Article Text

Abstract
Background High activity of poly(ADP-ribose) polymerase-1 (PARP1) in non-small cell lung cancer (NSCLC) cells leads to an increase in immunohistochemically detectable PAR, correlating with poor prognosis in patients with NSCLC, as well as reduced tumor infiltration by cytotoxic T lymphocytes (CTLs). Intrigued by this observation, we decided to determine whether PARP1 activity in NSCLC cells may cause an alteration of anticancer immunosurveillance.
Methods Continuous culture of mouse NSCLC cells in the presence of cisplatin led to the generation of cisplatin-resistant PARhigh clones. As compared with their parental controls, such PARhigh cells formed tumors that were less infiltrated by CTLs when they were injected into immunocompetent mice, suggesting a causative link between high PARP1 activity and compromised immunosurveillance. To confirm this cause-and-effect relationship, we used CRISPR/Cas9 technology to knock out PARP1 in two PARhigh NSCLC mouse cell lines (Lewis lung cancer [LLC] and tissue culture number one [TC1]), showing that the removal of PARP1 indeed restored cisplatin-induced cell death responses.
Results PARP1 knockout (PARP1KO) cells became largely resistant to the PARP inhibitor niraparib, meaning that they exhibited less cell death induction, reduced DNA damage response, attenuated metabolic shifts and no induction of PD-L1 and MHC class-I molecules that may affect their immunogenicity. PARhigh tumors implanted in mice responded to niraparib irrespective of the presence or absence of T lymphocytes, suggesting that cancer cell-autonomous effects of niraparib dominate over its possible immunomodulatory action. While PARhigh NSCLC mouse cell lines proliferated similarly in immunocompetent and T cell-deficient mice, PARP1KO cells were strongly affected by the presence of T cells. PARP1KO LLC tumors grew more quickly in immunodeficient than in immunocompetent mice, and PARP1KO TC1 cells could only form tumors in T cell-deficient mice, not in immunocompetent controls. Importantly, as compared with PARhigh controls, the PARP1KO LLC tumors exhibited signs of T cell activation in the immune infiltrate such as higher inducible costimulator (ICOS) expression and lower PD-1 expression on CTLs.
Conclusions These results prove at the genetic level that PARP1 activity within malignant cells modulates the tumor microenvironment.
- Tumor Microenvironment
- Lung Neoplasms
- Lymphocytes, Tumor-Infiltrating
- Immunologic Surveillance
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Poly(ADP-ribose) polymerase-1 (PARP1) is activated upon cisplatin-mediated DNA damage, and its hyperactivation mediates chemotherapy resistance in non -small cell lung cancer (NSCLC). Although high PARP1 activity is associated to poor prognosis of NSCLC, the impact of PARP1 on immunosurveillance has been elusive.
WHAT THIS STUDY ADDS
In our study, we show that cancer cell-specific knockout of PARP1, which is the most expressed member of this enzyme family in NSCLC, reverses cisplatin resistance. Moreover, in mouse models, knockout of PARP1 in NSCLC cells induces T lymphocyte-mediated tumor growth control coupled to signs of T cell activation in the local microenvironment.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE, OR POLICY
These results have implications for the mode of action of therapeutic PARP1 inhibitors and underscore a major impact of tumor cell metabolism on the cancer-immunity dialog.
Introduction
Many chemotherapeutic agents induce DNA damage via direct interactions with chromatin and chromatin-binding proteins. This applies also to platinum-based cytoxicant including cis-diamminedichloroplatinum(II) (CDDP, best known as cisplatin), which directly crosslinks DNA.1–3 Logically, CDDP resistance may involve the upregulation of DNA damage repair.4–7 Several years ago, we observed that a sizeable fraction of non-small cell lung cancer (NSCLC) cells that were selected for CDDP resistance by long-term culture in the continuous presence of CDDP were characterized by the enzymatic activation of poly(ADP-ribose) (PAR) polymerase-1 (PARP1),8 which is involved in the repair of CDDP-induced DNA lesions.9 Of note, this trait was conserved even after several months of culture in the absence of CDDP, reflecting an adaptive long-term alteration in tumor cell metabolism. Indeed, such cells contain higher levels of the PARP1 product PAR that can be detected by immunological methods.8 10
In NSCLC specimens, high level of immunohistochemically detectable PAR correlates with poor prognosis.11 Since the levels of PARP1 expression and PAR do not correlate among each other, it is necessary to measure PAR instead of PARP1 mRNA or protein expression to establish such a prognostic relationship.11 Like many other cancer types, NSCLC is affected in its prognosis by the density of the immune infiltrate.12 Accordingly, we observed that high PAR levels were associated with scarce tumor infiltration by CD8+ cytotoxic T lymphocytes (CTLs)13 suggesting that PARP1 activity might influence the cancer-immune dialog. However, this conjecture has not been explored in mechanistic terms. Nonetheless, pharmacological PARP1 inhibitors can be advantageously combined with immune checkpoint inhibitors targeting cytotoxic T-lymphocyte associated protein 4 (CTLA-4)14 or the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) interaction,15–17 as this has been reported in some clinical trials.
Here, we report the generation of PARP1 knockout cells from CDDP resistant PARhigh NSCLC cells. We show that this manoeuver reverses CDDP resistance, but renders the cells refractory to pharmacological PARP1 inhibition. Of note, this genetic manipulation profoundly affects the cancer-immune dialog, increasing the immune recognition of NSCLC in immunocompetent mice, hence improving their immunosurveillance. These findings have implications for the clinical anticorrelation between PAR levels and CTL infiltration.
Materials and methods
Cell culture conditions and chemicals. Lewis lung carcinoma (LLC) and tissue culture number one (TC1) murine lung cancer cell lines were purchased from the American Type Culture Collection (Rockefeller, Maryland, USA). PARhigh LLC and TC1 cell lines were obtained in vitro by prolonged culture of wild type LLC and TC1 cells with sublethal CDDP concentrations as previously described.8 13 The knockout of PARP1 or STING was induced by means of the CRISPR/Cas-9 technology, according to the manufacturer’s recommendations. Culture media and cell culture supplements were purchased from Gibco-Invitrogen Life Technologies (Carlsbad, Clifornia, USA). Wild-type LLC cells and their derivatives were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM). Wild-type TC1 cells and their derivatives were cultured in glutamax RPMI-1640 medium supplemented with non-essential amino acids. Media for cell culture were supplemented with 10% fetal bovine serum, 100 U/mL penicillin G sodium salt, 100 mg/mL streptomycin sulfate. Cells were routinely maintained at 37°C under 5% CO2 in T175, T75 and T25 flasks and seeded in appropriate supports (6, 12, or 96 wells plates) 24 hours before experimental determinations. All cells were amplified to generate liquid nitrogen stocks and, on thawing, never passaged for more than 1 month before use in experimental determinations. The cells were tested regularly for mycoplasma using MycoAlert Detection Kit (Lonza) and authenticated via PCR using nine short tandem repeat markers (IDEXX BioResearch). Niraparib and talazoparib were purchased from MedChemExpress and Selleck Chemicals, respectively. CDDP, DMXAA and methyl cellulose (viscosity 400 CP) came from Sigma-Aldrich.
CRISPR/Cas9-mediated knockout
CRISPR/Cas9-green fluorescent protein (GFP) plasmids targeting PARP1 (PARP1.a, target site: CGATGGGAAAGTCCCACACTGG; PARP1.b, target site: CGCCTGTCCAAGAAGATGGTGG), or specific for STING (target site: CTATAAGTCCCTAAGCATG), as well as control non-targeting plasmid were purchased from Sigma-Aldrich. Both PARhigh LLC cells and PARhigh TC1 cells plated in 6-well plates at a concentration of 0.25 million/well were transfected at 30%–40% confluence with CRISPR plasmids by means of the Fugene transfection reagent (Qiagen). Similarly, PARP1KO TC1 cells were transfected with CRISPR plasmid specific for STING. Twenty-four hours later, cells were trypsinized and transiently expressing GFP cells were sorted by cytofluorometry in 96-well plates (1 cell/ well).
Immunoblotting
Cells were collected, washed twice with cold phosphate-buffered saline (PBS) and lysed in a buffer containing 50 mM Tris HCl pH 6.8, glycerol 10%, 2% SDS, 10 mM DTT and 0.005% bromophenol blue. Protein extracts (30 µg/lane) were separated on precast 4% to 12% SDS-PAGE gels (Invitrogen), followed by electro transfer to Immobilon membranes (Sigma-Aldrich) and immunoblotting with antibodies specific for PAR (10 hours; Calbiochem, Merck KGaA), PARP1 (Cell Signaling Technology Inc.), STING (Cell Signaling Signaling Technology), pTBK1 or TBK1 (Cell Signaling technology). An anti-β-actin antibody (mAb to beta actin, abcam) was used to monitor equal lane loading. Finally, membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Southern Biotech), followed by chemiluminescence detection with the SuperSignal West Pico reagent and either CL-XPosure X-rayfilms (both from Thermo Scientific-Pierce) or the ImageQuantLAS 4000 Biomolecular Imager (GE Healthcare Life Sciences). Protein levels were quantified by densitometry using ImageJ software.
