Article Text

Abstract
Background Human immune cells, including monocyte-derived macrophages, can be engineered to deliver proinflammatory cytokines, bispecific antibodies, and chimeric antigen receptors to support immune responses in different disease settings. When gene expression is regulated by constitutively active promoters, lentiviral payload gene expression is unregulated, and can result in potentially toxic quantities of proteins. Regulated delivery of lentivirally encoded proteins may allow localized or conditional therapeutic protein expression to support safe delivery of adoptively transferred, genetically modified cells with reduced capacity for systemic toxicities.
Methods In this study, we engineered human macrophages to express genes regulated by hypoxia responsive elements included in the lentiviral promoter region to drive conditional lentiviral gene expression only under hypoxic conditions. We tested transduced macrophages cultured in hypoxic conditions for the transient induced expression of reporter genes and the secreted cytokine, interleukin-12. Expression of hypoxia-regulated genes was investigated both transcriptionally and translationally, and in the presence of human tumor cells in a slice culture system. Finally, hypoxia-regulated gene expression was evaluated in a subcutaneous humanized-mouse cancer model.
Results Engineered macrophages were shown to conditionally and tranisently express lentivirally encoded gene protein products, including IL-12 in hypoxic conditions in vitro. On return to normoxic conditions, lentiviral payload expression returned to basal levels. Reporter genes under the control of hypoxia response elements were upregulated under hypoxic conditions in the presence of human colorectal carcinoma cells and in the hypoxic xenograft model of glioblastoma, suggesting utility for systemic engineered cell delivery capable of localized gene delivery in cancer.
Conclusions Macrophages engineered to express hypoxia-regulated payloads have the potential to be administered systemically and conditionally express proteins in tissues with hypoxic conditions. In contrast to immune cells that function or survive poorly in hypoxic conditions, macrophages maintain a proinflammatory phenotype that may support continued gene and protein expression when regulated by conditional hypoxia responsive elements and naturally traffic to hypoxic microenvironments, making them ideal vehicles for therapeutic payloads to hypoxic tissues, such as solid tumors. With the ability to fine-tune delivery of potent proteins in response to endogenous microenvironments, macrophage-based cellular therapies may therefore be designed for different disease settings.
- Cell Engineering
- Cytokines
- Immunotherapy
- Macrophages
- Tumor Microenvironment
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Macrophages are efficiently recruited to solid tumors, where they persist and function.
Previous work demonstrates the use of lentivirally modified macrophages as a vehicle to deliver surface and secreted therapeutic proteins that reduce tumor burden.
We explored whether hypoxia responsive elements could be used as a proof of concept to conditionally express lentivirally encoded genes in response to microenvironmental conditions.
HRE regulated gene constructs resulted in lentivirally encoded protein expression in response to hypoxic conditions in vitro and in vivo.
Return to normoxic conditions reduced lentiviral gene expression to base line, indicating conditional expression of lentiviral genes in macrophages that may be regulated by environmental changes over time.
Introduction
Immunotherapy offers a more targeted approach to cancer treatment than radiation and chemotherapies and has shown significant promise for the eradication of many types of tumors. However, recent clinical trials that aim to use cellular immunotherapies for patients with solid tumors have exposed several limitations to these approaches. Aside from persistent issues that immunotherapies encounter in many types of cancer, including neoepitope loss and an immunosuppressive tumor microenvironment (TME), immunotherapies have additional hurdles to overcome when systemically administered, such as therapy persistence and tissue penetrance. These issues lead to increases in both dosing concentration and frequency for patients, which can cause unwanted side effects and off-target toxicities. Improved targeting of cell therapies that support local, sustained therapeutic delivery would overcome these obstacles.
Rapidly dividing tumor cells consume all the cellular nutrients available within the TME to maintain their growth, and consequently induce hypoxia. Cancerous cells have adapted to the hypoxic nature of the TME by altering their metabolism; rather than using oxidative phosphorylation to generate energy, these cells generate energy using the less efficient process of aerobic glycolysis. Tumor cells have further adapted to hypoxia by upregulating expression of glucose transporters to use the limited available glucose more efficiently.1 To compete for resources and eliminate tumor cells, cellular therapies will similarly have to adapt and respond to the hypoxic conditions present in the TME. Most types of immune cells are not capable of such adaptations. For example, lack of nutrients impairs and induces exhaustion of tumor-specific endogenous T cells and chimeric antigen receptor (CAR) T cells delivered to the TME2 3 contributing to unrestricted tumor growth. In contrast, tumor-associated macrophages (TAMs) can traffic to, infiltrate, and accumulate in many types of solid tumors, suggesting that there are macrophage-intrinsic adaptations to the nutrient restricted TME that support their survival and functions.
The hypoxic TME also induces transcriptional changes in many of the resident cells including myeloid derived suppressor cells (MDSCs), regulatory T cells, and TAMs which contribute to immunosuppression and support tumor growth. When cells are exposed to hypoxia, the transcription factor HIF1α, rather than being targeted for degradation as in normoxic conditions, translocates to the nucleus to join HIF1β, forming the heterodimer HIF-1. This complex then binds to hypoxia response elements (HREs) within promoter regions of target genes.4 5 The PD-L1 proximal promoter region contains an HRE regulated by HIF-1.6 PD-L1 is upregulated by both MDSCs and TAMs in hypoxic conditions and contributes to suppression of antitumor T cell immune responses.7 HIF-1 also binds FoxP3 to promote regulatory T cell formation,8 9 and therefore, suppression of effector T cell responses.
Given the suppressive, hypoxic TME in solid tumors, we sought to develop a cellular immunotherapy that would selectively produce proinflammatory factors in response to the molecular pathways induced in the TME. Because TAMs comprise a large proportion of the cells in the TME of solid tumors, and express high levels of HIF-1, macrophages are an ideal candidate to penetrate and persist in solid tumors. We have developed a platform of genetically engineered macrophages (GEMs) to deliver proteins that support T cell responses within the TME.10 GEMs engineered to constitutively secrete IL-1211 and bispecific T cell engagers12 induced T-cell activation, local proinflammatory cytokines and tumor cell death. However high concentrations of proinflammatory IL-12 may result in toxic effects in off-target tissues,13 14 therefore selective expression of transgenes is necessary.
In this study, we engineered macrophages using a lentiviral construct that contains HREs upstream of a minimal promoter to regulate target gene expression. We show that these macrophages express target genes and resulting proteins, including a secreted payload IL-12, in response to hypoxia in vitro and in the hypoxic subcutaneous xenograft glioblastoma tumor model. Monocyte-derived macrophages that were differentiated and lentivirally transduced with these inducible constructs also appear resistant to the polarizing and anti-inflammatory conditions found in hypoxic environments. On exposure to hypoxia, hypoxia-regulated GEMs downregulated genes associated with a M2-like, procancer phenotype, and increased expression of genes associated with active T cell immune responses. Hypoxia-responsive GEMs may therefore deliver therapeutic proteins locally within the TME and retain a proinflammatory macrophage phenotype to support T cell responses.
