hypothesized that TNF might be inhibiting the efficacy of vPD1/IL-12 in an acute setting. To test this, we asked whether acute, antibody-based TNF blockade might duplicate the improved efficacy seen in TNF-/- mice. The results demonstrated that cotreatment of mice with both vPD1/IL-12 and TNF-blocking antibody allowed for curative responses to occur in both the B16/F10 and MC38 models (figure 7). Finally, to understand the mechanism(s) through which TNF blockade enhanced vPD1/IL-12 therapy, we used flow cytometry to analyze the immune responses induced following treatment with either control MYXV or vPD1/IL-12 (figure 8A). Despite having highly distinct therapeutic efficacies, the phenotypic immune responses induced by both vGFP and vPD1/IL-12 were extremely similar with the only significant difference being a greater induction of regulatory T cells following treatment with vPD1/IL-12 (figure 8B). All other analyzed cell populations, including CD8+ T cells, were similar across both viral treatments. However, while most of the CD8+ T cells found in tumors treated with control MYXV were viable, in vPD1/IL-12-treated tumors, the majority of these cells appeared to have lost membrane integrity (figure 8C). To determine whether TNF played a role in this loss of intratumoral T-cell viability, we repeated our previous experiment treating mice with both vPD1/IL-12 and anti-TNF-blocking antibodies. Strikingly, the inclusion of TNF blockade significantly increased the percentage of viable CD8+ T cells found in vPD1/IL-12-treated tumors (figure 8D). A similar effect was seen in TNF-/- mice treated with vPD1/IL-12 (figure 8E). Taken together, these data demonstrate that the efficacy of vPD1/IL-12 therapy can be significantly enhanced by acute TNF blockade and suggest that this blockade functions by preventing intratumoral T cells from losing viability during treatment.

Antibody-based TNF blockade improves the efficacy of vPD1/IL-12 treatment. (A) Schematic representation of experimental design. C57/Bl6 mice were injected contralaterally with B16/F10 or MC38 cells. Tumors were allowed to establish and then either mock treated or treated with three IT injections of 1×107 FFU of vPD1/IL-12 (n=5–10 for all groups). Cohorts were then split in two and either mock treated or treated with 100 μg anti-TNF antibody IP every other day for duration of study. (B) Progression of individual B16/F10 tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (C) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (**p<0.01). Data in B and C are from a single experiment, which is representative of three independent experiments. (D) Progression of individual MC38 tumors after treatment. Data are displayed as the fold increase or decrease of tumor area compared with area at the initiation of treatment. (E) Overall survival of animals treated as indicated. Significance was determined using log-rank analysis (*p<0.05). Data in D and E are from a single experiment. FFU, foci forming units; IP, intraperitoneal; IT, intratumoral; MYXV, myxoma virus; SQ, subcutaneously; TNF, tumor necrosis factor; vPD1/IL-12, virus expressing programmed cell death protein 1 and interleukin 12.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immune changes induced by vPD1/IL-12 treatment. (A) Schematic representation of experimental design. Syngeneic C57/Bl6 mice were injected with B16/F10 cells SQ on the right flank. Tumors were allowed to establish and then either mock treated or treated with three IT injections of either vGFP or vPD1/IL-12. Eight days after initial treatment, mice were euthanized and tumors harvested for flow cytometry analysis. (B) Comparison of immune populations in tumors. Significance was assessed using a Student’s t-test (*p<0.05). (C) Comparison of CD8+ viability found in flow samples. Significance was assessed using a Student’s t-test (***p<0.001). (D) Syngeneic C57/Bl6 mice were injected with B16/F10 cells SQ on the right flank. Tumors were allowed to establish and then either mock treated or treated with three IT injections of either vGFP or vPD1/IL-12. Cohorts were then split in two and either mock treated or treated with 100 μg anti-TNF antibody IP every other day for duration of study. Eight days after initial treatment, mice were euthanized and tumors harvested to analyze IT T-cell viability. Significance was assessed using a Student’s t-test (**p<0.01). (E) C57/Bl6 or TNF-/- mice were injected with B16/F10 cells SQ on the right flank. Tumors were allowed to establish and then either mock treated or treated with three IT injections of either vGFP or vPD1/IL-12. Eight days after initial treatment, mice were euthanized and tumors harvested to analyze IT T-cell viability. Significance was assessed. IP, intraperitoneal; IT, intratumoral; MYXV, myxoma virus; N.S., not significant; SQ, subcutaneously; TNF, tumor necrosis factor; Tregs, regulatory T cells; vGFP, virus expressing green fluorescent protein; vPD1/IL-12, virus expressing programmed cell death protein 1 and interleukin 12.