Cytofluorometric quantification of cell death
For the simultaneous quantification of plasma membrane integrity and mitochondrial transmembrane potential (Δψm), both adherent and non-adherent cells were collected, washed, and costained for 30 min at 37°C in 300 µL of culture medium containing 1 µg/mL propidium iodide (PI) which only incorporates into dead cells exhibiting plasma membrane rupture, and 40 nmol/L 3,3’-dihexyloxacarbocyanine iodide DiOC6(3), a mitochondrial transmembrane potential sensitive dye (Molecular Probes-Invitrogen).18–20 Cytofluorometric acquisitions were carried out on a Miltenyi cytofluorometer (MACSQuant Analyzer V.10), and analyses were performed by using the FlowJo software V.10.4.2 (TreeStar) on gating on events exhibiting normal forward scatter and side scatter parameters.
Mouse housing, cancer models and treatments
Eight-week-old female C57Bl/6 were purchased from Envigo France. Mice were maintained in specific pathogen-free conditions, at 25°C, with 12 hours light/12 hours dark cycles and fed ad libitum with free access to water. All animals were used under an approved protocol by the local Ethics Committee of Cordeliers Research Center (C2EA 05 no B-75-06-12, protocol no 21661) in accordance with the EU Directive 63/2010. To establish subcutaneously transplanted tumors, 5×105 LLC cells (C9 or C12 PARhigh clones; P15 or P17 PARP1KO clones) or 3×105 TC1 cells (C5 or C7 PARhigh clones; P2 or P4 clones PARP1KO clones; PS2, PS4 PARP1KO STINGKO clones) were resuspended in 100 µL of PBS and inoculated into the right flank of C57Bl/6 mice under anesthesia with 2.5% isoflurane. For antibody mediated T cell depletion, mice received an intraperitoneal (i.p) injection of 100 µg anti-CD8a (clone YTS 169.4, BioXcell) and 100 µg anti-CD4 (clone GK1.5, BioXcell) antibodies or 200 µg of control isotype control (clone LFT-2, BioXcell) 2 days before tumor implantation. Then the antibodies were administered once a week. In order to inhibit the PD-1/PDL-1 interaction, anti-PD-1 (clone 29F.1A12, BioXcell) or isotype control (clone 2A3, BioXcell) was injected i.p. at 200 µg per mouse in 100 µL of PBS, 10 days after LLC cells injection. Then, the antibodies were administered twice a week.21 To test the therapeutic effect of PARP1 pharmacological inhibition, niraparib was dissolved in 0.5% methyl cellulose (Sigma-Aldrich) in sterile water and stirred overnight when tumors attained an average volume of ~20 mm3. Then, after the mice were randomized across the different groups, mice were administered 80 mg/kg niraparib orally in a volume of 100 µL per mice once daily for 5 days on and 2 days off per week. Control mice were administered 0.5% methyl cellulose. When used in combination, anti-PD-1 was injected 7 days after the first injection of niraparib, then twice a week. Tumor growth was monitored via repeated measurements of the tumor size using a digital caliper. The volume of tumors was calculated using the following formula: tumor size (mm3) = (length × width × height)/8×4/3 x π. Mean tumor growth curves were calculated by carrying over the last tumor size values of the mice that reached endpoint and interrupted when more than 50% of the group had reached endpoint. Mouse survival was carefully monitored. Tumor size exceeding 1500 mm3, tumor ulceration, weight loss superior to 20% as compared with the beginning of the treatment and poor body condition were considered as endpoints.
Ex vivo blood processing and evaluation of T cell depletion
PARhigh and PARP1KO tumors bearing mice were treated for 3 weeks with either control isotypes (CTL) or a combination of anti-CD8 and anti-CD4 antibodies (αCD8αCD4). Blood sampling was performed through retro-orbital sinus bleeding using a heparinized capillary tube. Blood was collected into a heparinized saline solution (30 U heparin/mL). Peripheral white blood cells were cleared of erythrocytes in 1X RBC lysis buffer (BioLegend) at room temperature for 10 min then washed twice in PBS. Surface staining of peripheral blood cells was performed with fluorescent antibodies: anti-CD-BV421 (145-2 C11), anti-CD8-PE (53-6.7) and anti-CD4-PerCP-Cy5.5 (RM4-5) purchased from BD Biosciences. Flow cytometry acquisition was performed on a MACSQuant Flow Cytometer (Miltenyi Biotec), and data analysis was conducted with the FlowJo software.
Ex vivo: tumor processing and phenotyping of immune infiltrate
LLC PARhigh and PARPKO tumors were harvested, placed ice in gentle MACS C tubes (Miltenyi Biotec) containing DMEM and processed as previously described.22 Briefly, tumors were dissociated with scissors, then enzymatically digested using Miltenyi Biotec mouse tumor dissociation kit and gentleMACS Octo Dissociator. Tumor homogenates were filtered through 70 µM MACS SmartStrainers (Miltenyi Biotec) and washed twice with ice-cold PBS. Then, after homogenization of bulk tumor cells in PBS at a concentration corresponding to 250 mg of the initial tumor weight /mL, 50 mg of the bulk tumor cell homogenates were stained with LIVE/DEAD Fixable Yellow dye (Thermo Fisher Scientific), and Fc receptors were blocked with anti-mouse CD16/CD32 (clone 2.4G2, BD Pharmingen). Surface staining of immune cells was performed with the following fluorochrome-conjugated antibodies: (1) ‘Myeloid cell’ panel: anti-CD45 APC-Fire750 (30 F-11, BioLegend), anti-Ly-6G PE (1A8, BD Pharmingen), anti-Ly-6C FITC (AL-21, BD Pharmingen), anti-CD11b V450 (M1/70, BD Pharmingen), anti-CD11c PE-Vio770 (REA754, Miltenyi Biotec), anti-CD80 PerCP-Cy5.5 (16–10 A1, BD Pharmingen), and anti-I-A/E (MHC-II) APC (M5/114.15.2, BioLegend); (2) ‘T-cell activation/exhaustion’ panel: anti-CD3 APC V450 (17A2, Thermo Fisher Scientific), anti-CD8 PE (53–6.7, BD Pharmingen), anti-CD4 PerCP-Cy5.5 (RM4-5, Thermo Fisher Scientific), anti-CD25 PE-Cy7 (PC61.5, Thermo Fisher Scientific), anti-inducible costimulator (ICOS) BV421 (7E.17G9, BD Pharmingen), and anti-PD-1 APC-Fire750 (29F.1A12, BioLegend). Cells were then fixed and permeabilized with anti-Foxp3 (eBioscience)/Transcription Factor Staining Buffer (Thermo Fisher Scientific). To complete the staining of the samples undergoing T cell immunophenotyping, cells were incubated with anti-Foxp3 FITC (clone FJK-16s, Thermo Fisher Scientific). Finally, stained samples were run through a BD LSR II flow cytometer and data acquired using BD FACSDiva 6.1.3 software (BD biosciences). Samples were then analyzed using FlowJo software.
Quantitative RT-PCR analysis of IFNA1 and IFNB1 mRNA expression
Total RNA was isolated using the Rneasy plus mini kit (Qiagen, Hilden, Germany) following manufacturer’s instruction. Genomic DNA was digested using the RNase-Free DNase Set (Qiagen) according to manufacturer’s guidance. Next, total RNA was reverse transcribed to cDNA using SuperScript IV VILO Master Mix (Thermo Fisher Scientific). A total of 1–2.5 µg of RNA were diluted in 16 µL of nuclease-free water and 4 µL of SuperScript IV VILO Master Mix were added. Reverse transcription was performed as previously described23: primer annealing at 25°C for 10 min, reverse transcription at 50°C for 10 min, and heat inactivation at 85°C for 5 min. Thereafter, IFNα1 (Mm03030145) or IFNβ1 (Mm00439552) were amplified using specific TaqMan Gene Expression assays (Thermo Fisher Scientific) using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) on the StepOnePlus RealTime PCR System (Applied Biosystems, Waltham, Massachusetts, USA). Running conditions were 50°C for 2 min, 95°C for 2 min followed by 40 cycles of target gene amplification (95°C for 1 s and 60°C for 20 s). Finally, threshold cycle (Ct) values of gene of interest were subtracted from Ct values of the housekeeping gene, PP1A, generating ΔCts; fold change of expression between PARhigh and PARP1KO samples was calculated using the formula: 2(-ΔΔCt).