Materials and methods
Generation of lentiviral constructs
All constructs were cloned into epHIV715 and epHIV7.210 lentiviral backbones, gifts from Dr. Michael Jensen (Seattle Children’s Research Institute). As previously described, in epHIV7 the Cytomegalovirus (CMV) promoter was replaced with Elongation Factor 1 Alpha (EF1α).10 In epHIV7.2, the EF1α promoter was replaced with a minimal EF1α (lacking the HTLV-1 domain) and the ampicillin resistance gene was replaced with kanamycin. All Gibson assembly was carried out using Hifi DNA Assembly Mix (New England Biolabs) and PCR amplification of template DNA was performed with Platinum SuperFi Green PCR (Invitrogen). Mutagenesis cloning was performed using Q5 Site Directed Mutagenesis Kit (New England Biolabs). Specific primers for Gibson assembly and mutagenesis are listed in online supplemental table 1 and resulting plasmid sequences following targeted mutagenesis are provided in online supplemental table 2.
Supplemental material
Supplemental material
epHIV7 lentiviral constructs
Luc2p (firefly luciferase, luc2, altered to contain PEST degradation sequences and a reduced half-life) was inserted into epHIV7 at BamHI and XbaI restriction sites by Gibson Assembly. Luc2p was PCR amplified from pGL4.32 (luc2P/NF-κB-RE/Hygro) (Promega). The following promoter regions were amplified by PCR: CMV Promoter (Addgene Plasmid #17 448), Ubiquitin C (Ub) (Addgene Plasmid #11 155), Phosphoglycerate Kinase 1 (PGK) (Addgene Plasmid #21 210), and phospho-Syk responsive promoter (pSYK). The pSyk promoter was designed in house to contain active promoter and enhancer regions for Syk signaling targets in human macrophages. Transgene vectors containing the specified promoters were generated using Gibson Assembly by inserting promoter regions into RruI/NheI restriction site of Luc2p epHIV7. The negative control construct contained the minimal promoter fragment from Herpes Simplex Virus Thymidine Kinase (HSV MiniTK) (Addgene Plasmid #26 731) and was incorporated into the Luc2p epHIV7 backbone by mutagenesis.
epHIV7.2 constructs
Constructs encoding truncated human CD19 (CD19t), a gift from Dr. Michael Jensen,16 eGFP:ffluc-t2a-CD19t were inserted into epHIV7.2 by gibson assembly. The CD19t-t2a-huIL12 and eGFP:ffluc-t2a-huIL12 constructs were synthesized by GeneArt and inserted into the epHIV7.2 backbone. The HRE Luciferase (Addgene Plasmid #26731), containing three repeated HREs and HSV MiniTK, was modified by mutagenesis to remove the Nhe1 restriction site in the HRE by introducing point mutation at 5115G>A and 5120C>A. The HREs and MiniTK promoter were inserted into epHIV7.2 transgene vectors by Gibson Assembly, amplifying the HRE MiniTK promoter by PCR. Backbones were digested at NheI and RruI restriction sites to replace the minimal EF1α promoter with the HRE MiniTK promoter. The MiniTK promoter was inserted into epHIV7.2 transgene vectors by mutagenesis.
Lentivirus production
293T cells (ATCC CRL-3216) were cultured in DMEM (Gibco) +10% FBS (Peak Serum)+10 mM HEPES (Gibco) +1X Glutamax (Gibco). For transfection, 293Ts were plated at 2×107 per 15 cm dish. Cells were rested for 4 hours and then each plate was transfected using the CalPhos Mammalian Transfection Kit (Clontech) and the following plasmids: pVPX, RSV-Rev, pMDL-X, pCMV-G, and the transgene vector.10 Supernatant containing virus was collected 48 hours post-transfection, filtered, and then concentrated at 108 000 x g for 90 min at 4°C. The virus pellet was then resuspended in DMEM and stored at −80°C. Concentration of lentiviral particles (LPs) was determined by detection of the virus associated lentivirus with the Quick Titer Lentivirus Kit (Cell Biolabs).
PBMC isolation
PBMCs were derived from Leukocyte Reduction System chambers obtained from healthy donors by BloodWorks Northwest. PBMCs were isolated by density gradient using Lymphoprep (Stemcell Technologies) in SepMate-50 tubes (Stemcell Technologies). Isolated PBMCs were cryopreserved in FBS (Peak Serum)+10% DMSO (Sigma).
CD14+ isolation and differentiation to macrophage
CD14+ monocytes were selected from thawed human PBMCs using the EasySep Human CD14 Positive Kit (StemCell). Monocytes were cultured in RPMI (Gibco) +10% FBS (Peak Serum)+10 ng/mL GM-CSF (R&D Systems) for 72 hours after which half the media was refreshed with addition of 10 ng/mL GM-CSF for the total volume. Seventy-two hours after the media change, macrophages were replated at a density of 53,000–66,000 cells/cm2 for transduction and downstream assays.
Macrophage transduction
To generate GEMs, on day 7 post-monocyte isolation, media was refreshed and lentivirus was added at 250 LPs or 500 LPs/cell.
Hypoxia chamber
Prior to addition to the hypoxia chamber, new media was added to the transduced macrophages. 20 mL of PBS (Gibco) was also added to the closed chamber to maintain humidity. The chamber was connected to a blood gas canister to flush out oxygenated air for 10 min at 20 L/min. After 10 min, the chamber was sealed and placed at 37°C (90% humidity, 5% CO2) for the designated incubation period.
RNA isolation
Cells were washed in PBS and lysed in RLT Buffer (Qiagen)+1% BME (Biorad) (500,000 GEMs/350 µL). RNA was isolated from cell lysates using RNeasy Mini Kit (Qiagen) per manufacturer’s instructions. RNase-Free DNase Set (Qiagen) was used for isolation of RNA for Nanostring.
Nanostring
RNA concentration was determined by Agilent TapeStation (Agilent). 25 ng RNA was added to Nanostring hybridization after normalizing using percent fragmentation. RNA was evaluated using Human Myeloid Innate Immunity Panel (Nanostring) and volumes were equilibrated with Nuclease Free Water (Ambion). Samples were hybridized with probes for 18 hours at 65°C and 30 µL of sample was loaded into nCounter Sprint Cartridge and run on nCounter Sprint. Data were analyzed using nSolver V.4.0 and Apple Numbers. Raw count data were subjected to background thresholding, two times the average negative control background. Positive and housekeeping gene normalization was determined using nSolver advanced analysis. Differential expression of data was determined by hypoxia as the predictor and transduction as a covariate.
cDNA Production and qPCR
cDNA was generated using SuperScript III First Strand Synthesis System for RT-PCR (Life Technologies) using Oligo DT to prime RNA. For qPCR, Power SYBR 2 x MM (Applied Biosciences) and Nuclease Free Water (Ambion) were added. cDNA samples were added (1:100) to the reaction and the total primer mix concentration was 0.5 µM. Primers used for amplification are listed in online supplemental table 1. Biorad QFX96 set protocol for 10 min/95°C, 15 s/95°C+1 min/60°C 40X, melt curve 55°C–95°C increment 0.5C. Cq data were analyzed on BioRad CFX Manager and Microsoft Excel. The β-actin housekeeping gene was used to normalize luciferase expression.