Discussion
The combination of OV and ICB has generated significant interest in recent years.24 25 This is largely due to oncolytic viruses’ ability to remodel the TME in ways which are thought to make it more amenable to immunotherapy. Like our current study, much of this literature has used recombinant viruses encoding immune-enhancing cytokines to generate more potent inflammatory responses. Indeed, while the specific combination of an MYXV encoding a PD1-inhibitor and IL-12 has never been published, numerous other groups have generated similar recombinant viruses in other oncolytic backbones.26 Most notably, an oncolytic herpes simplex virus (HSV) encoding both IL-12 and an anti-PD1 single chain variable fragment (scFv) was recently developed, which had significant efficacy in a variety of models.27 In contrast to our current work, however, the efficacy of this HSV construct appeared to be largely dependent on IFN-γ. Interestingly, inclusion of IL-12 and anti-PD1 modalities into other oncolytic backbones has been much less successful. A Newcastle disease virus encoding IL-12 and an anti-PD1 scFv displayed extremely limited therapeutic efficacy even in localized models.28 Similarly, an oncolytic adenovirus encoding these same transgenes was completely ineffective unless combined with adoptive cell therapy.29 These data suggest that even in oncolytic constructs encoding similar therapeutic transgenes, the impact of the viral backbone might play a major role in determining therapeutic outcomes.
In our current studies, vPD1/IL-12 represents a potent therapeutic agent in multiple models. Despite having some therapeutic efficacy in every model tested to date, however, tumors treated with vPD1/IL-12 clearly display distinct response patterns, which correlate with different mechanisms of therapeutic action. Models which display curative responses appeared to be predominantly T-cell dependent, since no complete responses were observed in either genetically T-cell-deficient animals (figure 4, online supplemental figures S4 and S5) or animals injected with T-cell-depleting antibodies (our unpublished observations). These results are consistent with most oncolytic paradigms suggesting that a large portion of OV’s therapeutic benefit is derived from the induction of antitumor T cells.4 In contrast, vPD1/IL-12’s efficacy in non-curative models appeared to be largely independent of T cells. Unfortunately, the mechanism(s) mediating delayed tumor growth in these models remains unknown. Critically, they do not appear to be caused by direct viral lysis of tumor cells since no virus could be detected in non-injected lesions (our unpublished observations) and no delays in tumor growth were observed in completely immune-deficient mice (online supplemental figure S6). Previous work has suggested several potential T-cell-independent mechanisms through which IL-12 can delay tumor growth including a role for natural killer cells30 31 and an inhibition of angiogenesis.32 33 However, these mechanisms have all been linked to IL-12’s ability to induce IFN-γ34–36 and the delays seen in our studies are clearly IFN-γ independent (online supplemental figure S6). More work is therefore needed to clarify the mechanism(s) mediating these non-curative responses as well as how they impact treatment in other oncolytic contexts.