In vitro analysis of PD-L1, MHC Class I, MHC Class II and CALR expression by flow cytometry
LLC and TC1 cells were seeded in 96-well plates (1000 cells per well) in 100 µL growth medium and let adapt for 24 hours before treatment. Cells were then treated by different concentrations of niraparib for another 72 hours. Post-treated cells were collected in 96-well V-shape plates (Greiner-bio-one). Cells were first stained with DAPI (Thermo Fisher Scientific) for 15 min at 4°C in the dark as an exclusion dye to select viable (DAPI-) cells, then washed and stained with an anti-mouse CD274 (PD-L1, MIH1, eBioscience), anti-mouse MHC Class I (H-2Kb, AF6-88.5.5.3, eBioscience), anti-mouse MHC Class II (I-A/I-E, M5/114.15.2, eBioscience) monoclonal antibody, anti-CALR (ab2907, Abcam) for another 25 min at 4°C in the dark. After staining, cells were fixed with 4% PFA and kept at 4°C. Flow cytometry acquisition was performed on a MACSQuant (Miltenyi Biotec), and data analysis was conducted with the FlowJo software.
Cytofluorometric analysis of polyploidy
LLC cells were seeded in 12-well plates (20 000 cells per well). After 24 hours adaptation, cells were treated with niraparib for 72 hours. Cells were then collected and fixed with cold 80% ethanol. Samples were kept at −20°C overnight and stained with FxCycle TM PI/RNase Staining Solution (Invitrogen) following all manufacturers instructions. Flow cytometry acquisition was performed on a MACSQuant (Miltenyi Biotec), and data analysis was conducted with the FlowJo software, as described.24 25
DNA damage foci measured by immunofluorescence detection of phosphorylated histone H2AX (γH2AX)
Cells were seeded into 384 wells black microplates (500 cells/well) and cultured in normal conditions or in the presence of niraparib (1.5, 3, 6, 12, 24 µM) for 72 hours. Alternatively, cells were seeded into 384 wells black microplates (1000 cells/well) and cultured in normal conditions or in the presence of CDDP (2.5, 5, 10, 20 µM) for 24 hours. After washing twice with PBS, plates were fixed with 4% PFA/PBS containing Hoechst 33 342 (2 μg/mL) for 10 min which was used for nuclear conterstaining, washed twice with PBS and incubated with quenching solution (2.67 g NH4Cl in 1 L PBS, pH 7.4) for 5 min. Thereafter, cells were washed with PBS, permeabilized with 0.1% Triton-X 100 for 10 min, and rinsed twice with PBS. Plates were blocked with 2% BSA in PBST (PBS, 0.01% Tween-20; v:v) for 30 min before incubation with anti-γH2AX (Ser139; clone JBW301) antibody (Sigma Aldrich) overnight at 4°C. Then the cells were rinsed twice with PBS and incubated with secondary antibody for 30 min at room temperature in the dark. After additional washing steps, 50 µL PBS were added to each well. Cells micrographs were acquired by means of an IXM XLS automated fluorescent microscope (Molecular Devices) equipped with a Plan Apo 20X objective using the DAPI and GFP filter sets, and subsequent image analysis was performed using the EBImage package form the R bioconductor repository. Briefly, nuclei masks were created using the Hoechst 33 342 signal, and used to measure the nuclear γH2AX intensity. γH2AX foci count was determined from the dots masks obtained by thresholding the top-hat-filtered GFP image.26
Sample preparation for metabolome analysis
LLC or TC1 cells were seeded in 6-well plates and cultured for 24 hours in complete medium. Forty-eight hours before extraction, medium was changed and cells were cultured either in complete medium alone or complete medium with niraparib (five replicates per condition). Subsequently, cells were washed five times with cold PBS before rinsing with water, and then scraped in 500 µL of methanol (90%)-water (10%). Then, 100 µL of chloroform were added. After centrifugation (10 000 g, 10 min, 4°C), the whole supernatant was evaporated at 40°C to obtain dried extracts. Three hundred µL of methanol were added on dried extract and split in two 150 µL fractions for gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) analyses respectively. For GC-MS assay, methanol solubilized aliquots were transferred to glass tubes and solvent was evaporated. A total of 50 µL of methoxyamine (20 mg/mL in pyridine) were added on dried extracts, then stored at room temperature in dark, during 16 hours. The day after, 80 µL of MSTFA were added and final derivatization occured during 30 min at 40°C. Samples were then transferred to vials and directly injected into GC-MS. After a second evaporation round, LC-MS dried extracts were solubilized in 300 µL of MilliQ water, centrifuged (10 min at 15 000 g, 4°C) and aliquoted in three microcentrifuge tubes (100 µL). Aliquots were transferred in ultra-high performance liquid chromatography (UHPLC) vials and injected into the UHPLC/MS or kept at −80°C until injection. Metabolomics was performed as previously described.27 28
Targeted analysis of cGAMP (2’,3’) by UHPLC coupled to a triple quadrupole (QQQ) mass spectrometer
Dry pellets obtained from LLC or TC1 cells cultured for 24 hours in complete medium were recovered with 500 µL of Methanol, vortexed 5 min (2500 rpm) and centrifugated (10 min, 15 000 g, 4°C). A total of 200 µL of supernatant were transferred in microtubes and evaporated. Dry samples were spiked with 150 µL of MilliQ water, vortexed and centrifugated. A total of 50 µL of water were transferred in injection vials and spiked with 5 µL of formic acid, before direct injection into LC-MS. Targeted analysis was performed on an LC 1290 system (Agilent Technologies) coupled to a Agilent 6470 mass spectrometer equipped with an electrospray source operating in positive mode. A total of 10 µL of sample were injected on an Agilent’s Column Zorbax XDB-C18 (4.6×50 mm, particle size 1.8 µm) heated at 40°C. The autosampler was kept at 4°C. The mobile phases were water (0.2% of acetic acid) (A) and acetonitrile (B). MRM transitions followed the previously decribed protocole.29
Bioinformatic analysis
The relative mRNA expression of PARP isoforms in human NSCLC cell lines, normal lung tissues, and lung tumor tissues were retrieved from RNA-seq data contained in the Cancer Cell Line Encyclopedia dataset (https://sites.broadinstitute.org/ccle/), the Genotype-Tissue Expression (GTEx, https://gtexportal.org/home/) project, and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). To analyze differential expression, data from TCGA and GTEx datasets were uniformly converted to TPM (transcripts per million) format by the Toil process.30
Statistical analysis
Unless otherwise specified, all in vitro, experiments were independently repeated at least three times, yielding comparable results. Data were analyzed with GraphPad Prism software or Microsoft excel (Microsoft) and statistical significance was assessed by means of unpaired Student’s t-test. In vivo, longitudinal analyses of tumor growth data were carried out by linear mixed-effect modeling on tumor sizes. Linear mixed-effects models were applied for longitudinal comparison of tumor growth curves with the TumGrowth web tool31 (https://github.com/kroemerlab/TumGrowth). For graphing, tumor growth data are represented in group-averaged tumor size alongside its SEM at each time point. Survival data are represented in Kaplan-Meier survival curves. Log-rank test was used to compute p values using GraphPad Prism software. The visualization and statistical analysis of gene expression data was based on R (V,3.6.3), ComplexHeatmap package (V.2.2.0), and ggplot2 package (V.3.3.3). P values were calculated by means of the Wilcoxon rank sum test. P values were considered significant when lower than 0.05. Bonferroni adjustment for multiple comparisons were used for metabolomic comparisons. Statistical significance was represented as following: */#p<0.05, **/##p<0.01, ***/###p<0.001.
Results
Expression of different PARP isoforms in NSCLC
At the mRNA level, cell lines derived from primary and metastatic NSCLC express PARP1 at much higher levels than any other members of the PARP gene family including those that possess enzymatic PARP activity (online supplemental figure S1A). Accordingly, in contrast to non-cancerous lung tissue (online supplemental figure S1B), primary NSCLC adenocarcinomas and squamous carcinomas exhibit a clear dominance of PARP1 expression over other members of the gene family (online supplemental figure S1C). Among the PARP isoforms endowed with the capacity of poly ADP-ribosylation (as opposed to mono ADP-ribosylation or other enzymatic activities), in particular PARP1, PARP2 and tankyrase/TNKS,32 only PARP1 was significantly upregulated in NSCLC adenocarcinomas and squamous carcinomas as compared with normal adjacent tissues (online supplemental figure S1D,E). We therefore decided to focus our analysis on PARP1, which appears to be the most NSCLC-relevant protein of its family.
Supplemental material
PARP1 knockout reverses CDDP resistance
Continuous culture of two murine NSCLC cell lines (LLC, and tumor cell-1 (TC1)) in the presence of CDDP led to the identification of cisplatin-resistant clones,13 which often demonstrate the overactivation of PARP1, resulting in the intracellular accumulation of PAR, as determined by immunoblot analyses (online supplemental figure S2A,B). We chose two clones (LLC R7 and TC1 R18) for transfection with a CRISPR/Cas9-GFP vector that either targets PARP1 or lacks a guidance RNA. The cells transiently expressing GFP were selected to derive clones that possess an elevated PARP1 activity, comparable to the parental clones (PARhigh cells), or that lack PARP1 expression and activity (PARP1KO cells) (online supplemental figure S2C, figure 1A,B). The knockout of PARP1 sensitized the cells to death induction by CDDP. Thus, a higher proportion of PARP1KO cells treated with CDDP tended to reduce their mitochondrial transmembrane potential (measured with DiOC6(3)) and to incorporate the vital dye PI than PARhigh cells (figure 1C–F).