293T culture
293T cells (ATCC CRL-3216) were cultured in DMEM +10% FBS+10 mM HEPES +1X Glutamax. Cells were plated at 100,000 per well of a 6 well plate and rested for 24 hours. 293Ts were transfected with HRE-Luciferase (Addgene Plasmid #26731) using MirusTransIT-LT1 (Mirus) and incubated for 24 hours. Transfected 293Ts were then placed into the hypoxia chamber for 15 or 24 hours. Cells were lifted and plated in a 96 well plate at 10,000 cells/well in 200 uL of media for luciferase assay. 20 µL of D-Luciferin (Perkin Elmer) at 1.4 mg/mL in PBS was added to each well. Cells were incubated at 37°C for 10 min, and luminescence was determined with the Perkin Elmer Victor 3.
Dimethyloxalylglycine and roxadustat assays
Dimethyloxalylglycine (DMOG, Sigma) and Roxadustat (Cayman Chemical) were resuspended in DMSO (Sigma) and diluted to specified concentrations in RPMI +10% FBS. Drugs were added to GEMs plated in 96 well plates and incubated for 6 hours prior to running luciferase assays.
Macrophage luciferase assay
GEMs were plated in white bottom 96 well plates at 10,000 cells/well. On day 7 post-transduction, media was replaced with 200 µL of RP10 with the addition of 20 µL of D-Luciferin (Perkin Elmer) at 1.4 mg/mL in PBS. Cells were incubated in 37°C incubator for 10 min, and luminescence was determined with the Perkin Elmer Victor 3.
Western blot
Cells were lysed in RIPA Buffer (ThermoFisher)+HALT Protease/Phosphatase Inhibitor (Thermofisher)+EDTA (500,000 macrophages in 50 µL of lysis buffer). Samples (20 µL of lysis sample +10 µL of Lammeli Buffer (Invitrogen) +1 µL BMe (BioRad)) were heated at 95°C for 10 min. Samples were run on a 4%–12% Bis Tris (Invitrogen) gel and transferred to a nitrocellulose membrane in NuPage transfer buffer (Invitrogen) +10% MeOH on ice. Membrane was blocked with 5% Milk in TBST and probed with Mouse anti-β-actin (CST, 8H10D10, 1:1000) and Goat anti-luciferase (Abcam, polyclonal, 1:1000) followed by secondary antibodies, Donkey anti-Goat 800 (Licor), and Goat Anti-Mouse 680 (Licor, 1:5000). Developed blots were analyzed using an Odyssey Infrared Imager. For quantification, β-actin was used to normalize the signal for luciferase expression.
Bioplex
Twenty-four hours post-incubation in the hypoxia chamber, media from GEMs was collected, spun down at 1000 x g at 4°C, and frozen at −80°C. IL-12 concentrations were determined by Human IL12p70 bioplex (Biorad) run with standard reagents following the manufacturer’s protocol. Samples were run on the Luminex instrument and washes were performed with an automatic plate washer. Samples that were expected to saturate the standard curve were diluted in culture medium 1:3 or 1:4.
Flow cytometry
Macrophages were lifted using TrypLE Express (Gibco) and cell scrapers. Once detached, cells were incubated in Human Fc block (Becton, Dickson and Company) at 0.05 mg/mL for 10 min at room temperature and diluted UV Live/Dead stain (Invitrogen, 1:500) was added for 20 min at 4°C. Cells were fixed using 2% paraformaldehyde (PFA) and run on BD LSR Fortesssa using BD FACDiva Software. Data were analyzed using FlowJo V.10.
GEM microscopy
GEMs were lifted using 0.25% Trypsin-EDTA (Gibco) and stained with 1 µM Celltracker Orange (Invitrogen) for 30 min at 37°C. Stained GEMs were then plated on chamber slides and rested for 4 hours to allow attachment. After GEMs were removed from the hypoxia chamber, cells were washed with PBS and fixed in 4% PFA on ice for 10 min. Fixed GEMs were then permeabilized with 0.1% Triton (Sigma) for 10 min at room temperature. Cells were incubated with polyclonal Chicken anti-GFP antibody (Novus) diluted 1:300 in blocking buffer (0.2% BSA + Goat Serum) for 1 hour at room temperature to detect GFP. Following washing, cells were incubated with Goat anti-Chicken secondary Alexa Fluor 488 (Fisher) diluted 1:500 in blocking buffer (0.2% BSA +Goat Serum) for 1 hour at room temperature. Cells were incubated with Hoescht Stain (Invitrogen) diluted 1:500 for 10 min at room temperature. Chambers were removed and samples were mounted with a cover slip and ProLong Gold mounting media (Invitrogen). Slides were Imaged on Nuance Microscope (Leica) and analyzed using Nuance software V.3.0.2.
Human slice culture
Patients were consented under FHCRC/University of Washington IRB protocol #1765. Culture of freshly resected human tumors was carried out as previously described.17 18 Immediately following surgical resection, 6 mm punch biopsies were placed in Cold Storage solution (Bridge to Life Ltd), and 250 µm axial slices were cut using a Vibratome (Leica Biosystems). Slices were placed atop a permeable PTFE membrane with 0.4 mm pores (Millicell; MilliporeSigma) in slice culture media at 37°C. The following day, 100,000 GEMs were added to each slice and cultures were placed in an incubator under hypoxic conditions (1% O2) at 37°C. Tumor slices were then removed from their wells and cultured in a 48-well plate containing EpCAM-Alexa 647 in 500 µL slice culture media for 3 hours on a rocker in a humidified, normoxic 37°C incubation chamber. 15 min prior to confocal imaging, Hoechst 33 342 (ImmunoChemistry Technologies) was added to the well. Slices were then transferred to µ-Slide 8-well clear bottom imaging slides (Ibidi) and imaged using a Leica SP8X confocal microscope with a 10×0.4 NA dry objective. Excitation was provided by a tunable White Light Laser system (499 nm, 577 nm, and 653 nm excitations) and UV laser (405 nm) at standard intensities. Three to four 20 µM z-stack series were obtained for each slice and maximum intensity projections were generated from acquired z-stacks. Hoechst+EpCAM+ and hypoxia-induced GFP+ cells were counted with ImageJ software by an investigator blinded to experimental conditions.
Mouse experiments
Mouse experiments were performed in accordance with Seattle Children’s Research Institute IACUC #13897. 10-week-old NSG male mice were purchased from Jackson Laboratories and injected with 1:1 ratio of 1×106 U87s and matrigel-matrix-gel (Corning) in 200 µL subcutaneously in the flank. Nineteen days after engraftment, 1×106 GEMs expressing CD19t, MiniTK eGFP:ffluc or HRE MiniTK eGFP:ffluc in 50 µL PBS were injected intratumorally. GEMs were detected using bioluminescent imaging with IVIS Spectrum (Caliper Life Sciences) 15 min after subcutaneous injection of D-Luciferin (Perkin Elmer). Bioluminescent images were analyzed using Living Image Software (Caliper Life Sciences). Tumor volume was monitored by caliper measurements, and mice were euthanized when tumor volume reached 2000mm3 or weight decreased by 20%. Tumor volumes were calculated using the following formula: volume= (width2 × length/2).