Consistent with the goal of encoding a master inflammatory regulator like IL-12 into an oncolytic virus, we observed that treatment with vPD1/IL-12 induced high levels of inflammatory cytokine expression within both treated and non-treated tumors highlighted by increased expression of both IFN-γ and TNF (figure 5). The induction of this type of inflammation is typically considered a goal of OV. Indeed, expression of TNF is considered enough of a positive indicator during OV that numerous groups have directly encoded this cytokine into their oncolytic backbones, which has typically improved therapeutic efficacy.37–40 Our most striking finding is, therefore, that blockade of TNF significantly enhances the therapeutic efficacy of vPD1/IL-12 in multiple models. In contrast to the existing oncolytic literature, these results are much more in line with recent observations on the role of TNF in ICB therapy.41–44 These studies have shown that blockade of TNF can enhance the efficacy of both PD1 and CTLA4 blockade in preclinical models.42 43 Additionally, concurrent therapy of human patients with TNF blockade and ICB therapy can both increase therapeutic efficacy as well as reduce the development of immune related adverse events.44 Unfortunately, the reason TNF plays such distinct roles in different tumor settings remains unclear. It is possible that the impact of TNF is model dependent, however, our results involving TNF blockade were consistent among the two models tested (figures 6 and 7). Alternatively, the various forms of TNF might play distinct roles during tumor therapy. This hypothesis is partially supported by previous observations that part of the immune suppressive function of myeloid derived suppressor cells (MDSCs) is mediated by membrane-bound TNF.45 Importantly, this hypothesis also has significant clinical implications since the various Food and Drug Administration (FDA)-approved TNF inhibitors show distinct molecular properties.46–51 Most notably, the soluble TNF receptor 2-based etanercept has been shown to bind to soluble TNF but not membrane-bound TNF.49 52 It is therefore critical to better understand the molecular details mediating TNF’s ability to inhibit various forms of immunotherapy to rationally apply the appropriate form of TNF blockade.
Finally, in the previous studies combining TNF blockade and ICB it was suggested that TNF inhibited antitumor immunity by inducing T-cell exhaustion through upregulation of the TIM3 checkpoint protein.43 53 While we did not examine a potential role for T-cell exhaustion in our studies, in both TNF-/- mice and mice treated with TNF-blocking antibodies, intratumoral T cells display significantly increased viability. In this context, both direct engagement of TNF receptors on T cells by TNF itself54 55 as well as engagement of TIM3 with its primary ligand galectin-956 57 have been shown to induce T-cell apoptosis. Interestingly, treatment with TNF-blocking antibodies had a more pronounced effect on T-cell viability than genetic ablation of TNF did (figure 8D,E). This could be the result of T cells developing in slightly different contexts, but might also be explained by simple experiment-to-experiment variation. Further work is, therefore, needed to determine the mechanism(s) through which the presence of TNF reduces intratumoral T-cell viability.
In conclusion, our data demonstrate that the induction of TNF during OV can play an immune regulatory role and that blockade of this TNF during MYXV-based treatment can result in potent therapeutic synergy including the complete eradication of normally non-responsive, disseminated disease. Additionally, since five TNF inhibitors are already FDA approved, this work also establishes a clear path for the clinical translation of localized OV against various forms of metastatic cancer.
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies were approved by, and conducted under the supervision of, the institutional animal care and use committees at the Medical University of South Carolina and the University of New Mexico Health Sciences Center.
Acknowledgments
We thank Dr Chrystal Paulos for valuable discussion throughout the development of this project. We also thank Bulent Arman Aksoy for his contribution to the bioinformatics analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MV-C, CG, and MYB performed experiments, analyzed data, and assisted in manuscript preparation. PD performed bioinformatics analysis; EB designed experiments, oversaw project, and prepared manuscript and acts as the guarantor of the work.
Funding This work was funded by grants to E.B. from: the National Insitutes of Health (National Cancer Institue - R01CA194090, and National Institute of Allergy and Infectious Disease - R21AI142387) and the American Cancer Socieity (RSG-17-047-01-MPC), as well as grants to the Hollings Cancer Center (87455-10 and P30-CA138313), and the UNM Comprehensive Cancer Center (P30-CA118100).
Competing interests EB has intellectual property rights to the virus discussed in this paper.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.