Supplemental material
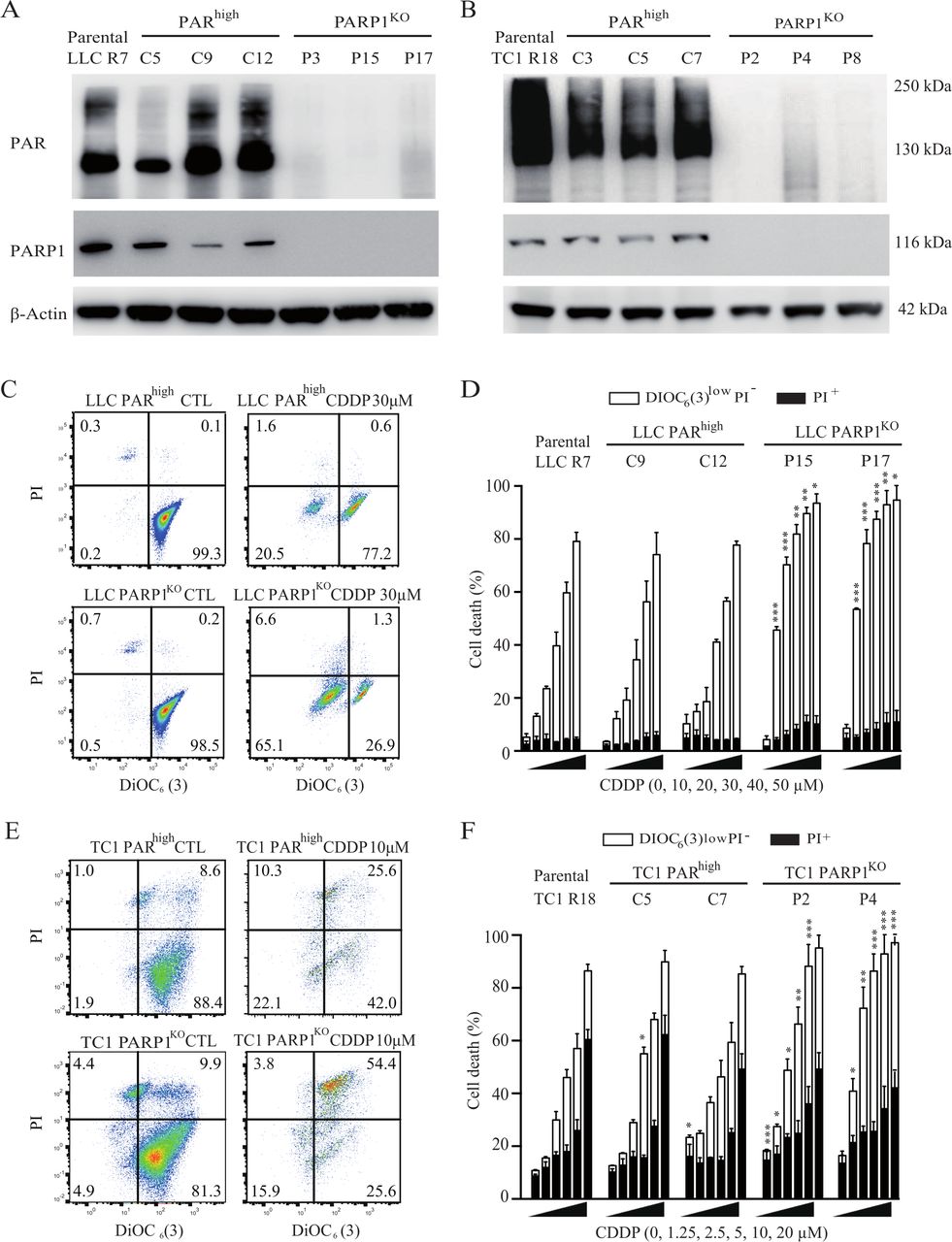
PARP1 hyperactivity is involved in cisplatin resistance. (A, B) Cisplatin-resistant R7 LLC (in A) and R18 TC-1 (in B) NSCLC clones were transfected with a CRISPR/Cas9-GFP vector that either targets PARP1 or lacks a guidance RNA. Parental clones and their CRISPR/Cas9 derivatives were processed for the immunoblotting-based assessment of PAR-containing proteins. Actin levels were monitored to ensure equal loading of lanes. (C–F) Parental cells and their CRISPR/Cas9 derivatives were maintained in control conditions or treated with the indicated concentrations of cisplatin for 72 hours. Then, cells were subjected to the cytofluorometric analysis of cell death-related parameters on costaining with the vital dye propidium iodide (PI) and the mitochondrial membrane potential-sensing dye DiOC6 (3). C and E illustrate representative dot plots of LLC and TC1 cells respectively (numbers refer to the percentage of cells found in each quadrant), whereas D and F show quantitative data. White and black columns illustrate the percentage of dying and dead cells respectively (means±SEM, n=3). *P<0.05; **p<0.01; ***p<0.001 (Student’s t-test), compared with equally treated parental R clones. GFP, green fluorescent protein; LLC, Lewis lung cancer; NSCLC, non-small cell lung cancer; PARP1, poly(ADP-ribose) polymerase-1.
Altogether, these results establish that PARP1 overactivation is causally involved in CDDP resistance.
PARP1 knockout increases natural immunosurveillance
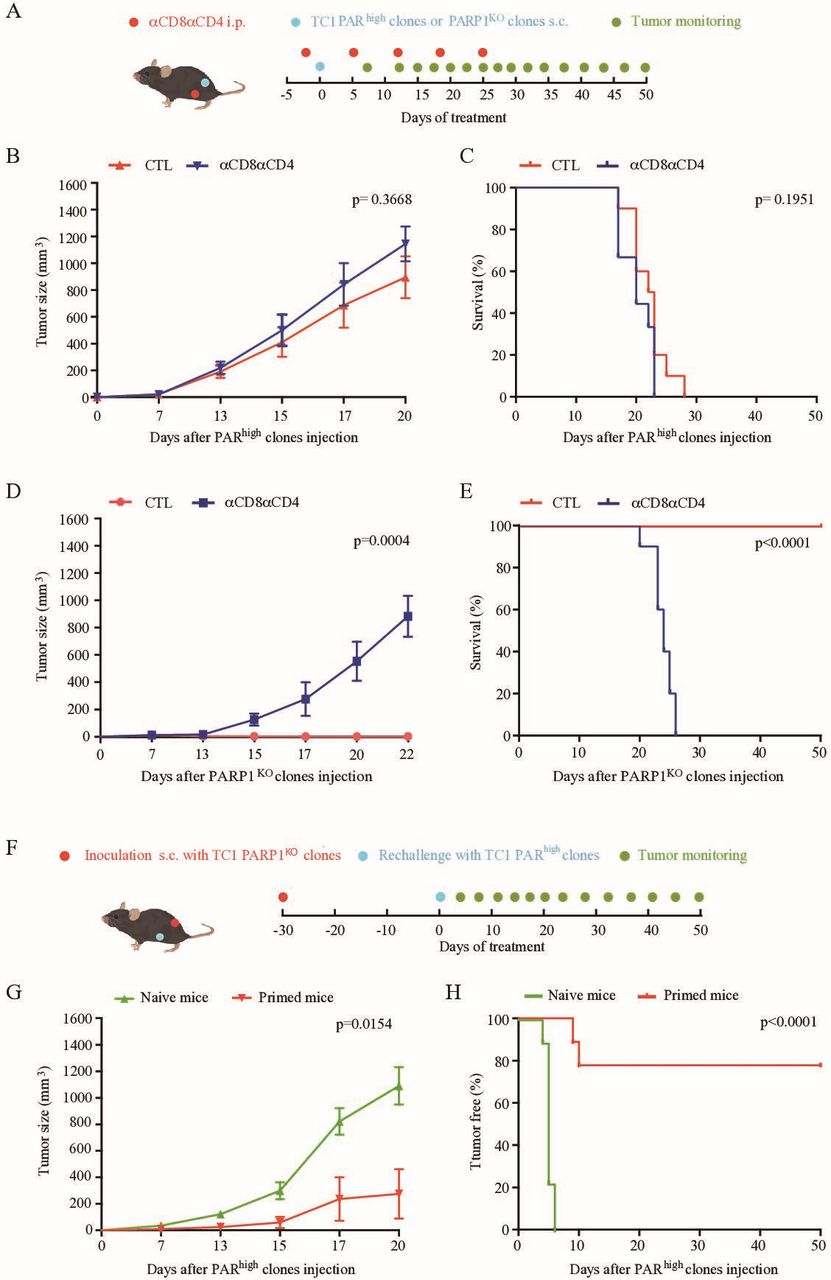
We previously demonstrated that, in patients with NSCLC, PARhigh tumors are less infiltrated by CD8+ T lymphocytes than PARlow tumors.13 For this reason, we investigated whether PARP1 activity may affect anticancer immunosurveillance in mice. Representative PARhigh and PARP1KO LLC clones were inoculated into the flanks of histocompatible C57Bl/6 mice, which were injected with either control isotypes or antibodies that deplete CD4+ and CD8+ T cells (figure 2A, online supplemental figure S3). PARhigh LLC tumors grew indistinguishably on immunocompetent and T cell-depleted mice (figure 2B,C, online supplemental figure S4A). However, the growth of PARP1KO LLC tumors was accelerated by depletion of T lymphocytes, indicating that such cancers are under immunosurveillance (figure 2D,E, online supplemental figure S4B). Accordingly, cytofluorometric analyses demonstrated that the immune infiltrate of PARhigh and PARP1KO LLC tumors were different. Although the absolute numbers of tumor-infiltrating CD45+ leukocytes and CD3+ cells did not change, the percentage of cells exhibiting the activation marker ICOS was increased among CD8+ (but not CD4+Foxp3- cells) and reduced among regulatory T cells (Tregs, defined as CD4+Foxp3+ cells, online supplemental figure S5 and figure 2F) from PARP1KO LLC tumors as compared with PARhigh tumors. Moreover, the percentage of CD8+ and CD4+Foxp3- T cells expressing the exhaustion marker PD-1 was reduced in PARP1KO LLC tumors in comparison to PARhigh tumors (figure 2F). These changes in ICOS and PD1 expression indicated as percentage of positive cells (figure 2F) were paralleled by similar shifts in the mean fluorescent intensity for such positive cells (online supplemental figure S6), underscoring the robustness of the results. Although upregulation of PD-1 might favor the action of PD-1 blockade, injection of an anti-PD-1 antibody failed to affect the growth of both PARP1KO and PARhigh LLC tumors (online supplemental figure S7). Of note, PARP1KO tumors were more infiltrated in activated dendritic cells (CD45+CD11c+MHCIIhigh cells, online supplemental figure S8) as compared with their PARhigh counterparts (figure 2G). In contrast, the frequency of CD45+CD11b+Ly6G−Ly6Clow/intermediate myeloid cells were not different among the two groups (figure 2G).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material

PARP1 knockout in LLC cancer cells impacts local immunosurveillance. (A) Experimental schedule of the implantation and treatment of syngeneic subcutaneous lung cancer in C57Bl/6 mice. 5×105 PARhigh cells or PARP1KO LLC cells were subcutaneously grafted into the flank of C57BL/6 mice. A combination of anti-CD8 and anti-CD4 (αCD8αCD4) neutralizing antibodies were intraperitoneally administrated 2 days before injection of tumor cells, then weekly. PARhigh CTL, PARhighαCD8αCD4 and PARP1KO CTL, n=10 mice; PARP1KOαCD8αCD4, n=9 mice. (B–E) Mean tumor growth curves and Kaplan-Meier survival curves of mice subcutaneously grafted with PARhigh cells (B, C) or PARP1KO cells (D, E). Tumor growth is reported as means±SEM. P values were calculated as compared with control (CTL) group (Wald test, type 2 ANOVA). For Kaplan-Meier, p values were determined by means of the log-rank test. (F, G) Characterization of the immune infiltrate in mouse lung cancers derived from PARhigh and PARP1KO LLC cells. Immunocompetent C57BL/6 mice were injected subcutaneously with PARhigh and PARP1KO clones (10 and 15 mice, respectively). In (F), dot plots illustrate the percentage of ICOS+ and PD-1+ cells among CD8+, CD4+ and T regulatory cells (Tregs) cells. In (G), percentage of CD11c+MHCIIhigh cells (among CD11c+) cells and CD11b+Ly6G-Ly6Clow/intermediate (among CD45+ cells). Means±SEM, p values were calculated by Student’s t test as compared with PARhigh immune infiltrates. ANOVA, analysis of variance; CTL, cytotoxic T lymphocytes; ICOS, inducible costimulator; i.p, intraperitoneal; LLC, Lewis lung cancer; PAR, poly(ADP-ribose).