Results
Constitutive expression of high concentrations of proinflammatory proteins following systemic administration of cellular immunotherapies can lead to destruction of healthy tissue and organs.13 19 To minimize off-target effects of proinflammatory payloads from engineered macrophages, we determined which constitutive promoters resulted in lower amounts of target protein. We transduced human monocyte-derived macrophages with lentiviral constructs encoding the short half-life luciferase (Luc2P) under the control of EF1α, Ubiquitin, CMV, PGK, a phosphorylated Syk responsive promoter (pSyk) and MiniTK (figure 1A). We found that MiniTK, CMV, and pSyk resulted in the lowest amounts of luciferase (figure 1B). These alternative, lower yielding promoters could potentially limit concentrations of payloads delivered by genetically engineered cells and thereby potentially decrease adverse events compared with the standard EF1α promoter.
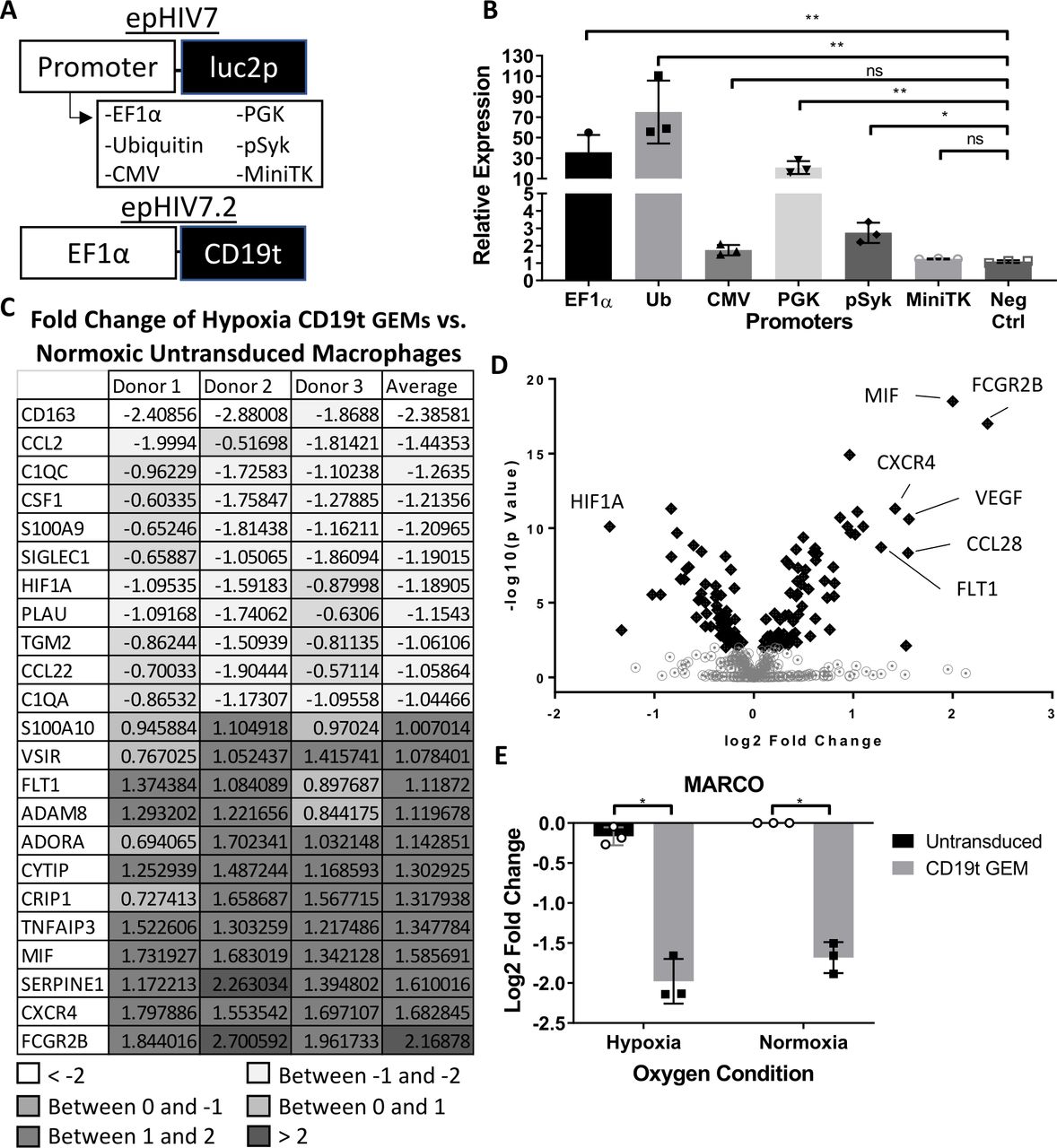
GEMs modulated by mini thymidine promoter are low yielding and express proinflammatory genes under hypoxic conditions. (A) Transfer gene models of (top) constitutive promoters (EF1α, ubiquitin, CMV, PGK, pSyk and MiniTK) modulating firefly luciferase with PEST degradation sequence, luc2p, in the epHIV7 plasmid backbone and (bottom) minimal EF1α promoter modulating CD19t (truncated CD19) in epHIV7.2 plasmid backbone. (B) Luciferase assay results of GEMs transduced with luc2p epHIV7 with different promoters: EF1a, Ubiquitin, PGK, CMV, pSYK, or MiniTK. CD19t epHIV7.2 GEM was a negative control. (n=3 independent donors, 6 technical replicates per donor). Relative expression of each GEM group luciferase activity compared to an untransduced, control macrophages. (C) Nanostring Log2 fold change of hypoxic CD19t GEMs relative to untransduced macrophages in normoxia. Genes listed are selected based on average log2 fold change greater than 1 or less than −1 (n=3 independent donors). (D) Volcano plot of all GEMs and untransduced macrophages exposed to hypoxia compared to normoxia. Gene log2 fold change graphed against the –log10 of the p value. Genes with a p-value < 0.01 are shown with black diamonds. (E) Nanostring analysis of the gene MARCO with log2 fold change of CD19t GEMs relative to normoxic, untransduced macrophages (n=3 independent donors). Paired t-test, ns: p>0.005, *p<0.05, **p<001. CMV, cytomegalovirus; EF1α, elongation factor 1 alpha; GEMs, genetically engineered macrophages; ns, not significant.
Cells engineered with a lentiviral construct driven by a hypoxia-response promoter could further regulate delivery of therapeutic proteins by tumor-specific delivery within the hypoxic TME. Macrophages within the hypoxic TME are known to transition into TAMs that support tumor growth and survival, therefore, we investigated the transcriptional changes in GEMs under hypoxia. We transduced human monocyte-derived macrophages with a lentivirus encoding a non-signaling truncated human CD19 (CD19t) tag under a constitutive EF1a promoter (figure 1A) and placed the engineered cells in 1% oxygen conditions or normal oxygen levels for 24 hours. Following incubation, we found that many of the over 700 genes tested remained unchanged. Unexpectedly, we discovered decreased expression of genes associated with the M2, protumor phenotype including CD163, CSF1, S100A9, CCL22, and PLAU20–23 (figure 1C) following exposure to hypoxia. Genes associated with complement activation (C1QC and C1QA), which has also been shown to promote tumor growth in settings of chronic inflammation,24 were also decreased (figure 1C). Antitumor, proinflammatory genes associated with T cell activation (VSIR, ADORA3, and MIF) and phagocytosis (FCGR2B) were increased (figure 1C). As expected, the angiogenesis-related gene, FLT1, was upregulated, while HIF1A decreased (figure 1C). As shown by other groups, the decrease in HIF1A may be due to transcriptional regulation under prolonged hypoxic conditions.25 26 To determine the most variable genes and transcriptional profile of hypoxia-exposed GEMs, all hypoxia-treated samples were analyzed in comparison to untransduced, control macrophages under normoxia. Many similar transcriptional changes were detected for untransduced macrophages under hypoxic conditions (online supplemental figure 1A) and fewer changes were detected in lentivirally transduced macrophages (online supplemental figure 1B) suggesting that hypoxia is the main factor inducing these transcriptional changes. Hypoxia-specific genes including VEGFA, FLT1, and HIF1A (figure 1D) were among the top differentially regulated genes. The scavenger receptor MARCO, expressed on TAMs and linked to poor prognosis in breast cancer,27 28 was the most downregulated gene in transduced macrophages (figure 1E). These data suggest that GEMs may maintain a stable, proinflammatory phenotype under the hypoxic conditions present in the tumor. Together, our results show that GEMs do not polarize to protumor phenotypes following exposure to hypoxia and instead may support active antitumor immune responses.
Supplemental material
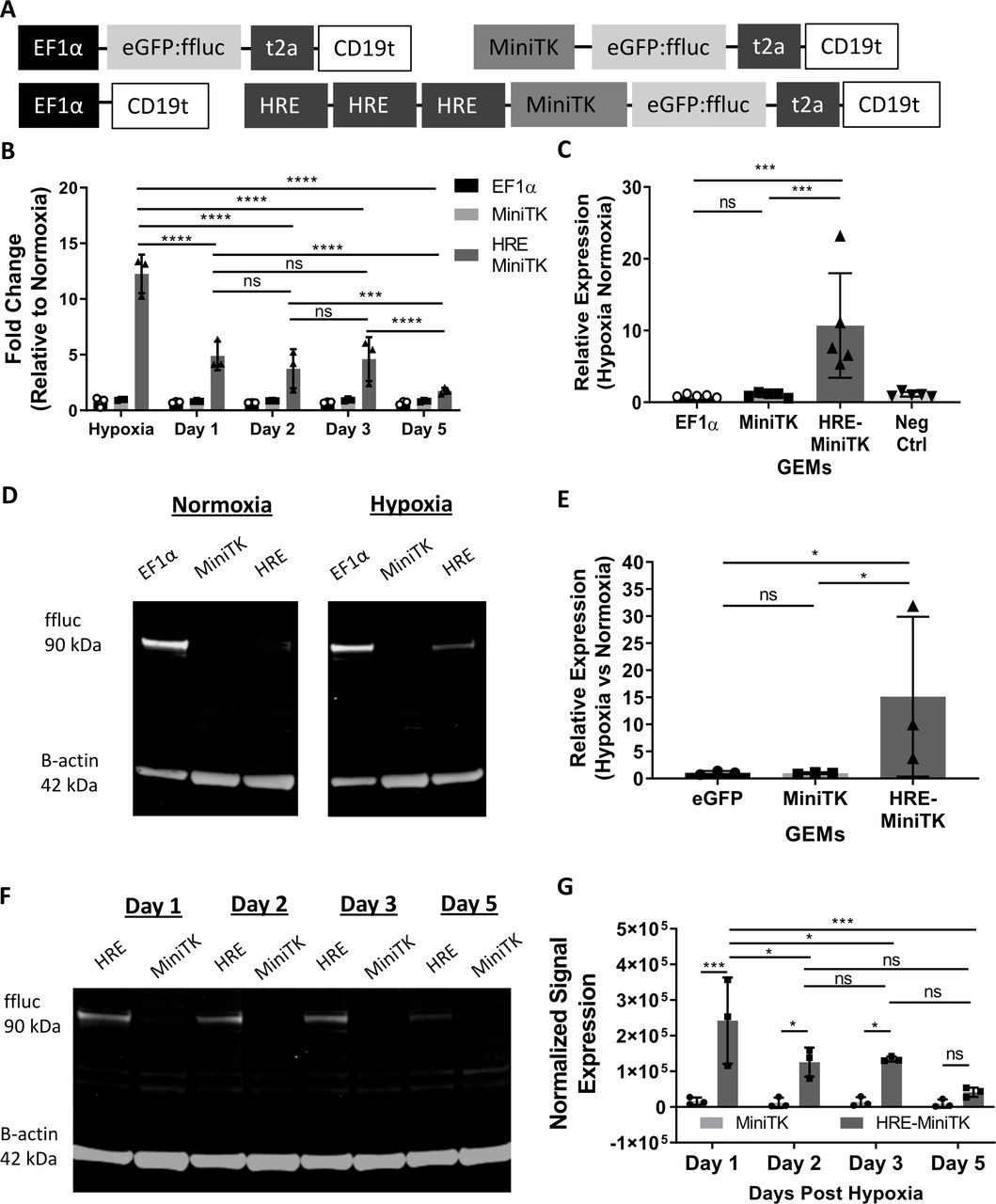
We then determined if macrophages transduced with lentiviruses encoding HREs upstream of the MiniTK promoter had hypoxia-specific regulation of reporter genes. Using transfected 293T cells as a model cellular system, we determined that 293T cells transfected with a luciferase plasmid regulated by HRE-MiniTK only slightly upregulated luciferase activity following 15 hours of exposure to hypoxia (online supplemental figure 2A), but following 20 hours of hypoxia there was an increase in luciferase activity with hypoxia compared with cells under normoxic conditions (online supplemental figure 2B). In all subsequent experiments, macrophages were placed under hypoxia for 24 hours prior to measuring experimental results. Macrophages were transduced with a lentivirus encoding eGFP:ffluc with three HREs upstream of MiniTK. CD19t regulated by the constitutive EF1α promoter (EF1α -CD19t) and a lentivirus encoding eGFP:ffluc under a MiniTK promotor lacking HREs were negative controls (figure 2A). GEMs transduced with the lentivirus encoding EF1α-regulated eGFP:ffluc constitutively express eGFP:ffluc and were a positive control (figure 2A). GEMs transduced with the HRE-regulated construct showed increased luciferase mRNA following 24 hours of hypoxic treatment compared with normoxia (figure 2B). Increased luciferase mRNA under hypoxic conditions was not detected with MiniTK-regulated or EF1α-regulated controls (figure 2B). Following removal from hypoxic conditions, the fold increase in expression of luciferase transcription relative to GEMs transduced with the HRE-regulated luciferase lentivirus in normoxia decreased over the course of 5 days (figure 2B) indicating that on resolution of hypoxic conditions, the HRE-regulated promoter had decreased activity. Luciferase activity measured at the protein level and determined by a luminescence assay led to similar results (figure 2C). The HRE-regulated construct had an average of 10.7-fold increase in luminescence compared with HRE-MiniTK GEMs under normoxic conditions, while MiniTK-regulated or EF1α-regulated controls had similar luminescence in each oxygen condition (figure 2C). To directly detect luciferase protein, we measured luciferase from the cell lysates of GEMs with an anti-luciferase antibody (figure 2D-G). Consistent with luciferase assay results, we found that expression of luciferase was increased under hypoxic conditions in the macrophages transduced with the HRE-regulated luciferase (HRE, figure 2D, top right) relative to normoxic conditions (HRE, figure 2D, top left), while control constructs (EF1α and MiniTK, figure 2D,E) had similar luciferase expression in both oxygen conditions. To determine the kinetics of luciferase protein expression following hypoxia treatment, we measured luciferase expression over time relative to the MiniTK control at similar time points. Luciferase protein decreased over time following initial hypoxia treatment in the HRE-regulated GEMs, while macrophages transduced with the eGFP:ffluc construct regulated by MiniTK had stable luciferase expression over similar time points (figure 2F,G). Hypoxia-regulated control of luciferase constructs in GEMs was further verified by DMOG and Roxadustat, drugs that mimic the molecular pathways induced in cells during conditions of hypoxia by inhibiting the degradation of the transcription factor HIF1α (online supplemental figure 3A, B). These results suggest that GEMs encoded with an HRE-regulated eGFP:ffluc specifically upregulate luciferase transcriptionally and translationally under hypoxic conditions, and expression decreases over time once oxygen conditions returns to basal levels.