We also investigated the growth of PARhigh and PARP1KO TC1 clones in immunocompetent versus T cell-deficient mice (figure 3A). Again, the growth of PARhigh tumors was not affected by the depletion of T cells. In sharp contrast, PARP1KO TC1 clones were unable to form tumors in immunocompetent mice, but grew on T cell-depleted mice (figure 3B–E, online supplemental figure S9A,B). When immunocompetent mice had rejected the implantation of PARP1KO TC1 cells (figure 3F), they subsequently also became resistant against PARhigh cells inoculated into the opposite flank (figure 3G,H, online supplemental figure S9C). These results demonstrate that PARP1KO TC1 cells cause an anticancer immune response leaving a memory of the antigenic encounter.
Supplemental material

Knockout of PARP1 on TC1 cancer cells abolishes tumor formation in immunocompetent mice. (A) Schedule of the implantation and treatment of syngeneic subcutaneous TC1 cancer cells in naïve immunocompetent C57Bl/6 mice. 3×105 PARhigh cells or PARP1KO TC1 cells were subcutaneously grafted into C57BL/6 mice (PARhigh CTL, PARhighαCD8αCD4 and PARP1ko CTL n=10 mice; PARP1KOαCD8αCD4, n=9 mice). Control isotypes or a combination of anti-CD8 and anti-CD4 neutralizing antibodies (αCD8αCD4) were intraperitoneally administrated 2 days before injection of tumor cells and then weekly. (B, C) Mean tumor growth and Kaplan-Meier survival curves of mice subcutaneously grafted with PARhigh cancer cells, respectively. (D, E) Mean tumor growth and Kaplan-Meier curves respectively of mice subcutaneously grafted with PARP1KO cells. (F–H) 3×105 PARhigh TC1 cells were injected into the left flank of immunocompetent C57BL/6 naïve tumor free mice or tumor free mice inoculated 1 month before with PARP1KO TC1 cells in the right flank (F). The evolution of tumor incidence over time was reported as tumor growth (G) and Kaplan-Meier (H) curves (naive and primed groups, n=8 and 9 mice, respectively). Error bars indicate SEM. P values were calculated as compared with CTL group (Wald test, type 2 ANOVA). For Kaplan-Meier, p values were determined by means of the log-rank test. ANOVA, analysis of variance; CTL, cytotoxic T lymphocytes; i.p, intraperitoneal; PARP1, poly(ADP-ribose) polymerase-1.
In sum, these results demonstrate that the level of PARP1 activity modulates immunosurveillance. Cell-autonomous inhibition of PARP1 enhances the immune recognition of NSCLC cells.
On-target and off-targets effects of PARP inhibitors
We next wondered whether pharmacological PARP1 inhibition with niraparib, which is clinically approved for the treatment of platinum-resistant cancer,33 could kill specifically PARhigh cancer cells. While PARhigh LLC cells died after a latency (7 days) in culture with niraparib (3 to 12 µM), PARP1KO LLC cells only showed a marginal response at the highest niraparib dose (12 µM) (figure 4A,B), and similar effects were found when comparing the niraparib effects on PARhigh and PARP1KO TC1 cells (figure 4C,D), or when replacing niraparib by talazoparib, another clinically approved PARP1 inhibitor (online supplemental figure S10). Thus, the knockout of PARP1 by genetic methods largely abolishes the cytotoxic effects of pharmacological PARP1 inhibitors. The culture of PARhigh LLC cells with PARP1 inhibitors for 3 days led to an increase in DNA damage foci measured by immunofluorescence detection of phosphorylated histone H2AX (γH2AX), and this effect was attenuated for PARP1KO cells that lack the principal niraparib target (online supplemental figure S11A,B). Exposure of PARhigh LLC cells to niraparib caused an increase in nuclear size (online supplemental figure S11A) that reflects an increase in ploidy, as determined by cytofluorometric analyses of ethanol-fixed, RNAse-digested and PI-stained cells (online supplemental figure S11C,D). Again, the response of PARP1KO cells to niraparib was much attenuated with respect to polyploidization (online supplemental figure S11). In sharp contrast, addition of CDDP to PARP1KO LLC cells induced more γH2AX foci than in PARhigh cells (online supplemental figure S11E), in accord with the fact that PARP1KO cells are more susceptible to CDDP-induced DNA damage.
Supplemental material
Supplemental material

Niraparib preferentially kills cisplatin resistant PARhigh NSCLC cells. Mouse PARhigh and PARP1KO LLC (A, B) or TC1 (C, D) cells were maintained in control conditions or treated with the indicated concentrations of niraparib for 7 days prior to the cytofluorometric assessment of apoptosis-related variables on costaining with propidium iodide (PI) and DiOC6(3). Representative dot plots are shown in A and C whereas B and D show quantitative data (mean±SEM; n=3). *P<0.05; **p<0.01; ***p<0.001 (Student’s t test) as compared with equally treated PARhigh cells. LLC, Lewis lung cancer; NSCLC, non-small cell lung cancer; PARP1, poly(ADP-ribose) polymerase-1.