Hypoxia induces increased transfer gene expression in GEMs transduced with HRE MiniTK eGFP:ffluc-t2a-CD19t transcriptionally and translationally and expression is reduced following removal of GEMs from hypoxia. (A) Transfer gene design of eGFP:ffluc-t2a-CD19t modulated by minimal EF1α promoter, MiniTK promoter and HRE-MiniTK (3x HRE enhancers + MiniTK) promoter. (B–G) Macrophages are transduced at 500LP/cell. (B) qPCR of GEMs exposed to hypoxia and 1, 2, 3, and 5 days post-exposure to hypoxia. Luciferase ddCq calculated comparing β-actin expression and fold change calculated relative to GEMs incubated in normoxia (n=3 independent donors). (C) Luciferase activity of GEMs exposed to hypoxia for 24 hours. Counts per second (CPS) values are represented as relative expression compared to GEMs incubated in normoxia. Bars represent mean ± SD (n=3 independent donors run in 6 technical replicates). (D) Representative western blot of luciferase of eGFP:ffluc-t2a-CD19t GEMs modulated by EF1α, MiniTK and HRE-MiniTK GEMs exposed to hypoxia and normoxia. (E) Western blot band signal analysis of GEMs (n=3 independent experiments). (F-G) Western blot of eGFP:ffluc-t2a-CD19t GEMs modulated by MiniTK and HRE-MiniTK GEMs recovering 1, 2, 3, and 5 days post-exposure to 24 hours in hypoxia. (G) Western blot band signal analysis of GEMs (n=3 independent experiments). Band signal quantified by Image Studio software, normalized to β-actin and represented as (E) relative expression compared to its respective normoxic control or (G) normalized signal by β-actin. (B,G) Two-way analysis of variance with Tukey multiple comparison test: ns>0.05, *p<0.05, **p<0.01, ***p<0.001,****p<0.0001. (C, E) Paired t-test: ns>0.05, *p<0.05,**p<0.01, ***p<0.001. GEMs, genetically engineered macrophages; HRE, hypoxia response element.
The ability to regulate expression of proinflammatory therapeutic proteins by factors present within the TME offers an alternative to traditional, constitutively expressed proinflammatory genes. The proinflammatory cytokine IL-12 supports both innate and adaptive immune responses and has shown to be an effective treatment for many cancer types29 30; however, systemic delivery of large amounts of IL-12 can lead to unintended consequences outside of the tumor.13 14 IL-12 lentiviral constructs encoded with an HRE-regulated promoter may allow for tumor-specific, conditional expression of IL-12. Therefore, we designed and tested an HRE-regulated IL-12 construct (HRE-MiniTK-CD19t-T2A-hIL12p40p35, encoding the fusion of IL-12 subunits p40 and p35) under hypoxic conditions (figure 3A–C). GEMs encoding the HRE-regulated IL-12 lentiviral construct showed that, across three different donors, IL-12 mRNA expression was upregulated on average 4.3-log2 fold relative to normoxic, untransduced macrophages, while CD19t and IL-12 control GEMs did not change under similar comparison (figure 3B). Similar to results with GEMs encoding the HRE-regulated luciferase, macrophages transduced with the HRE-regulated IL-12 had decreased IL-12 secretion following resolution of hypoxia (figure 3C). IL-12 expression from the HRE-MiniTK-eGFP:ffluc-t2a-huIL-12p40p35 (figure 3D) was also induced under hypoxic conditions at the protein level (figure 3E). Increased expression of GFP encoded as a tag within the same IL-12 construct (HRE-MiniTK-eGFP:ffluc-t2a-huIL12p40p35) (figure 3D) following hypoxic conditions and relative to GEMs under normal oxygen conditions was also detected, further confirming hypoxia-regulated expression (figure 3F,G). Collectively these results show that HREs encoded within lentiviral constructs and used to generate GEMs allow for hypoxia-regulated expression of the therapeutic target IL-12.

IL12 secretion increases in HRE MiniTK CD19t-t2a-huIL12p40p35 and HRE MiniTK eGFP:ffluct2a-huIL12p40p35 GEMs following hypoxic conditions. (A) Transfer gene design of CD19t and CD19t-t2ahuIL12p40p35 modulated by EF1α, MiniTK, and HRE-MiniTK. (B) Nanostring gene expression of CD19t-t2a-huIL12p40p35 GEMs (transduced at 500LP/macrophage) in hypoxia represented as log2 fold change relative to normoxic GEM control (n=3 independent donors). (C) Production of IL-12 by CD19t-t2a-huIL12p40p35 GEMs (transduced at 250LP/macrophage) after incubation in hypoxia, normoxia, 1, 2, 3, 4, 5, 7, 14, and 21 days post-hypoxia exposure. 24 hour collection of supernatant measured by huIL12p70 bioplex. Bars represent mean ± SD (n=3 independent donors, 2 technical replicates). (D) Transfer gene design of eGFP:ffluc-t2a-huIL12p40p35 modulated by minimal EF1α, MiniTK, and HRE-MiniTK. (E) Production of IL-12 by eGFP:ffluc-t2a-huIL12p40p35 GEMs (transduced at 250LP/macrophage) after 24 hour incubation in hypoxia or normoxia measured by huIL12p70 bioplex of collected supernatant (n=3 independent donors, 2 technical replicates). (F) Representative flow plots of GEMs in hypoxia and normoxia. Zebra plots shown as GFP by FSC-H. (G) % GFP expression of eGFP:ffluc-t2a-huIL12p40p35 GEMs by flow cytometry in hypoxia and normoxia (n=3 independent donors). (B, G) One-way analysis of variance and Tukey multiple t-test: ns>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (C, E) Two-way analysis of variance (ANOVA) and Tukey multiple t-test: ns>0.05, *p<0.05, **p<0.01, ***p<0.001. FSC-H, Forward scatter; GFP, green fluorescent protein; EF1α, elongation factor 1 alpha; GEM, genetically engineered macrophage; HRE, hypoxia response element; ns, not significant.
Hypoxia-regulated expression of proteins by human macrophages transduced with HRE-regulated, lentivirally encoded genes was further validated and visualized by microscopy. Cell tracker orange labeled GEMs expressing HRE MiniTK eGFP:ffluc-t2a-CD19t were placed under hypoxic conditions and then induction of GFP was evaluated on day 7 post-transduction following 24 hours of exposure to hypoxia with an anti-GFP antibody (figure 4A). GEMs with the EF1α-eGFP:ffluc construct had similar percentages of GFP positive macrophages in both normoxia and hypoxia conditions (figure 4A,B: top panel), while those with the HRE-MiniTK-eGFP:ffluc lentiviral construct only had GFP positive cells following incubation in hypoxic conditions (figure 4A,C: bottom panel). GEMs encoding the MiniTK promoter did not have many GFP+ GEMs in either condition (figure 4A,C: middle panel). Hypoxia-regulated expression of target genes by GEMs was also investigated in the presence of patient tumor cells. Precision-cut 250 µm slices of resected colorectal carcinoma liver metastasis tumor were cocultured with GEMs on day 17 following transduction and placed in hypoxic conditions for 24 hours (figure 4D). In the slice culture, GEMs expressing GFP under the control of the HRE-regulated MiniTK promoter had more GFP positive macrophages on average per image compared with both GEMs with GFP under the control of the MiniTK promoter and also those encoding GFP under the control of the constitutive EF1α promoter (figure 4E–H). Thus, hypoxia-regulated expression of target proteins by GEMs is detected by microscopy in a model TME human slice culture system.

HRE MiniTK eGFP:ffluc-t2a-CD19t GEMs increased GFP expression when cultured under hypoxic conditions as captured by microscopy in vitro and in human colorectal cancer liver metastasis slice coculture. (A) Representative Nuance microscopy images of eGFP:ffluc-t2a-CD19t GEMs modulated by minimal EF1α, MiniTK, and HRE MiniTK stained with CellTracker Orange dye for 30 min then incubated 24 hours in hypoxia. Images shown are GEMs stained at 1µM CellTracker Orange, Hoechst, and chicken anti-GFP antibody. (B) %GFP/Hoechst positive EF1α GEMs (n=12 field/group, 40X) (C) %GFP/Hoechst positive MiniTK and HRE MiniTK GEMs (n=12 fields/group, 40X). (D) Slice coculture experimental design. The 250 µm thick slices from colorectal carcinoma resection were cultured with 1x 105 EF1α, MiniTK, or HRE MiniTK eGFP:ffluc-t2a-CD19t GEMs. (E–G) Representative microscopy images of colorectal slices containing GEMs. Tumor slices stained with EpCAM-Alexa 647 (Red) and Hoechst 33342 (Blue). GEMs express GFP (green). Representative images for (E) EF1α (F) MiniTK (G) HRE MiniTK GEMs are shown. (H) Percentage of GFP+ cells quantified as %GFP/EpCAM positive cells (n=3–5 high-power fields/group, (10X) Bars represent mean±SD). Two-way analysis of variance and Tukey multiple comparison t-test: ns>0.05, ****p<0.0001. EpCAM, epithelial cell adhesion molecule; GFP, green fluorescent protein; PBMC, peripheral blood mononuclear cell; GM-CSF, granulocyte macrophage colony stimulating factor; EF1α, elongation factor 1 alpha; GEMs, genetically engineered macrophages; HRE, hypoxia response elements; ns, not signficant.
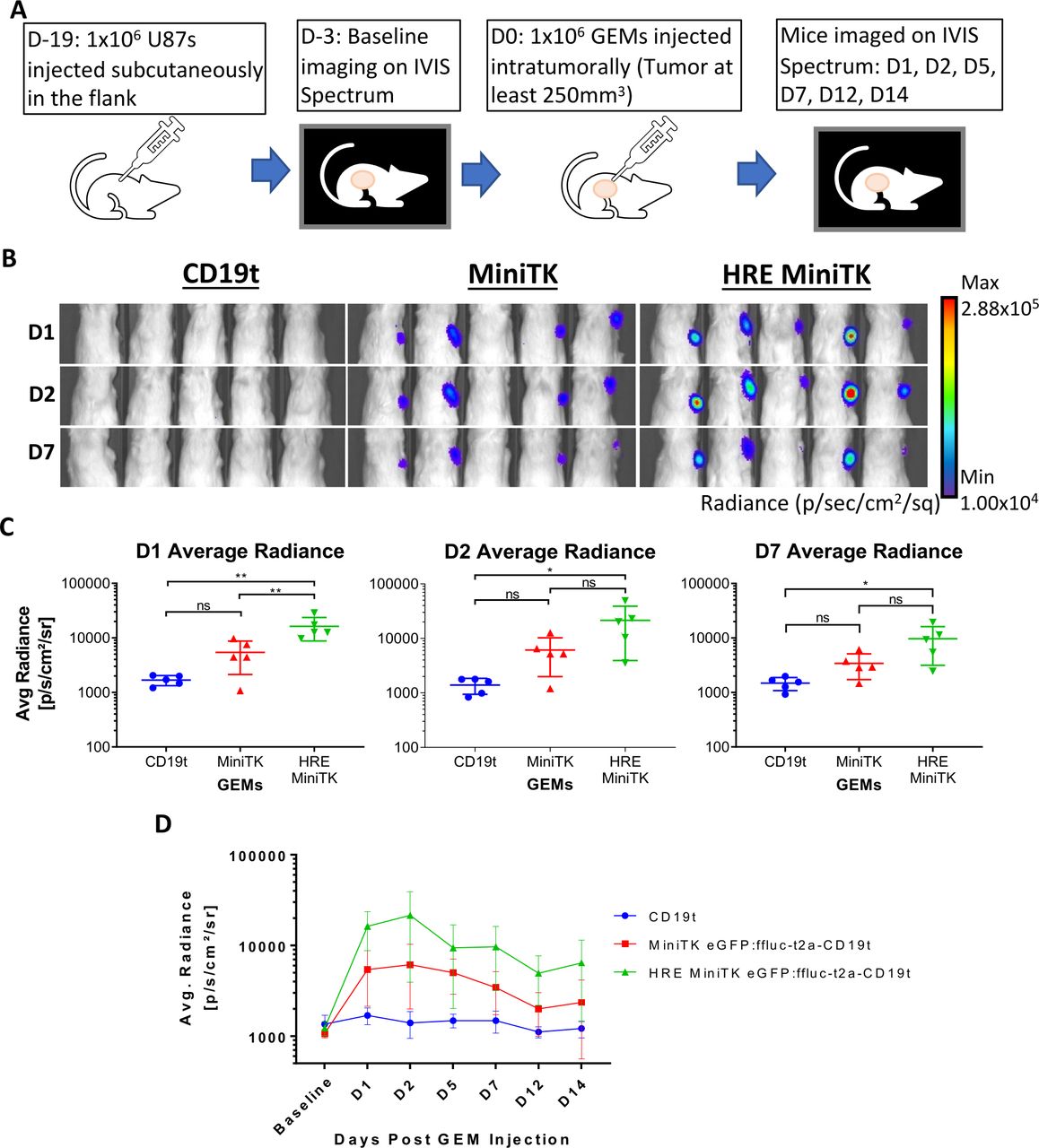
Finally, the HRE-MiniTK GEMs were tested in the hypoxic xenograft model of human glioblastoma. Mice were injected subcutaneously with 1 million U87 glioma cells in their flank and then 18 days later mice received an intratumoral injection of 1 million GEMs expressing either MiniTK-regulated or HRE-MiniTK-regulated luciferase (figure 5A). Luminescent images of mice at day 1, day 2, and day 7 post-GEM injection were analyzed measuring average radiance (p/s/cm2/sr) (figure 5B–D). Luminescent signal in the HRE-MiniTK eGFP:ffluc-t2a-CD19t mice was significantly greater (p<0.01) at day 1 than those mice that received the MiniTK eGFP:ffluc-t2a-CD19t GEMs and compared with CD19t controls on days 1, 2, and 7 (figure 5B,C). These results suggest that in addition to in vitro regulation of gene delivery by incubation in 1% oxygen conditions, GEMs can also be induced to deliver transgenes within hypoxic glioma tumors in vivo. Therefore, we have established a model in which GEMs can be designed to respond to tumor-intrinsic factors, allowing for tighter and tumor-specific delivery of different payloads.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HRE MiniTK eGFP:ffluc-t2a-CD19t GEMs increase luciferase expression in hypoxic subcutaneous glioma model. (A–D) NSG Mice with subcutaneous U87-MG tumor were injected intratumorally with 1 x 106 HRE MiniTK eGFP:ffluc-t2a-CD19t, MiniTK eGFP:ffluc-t2a-CD19t, or CD19t GEMs (n=5 per group). For imaging, mice are injected subcutaneously with D-luciferin and imaged for luciferase expression by IVIS Imager 15 min post D-luciferin injection. (A) In vivo schematic and timeline. (B) Representative images of luminescent signal in mice from day 1 (D1), D2, and D7 post-GEM injection. (C) Average radiance (p/s/cm²/sr) measured at D1, D2, and D7 post-GEM injection from left to right. Bars represent mean±SD. One-way analysis of variance and Tukey multiple comparison t-test: ns>0.05, *p<0.05, **p<0.01 (D) Average radiance (p/s/cm²/sr) is shown for each mouse group that received GEMs at baseline, day 1 (D1), D2, D5, D7, D12, and D14 post-GEM injection. (C, D) Results for mice receiving CD19t GEMs are shown in blue, MiniTK GEMs are shown in red, and HRE MiniTK GEMs are shown in green. GEMs, genetically engineered macrophages; HRE, hypoxia response element.
Discussion