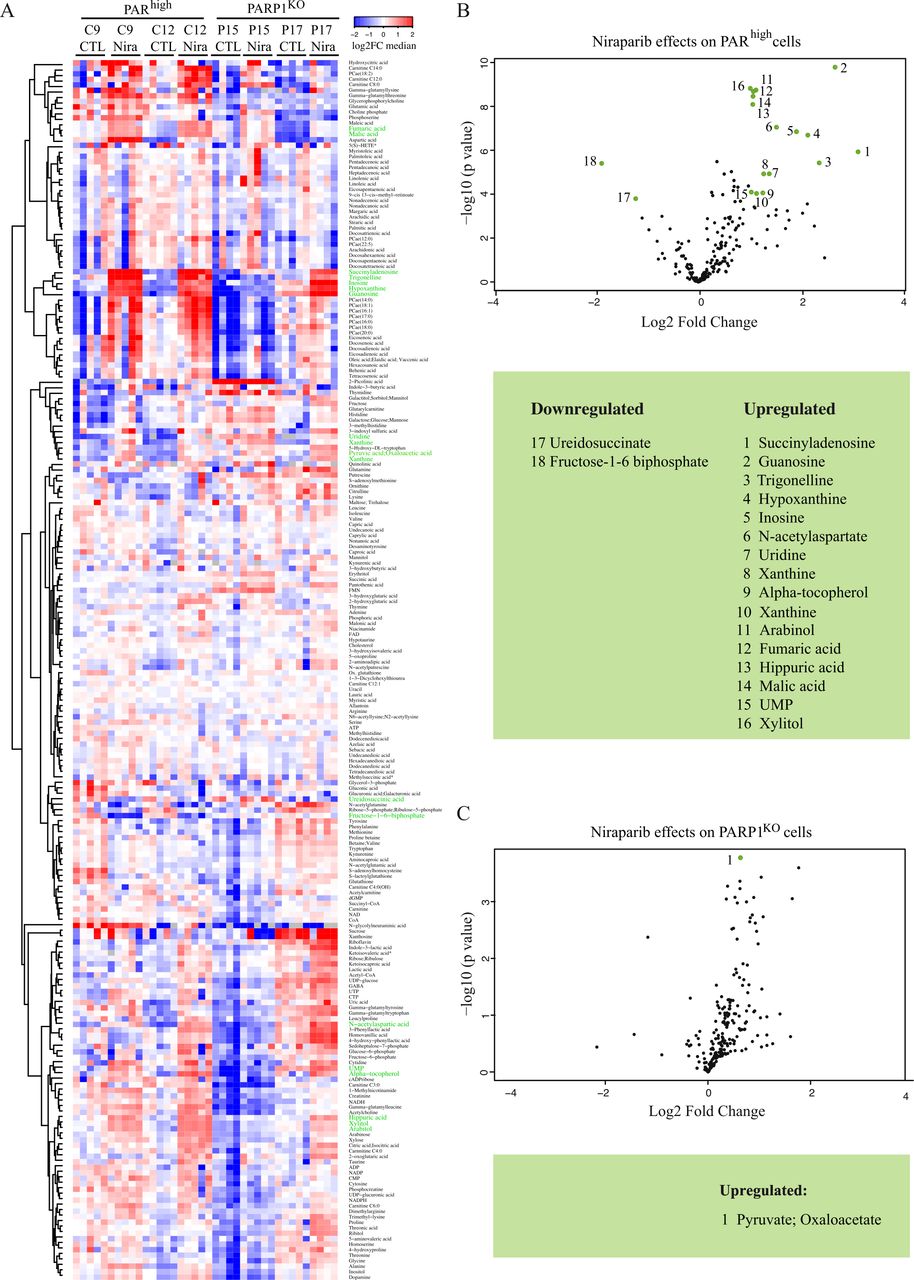
Mass spectrometric metabolomics confirmed the preferential response of PARhigh LLC cells to niraparib (figure 5A). Of note, only two metabolites were significantly changed by the PARP1 knockout, namely NADH and γ-glutamylthreonine, which both were reduced as compared with PARhigh cells (online supplemental figure S12). Volcano plots revealed that PARhigh cells responding to niraparib upregulated numerous nucleoside-relevant metabolites (such as guanosine, hypoxanthine, inosine, succinyladenosine, uridine, uridine monophosphate, xanthine) as well as Krebs cycle intermediates (fumarate, malate) (figure 5B). In contrast, only two metabolites (fructose-1–6-biphosphate, ureidosuccinate) were significantly downregulated by PARhigh LLC cells (figure 5B). Moreover, PARP1KO cells showed a rather minor metabolic response to niraparib (figure 5C), in accord with the interpretation that PARP1 is the only relevant target of niraparib.
Supplemental material

Metabolic response of PARhigh LLC lung cancer cells to niraparib. PARhigh and PARP1KO LLC cells were cultured for 72 hours in normal condition or in the presence of niraparib (6 µM). (A) Heatmap represents metabolite differences between control condition and the presence of niraparib (Nira) shown as a color gradient (log2 scale). Five replicates per condition. Metabolites were clustered by means of the Ward method on the Euclidean distance matrix. In green, the name of metabolites whose level vary the most in the presence of niraparib. (B, C) Volcano plots identifying the metabolites whose levels vary significantly during treatment with niraparib in PARhigh (B) and PARP1KO (C) cells, as compared with control (CTL) conditions. Niraparib and control metabolite levels were compared using t tests with Bonferroni adjustment for multiple comparisons. In B, green dots represent the metabolites which increase or decrease the most in PARhigh cells during the treatment with niraparib (adjusted p<0.05 and fold change >2). These metabolites are ordered in a table according to the descending fold-change. In C, only pyruvate/oxaloacetate increases significantly in PARP1KO cells treated with niraparib as compared with CTL (adjusted p=0.0373 and fold change=1.587). CTL, cytotoxic T lymphocytes; LLC, Lewis lung cancer; PAR, poly(ADP-ribose).
We also investigated whether PARhigh and PARP1KO NSCLC cells might differ in their immunological characteristics,34 determining the expression levels of PD-L1, MHC class I (H2Kb), MHC class II (I-A/Eb) molecules and calreticulin (CALR) by cytofluorometric staining. While there was no major difference in the mean fluorescence intensity between PARhigh and PARP1KO LLC and TC1 cells, niraparib tended to increase the expression of these markers of immunogenicity more efficiently in PARhigh than in PARP1KO cells (online supplemental figures S13, S14).
Supplemental material
Supplemental material
Altogether, these results demonstrate that clinically used PARP1 inhibitors have minor effects on cancer cells lacking PARP1, confirming that these treatments have scarce off-target effects.