In this study, human monocyte-derived macrophages were engineered to conditionally express proteins in the hypoxic environment characteristic of solid tumors. Hypoxia-restricted delivery of therapeutic payloads by macrophages, which are known to traffic to and persist within solid tumor sites,31 32 may help to eliminate the off-target toxicities of potent antitumor factors compared with traditionally, constitutively delivered immunotherapies.
Hypoxia has impeded many cancer treatments. Hypoxic tumor signatures and stabilization of HIF-1α in cancer patient samples have shown prognostic value and negatively correlates with patient survival.33 34 Hypoxic cells are less sensitive to radiation35 and hypoxia also limits both endogenous and engineered T and NK cell responses.3 36 37 Hypoxia-targeting drugs,38–40 CAR T cells,41 and oxygen treatment42 are all being evaluated for antitumor effects. Rather than specifically targeting hypoxia, we have generated an engineered cell that persists within the hypoxic solid TME10 and capitalizes on the active hypoxia signaling pathways within the TME to deliver treatment and aid in tumor elimination.
Within the heterologous TME immunosuppressive TAMs have been shown to accumulate HIF-1, localize to regions of hypoxia, and support tumor growth.43–45 Therefore, some studies have investigated targeting macrophages for elimination or repolarization to a proinflammatory phenotype.46 47 In this study, we found that despite a 24-hour incubation in hypoxic conditions, GEMs did not polarize to an immunosuppressive phenotype, but rather increased genes associated with proinflammatory pathways and active T cell responses (figure 1). Future studies will investigate whether GEMs in the presence of hypoxic tumor cells or exposed to hypoxia for longer durations transition to a more immunosuppressive phenotype, despite being engineered to secrete proinflammatory cytokines. Macrophages engineered to express CARs have been shown to aid in tumor elimination and commit to a proinflammatory phenotype both in vivo and in vitro,48 and CAR macrophages are currently being evaluated in clinical trials for the treatment of solid tumors. Therefore, macrophages engineered to conditionally produce chimeric receptors, proinflammatory cytokines, or tumor-specific antibodies may help to maintain a proinflammatory phenotype within the TME and support local antitumor immune responses.
Hypoxia-regulated cellular immunotherapies have the potential to specifically deliver potent treatments within solid tumors. IL-12 has shown some efficacy when used as a monotherapy in clinical trials,49 but systemic delivery has resulted in adverse events.13 50 We previously demonstrated that intravenously injected GEMs traffic to and are enriched in solid tumor; however, they initially transit through the lungs, thereby providing an opportunity for off-target effects.11 In this study, we show that IL-12 can be delivered locally in a hypoxic environment by GEMs, and IL-12 production resolves following transfer to normal oxygen conditions (figure 3). Furthermore, our in vivo studies show that GEMs delivered to a subcutaneous tumor can selectively express luciferase under hypoxic conditions (figure 5). The ability to regulate delivery of IL-12 may help to prevent the damage to healthy tissue outside the TME which limited results in previous clinical trials.
Hypoxia-specific production of a therapeutic payload by engineered cells may allow for expression of antitumor factors in response to the tumor-intrinsic suppressive signaling pathways within solid tumors. Expression of antitumor factors by GEMs may be further amplified by combining active tumor-specific promoters with engineered surface receptors. In these systems, activation of a tumor-specific promoter could result in the production of receptors designed to initiate proinflammatory signaling pathways on binding tumor-specific ligands or soluble proteins. Since many different tumor types share similar suppression mechanisms, macrophages engineered to respond to common inhibitory factors may be effective in a wide variety of tumor types.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by FHCRC/University of Washington IRB protocol #1765.
Acknowledgments
The authors thank Yoshinobu Koguchi and Katherine Brempelis for thoughtful manuscript review; the Jensen lab for gift of cell lines, lentiviral backbones, and advice with methods associated with development of subcutaneous mouse studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @LiebermanNicole, @pancsurg
HKC and JLG contributed equally.
Contributors HC and JLG designed and performed studies, analyzed and interpreted data. HC generated figures and drafted methods. JLG drafted manuscript text. LRM performed experiments, analyzed data, and supported mouse studies. KPL performed human slice culture experiments and analyzed and interpreted associated data. TNM analyzed data from human slice culture imaging experiments. NAPL designed luciferase experiments with different promoter constructs. AD generated human PBMCs used in all studies. CAC conceived the idea, advised on study design and manuscript preparation. All authors read and approved final manuscript. The Guarantor is CAC.
Funding This work was supported by Steven Higgins Brain Tumor Fund, the Aldarra Foundation, and Stand Up to Cancer.
Competing interests CAC is an inventor on a patent US20170087185A1, Genetic engineering of macrophages for immunotherapy. CAC receives research support and is on the scientific advisory board for BlueRock Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.