Effects of PARP inhibitors on PARP1-deficient tumors in vivo
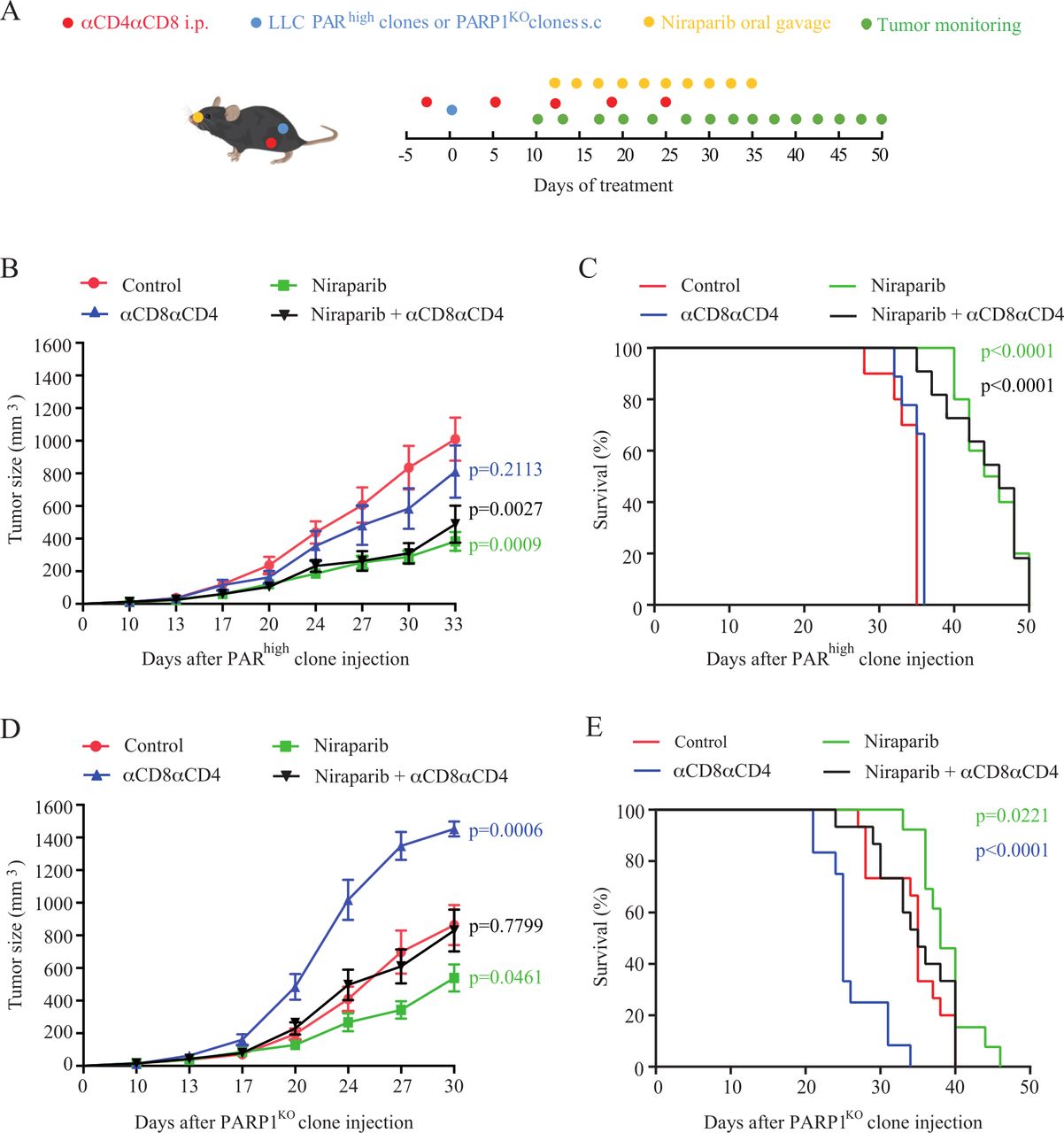
Since pharmacological PARP1 inhibition has little or no effects on PARP1KO cells, it becomes possible to investigate potential effects of PARP inhibitors on immunosurveillance that occur independently from their inhibitory effects on PARP1 expressed by malignant cells (figure 6A). PARhigh tumors implanted in immunocompetent mice reduced their growth on treatment with niraparib, and this effect was similar in T cell-depleted mice (figure 6B, online supplemental figure S15A). Moreover, niraparib similarly extended the survival of immunocompetent and immunocompromised mice bearing PARhigh tumors (figure 6C), suggesting that the tumor growth-reducing effect of niraparib does not require the action of T lymphocytes. Of note, niraparib also reduced the growth of PARP1KO cancers and extended animal survival. In this case, the magnitude of tumor growth reduction was affected by T cell depletion, and the acceleration of death by injection of anti-CD4 and anti-CD8 antibodies was reversed by niraparib (figure 6D,E, online supplemental figure S15B). These latter results suggest that niraparib can mediate anticancer effects that do not require PARP1 expression in tumor cells and depend on T cells. The anticancer activity of niraparib on both PARP1KO and PARhigh was not enhanced by PD-1 blockade (online supplemental figure S16).
Supplemental material
Supplemental material

Effect of niraparib on tumors derived from PARhigh and PARP1KO LLC cells. (A) Experimental schedule of the implantation and treatment of syngeneic subcutaneous lung cancer in C57Bl/6 mice. PARhigh cells and PARP1KO LLC cells were implanted into C57BL/6 mice with n mice per group and treated with vehicle or niraparib (80 mg/kg) along with an isotype control or a combination of anti-CD8 and anti-CD4 (αCD8αCD4) neutralizing antibodies. PARhigh CTL, PARhigh niraparib, PARhigh niraparib+αCD8αCD4, n=10 mice; PARhighαCD8αCD4, n=9 mice; PARP1KO CTL and PARP1KO niraparib+αCD8αCD4, n=15 mice; PARP1KO niraparib, n=13 mice; PARP1KOαCD8αCD4, n=12 mice. (B–E) Mean tumor growth curves and Kaplan-Meier survival curves of mice subcutaneously grafted with PARhigh cells (B, C) or PARPKO cells (D, E). Tumor growth is reported as means±SEM over time. P values were calculated as compared with control (CTL) group (Wald test, type 2 ANOVA). For Kaplan-Meier, p values were determined by means of the log-rank test. ANOVA, analysis of variance; i.p, intraperitoneal; LLC, Lewis lung cancer; PAR, poly(ADP-ribose).
Relationship between immununosurveillance of PARP1KO cells and cGAS/STING pathway activation
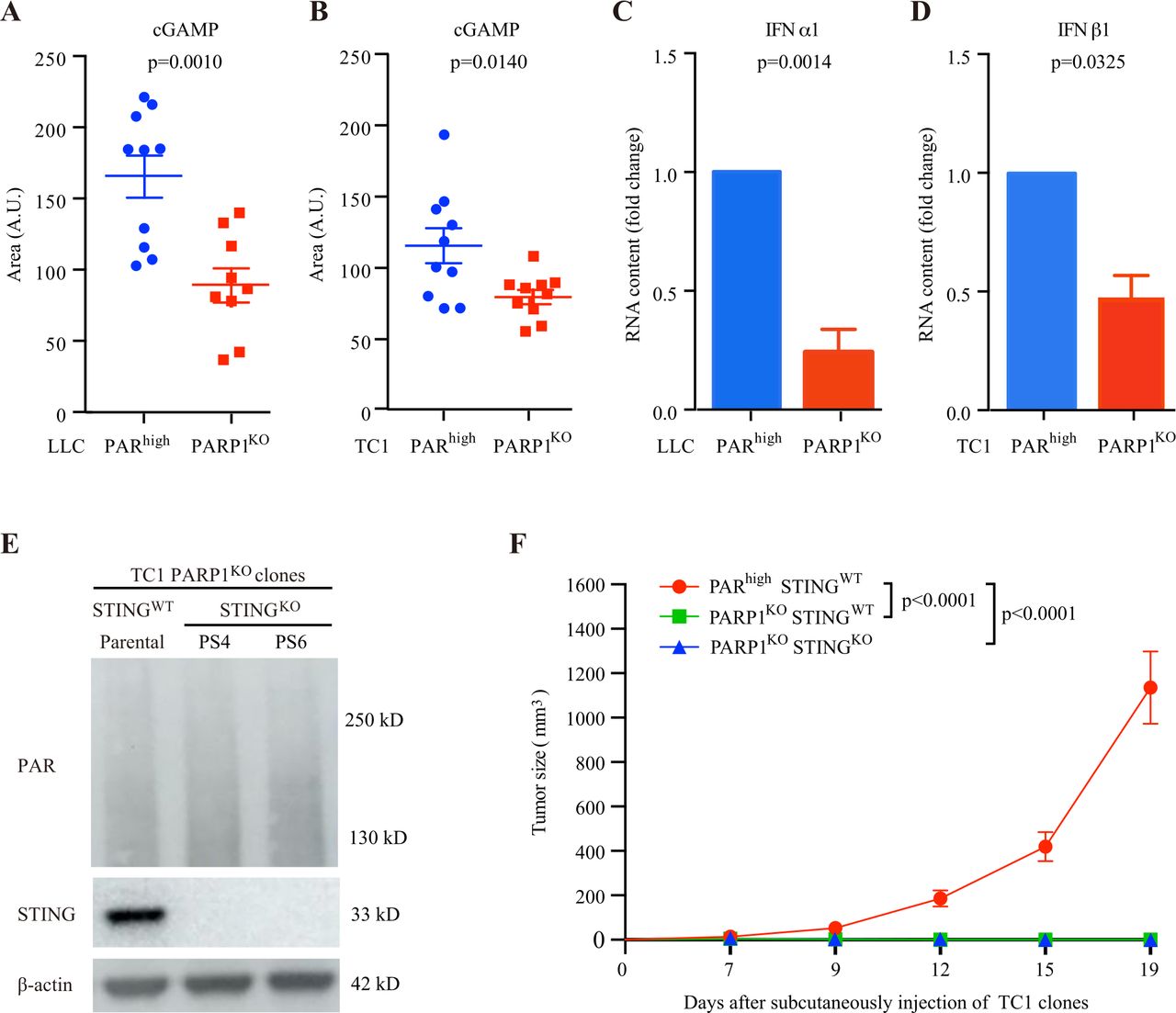
Subversion of the DNA damage response by inhibition of PARP reportedly activates the cyclic GMP-AMP (cGAMP) synthase/stimulator of interferon (IFN) genes (cGAS/STING) innate immune pathway in multiple cell types secondary to the leakage of DNA into the cytosol.35–39 This pathway leads to the induction of type-1 IFN expression, possibly explaining the recruitment of T cells in tumors.40 41 Thus, the knockout of STING in cancer cells reduces or abolishes the therapeutic response to PARP inhibition.35–41 Of note, both PARhigh and PARP1KO NSCLC cells exhibited a low baseline level of TBK1 phosphorylation on serine 172, yet indistinguishably responded to the synthetic STING activator DMXAA by increasing TBK1 phosphorylation, demonstrating that they are, in principle, capable of activating the STING pathway (online supplemental figure S17). Moreover, in both mouse models of NSCLC (LLC and TC1), the knockout of PARP1 caused a decrease rather than an increase in intracellular cGAMP concentrations, indicating that the cGAMP-producing enzyme cGAS was not activated by the genetic inhibition of PARP1 (figure 7A,B). In addition, PARP1KO cells failed to manifest an increase in the expression of the mRNA coding for the type-1 IFN alpha-1 or beta 1 (IFNA1, IFNB1) (figure 7C,D). Finally, the knockout of STING (figure 7E) failed to reverse the growth inhibition of PARP1KO TC1 cells in vivo, in C57Bl/6 mice (figure 7F). These results demonstrate that, in our specific system (which focuses on cisplatin-resistant NSCLC), overactivation of the cGAS/STING cannot explain the immune‐dependent growth inhibition of PARP1KO cells.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
cGAS/STING pathway signaling and immune-dependent control of PARP1KO cells. (A,B) Levels of cGAMP in LLC (A) and TC1 (B) clones, cultured for 24 hours in normal culture medium. Mean±SEM Ten replicates per group except in LLC PARP1KO group (n=9–10 replicates). Data are shown as area of the metabolite peak. A.U: arbitrary unit. P values were calculated using the Student’s t-test. (C, D) IFNA1 and IFNB1 expression was assessed in PARhigh and PARP1KO LLC (C) and TC1 (D) cells by quantitative PCR using specific fluorescently labeled primer-probes sets. GAPDH was used as house-keeping gene. Fold changes of three independent experiments. Means±SEM. P values were calculated using the Student’s t-test. (E) PARPKOSTINGWT TC1 parental cells and their PARP1KOSTINGKO derivatives (PS4 and PS6 clones) were cultured in normal culture medium and processed for the immunoblotting-based assessment of PAR-containing proteins and STING expression. Actin levels were monitored to ensure equal loading of lanes. (F) PARhighSTINGWT, PARP1KOSTINGWT and PARP1KOSTINGKO TC1 cells were implanted subcutaneously into C57BL/6 mice (n=10 mice per group). Tumor growth was routinely monitored with a standard caliper and is reported as means±SEM. P values were calculated as compared with PARhighSTINGWT group (Wald test, type 2 ANOVA). Tumor growth is reported as means±SEM. ANOVA, analysis of variance; LLC, Lewis lung cancer; PAR, poly(ADP-ribose).
Discussion
PARP1 is an enzyme involved in the DNA damage response, and its activity is strongly induced by platinum-based chemotherapeutics.42–45 We have shown in the past that PARP1 activity is durably increased in cancer cells that have been selected by cultured in the presence of normally lethal CDDP doses, causing an increase in the PARP1 product PAR. Of note, in patients with NSCLC, a high abundance of PAR indicates poor prognosis, suggesting that PARP1 activity indeed is clinically relevant.11 Here, we show that knockout of PARP1 eliminates PAR accumulation in cisplatin resistant NSCLC cells, indicating that it is indeed only PARP1 (and none of the other PARP isoforms) that accounts for PAR accumulation. Moreover, the deletion of PARP1 reverses CDDP resistance, formally proving at the genetic level the importance of PARP1 activity.
Importantly, PARhigh cells were susceptible to cytotoxic effects of PARP1 inhibitors that were not observed with PARP1KO cells, supporting that the pharmacological inhibitors that we characterized here (niraparib, talazoparib) are indeed highly specific for PARP1, lacking major off-target effects. In accord with this interpretation, niraparib caused DNA damage, polyploidization, metabolic effects (with a prominent upregulation of nucleosides, perhaps reflecting stalled DNA synthesis) and the upregulation of immune-relevant surface molecules (PD-L1, MHC class I and II, calreticulin) when added to PARhigh cells, but had no such effects on PARP1KO cells, again supporting its specificity for PARP1. It is important to note that PARP1KO cells lacked signs of spontaneous DNA damage, had a normal ploidy, were metabolically close-to-undistinguishable from their PARhigh counterparts, and did not show any upregulation of immune-relevant surface molecules. Hence, the long-term effects of the knockout of PARP1, which is followed by a phase of clonal selection, is not the same as the pharmacological inhibition of PARP1. This may reflect kinetic effects, namely chronic (genetic) inhibition allowing for adaptive mechanisms to come into action versus acute (pharmacological) inhibition. Moreover, enzymatic inhibition of PARP1 may cause the protein to stall DNA replication,46–48 a phenomenon that would not occur in the absence of PARP1. Thus, the absence of PARP1 caused by the knockout of the gene may have different cellular effects than the drug-mediated inhibition of PARP1.
In this context, it appears intriguing that pharmacological PARP1 inhibition does reduce the growth of PARhigh tumors in a fashion that apparently is mediated by cell-autonomous effects since depletion of T cells (which are required for tumor growth reduction by anthracyclines or oxaliplatin49) does not interfere with this therapeutic effect. In sharp contrast, knockout of PARP1 did change the cancer-immune dialog in the sense that PARP1KO tumors accelerated their growth on depletion of T cells (by combined injection of anti-CD4 and anti-CD8 antibodies), a phenomenon not seen for PARhigh tumors. An interesting difference was observed between LLC and TC1 NSCLC lines, knowing that TC1 are transduced with the E7 protein of human papilloma virus, rendering them highly antigenic.50 PARP1KO TC1 cells were unable to form tumors in immunocompetent mice and only did so after depletion of T cells. Mice that had been exposed to PARP1KO TC1 cells visibly were immunized against TC1 epitopes since they became resistant against the inoculation of PARhigh TC1 cells that readily form tumors in naïve immunocompetent recipients. PARP1KO LLC cells did form macroscopic cancers in immunocompetent mice, allowing to compare their immune infiltrate with that of PARhigh LLC tumors. Of note, PARP1KO LLC tumors were more infiltrated in dendritic cells with high levels of MHC class-II expression than those found in PARhigh LLC tumors. Moreover, T cells infiltrating PARP1KO LLC tumors exhibited signs of enhanced activation (enhanced percentage of ICOS+ cells among CD8+ T cells) and reduced exhaustion (reduced percentage of PD-1+ cells among both CD4+ CD8+ T cells) when compared with T lymphocytes recovered from PARhigh LLC tumors. Moreover, a smaller percentage of Tregs infiltrating PARP1KO LLC tumors expressed ICOS compared with Tregs infiltrating PARhigh LLC tumors. These results strongly suggest that the levels of PARP1 activation shape the tumor microenvironment, establishing a causal link. Thus, the clinical observation that PARlow NSCLCs contain more CD8+ T cells than PARhigh NSCLC does not only reflect a correlation, but likely reflects a cause-and-effect relationship.13 However, no precise mechanistic explanation can be provided for the enhanced immunogenicity of PARP1KO (or PARlow) NSCLC. Indeed, when compared with their PARhigh counterparts, PARP1KO cells did not exhibit any major changes in the immunogenic surface molecules studied nor in their metabolism. Future studies, for instance of the surface proteome or the immunopeptidome, must address this question.
Another observation requiring further investigation concerns the capacity of niraparib to reduce the growth of PARP1KO tumors in mice. This effect cannot be explained by cell-autonomous effects because, at least in cell culture, PARP1KO cells are resistant against the DNA damage-inducing and cytotoxic effects of niraparib. In contrast to our expectations, this effect seems to involve T lymphocytes. At this point, the mode of action of niraparib on PARP1KO NSCLC remains enigmatic. Future studies must determine which host cell type (including fibroblasts or leukocytes other than T lymphocytes) or processes (such as angiogenesis) may be affected by niraparib to explain its paradoxical effects on PARP1KO NSCLC.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
- Data supplement 14
- Data supplement 15
- Data supplement 16
- Data supplement 17
Footnotes
Twitter @AdrienJoseph9
Contributors PJ, AJ, AL, JM, JP, and SL performed animal experiments. PJ, AJ, and AL generated cell lines and performed in vitro experiments on cultured cells and immunoblots. ES performed immunoblots. SL performed bioinformatic analyses. Immunofluorescence images were analyzed by AS, GC, ML and OK. SD, FA, and NN performed metabolome analyses. GK and MC conceived the project and wrote the manuscript. MC acts as guarantor. All of the authors critically reviewed and substantially improved the manuscript.
Funding GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)-Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association 'Ruban Rose'; Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-PhenoScreen; European Joint Programme on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); the Leducq Foundation; a Cancer Research ASPIRE Award from the Mark Foundation; the RHU Torino Lumière; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. AJ was supported by a fellowship from “Fondation ARC pour la recherche sur le cancer”. AL was supported by a fellowship from Institut Thématique Multi-Organisme Cancer (ITMO Cancer) du Plan Cancer 2014-2019. ES was supported by the University of Las Palmas de Gran Canaria (ULPGC), financed by the Ministry of Universities, granted by Order UNI/501/2021 of May 26, and by the European Union -Next Generation EU Funds.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.