Article Text

Abstract
The development of cancer vaccines to induce tumor-antigen specific immune responses was sparked by the identification of antigens specific to or overexpressed in cancer cells. However, weak immunogenicity and the mutational heterogeneity in many cancers have dampened cancer vaccine successes. With increasing information about mutational landscapes of cancers, mutational neoantigens can be predicted computationally to elicit strong immune responses by CD8 +cytotoxic T cells as major mediators of anticancer immune response. Neoantigens are potentially more robust immunogens and have revived interest in cancer vaccines. Cancers with deficiency in DNA mismatch repair have an exceptionally high mutational burden, including predictable neoantigens. Lynch syndrome is the most common inherited cancer syndrome and is caused by DNA mismatch repair gene mutations. Insertion and deletion mutations in coding microsatellites that occur during DNA replication include tumorigenesis drivers. The induced shift of protein reading frame generates neoantigens that are foreign to the immune system. Mismatch repair-deficient cancers and Lynch syndrome represent a paradigm population for the development of a preventive cancer vaccine, as the mutations induced by mismatch repair deficiency are predictable, resulting in a defined set of frameshift peptide neoantigens. Furthermore, Lynch syndrome mutation carriers constitute an identifiable high-risk population. We discuss the pathogenesis of DNA mismatch repair deficient cancers, in both Lynch syndrome and sporadic microsatellite-unstable cancers. We review evidence for pre-existing immune surveillance, the three mechanisms of immune evasion that occur in cancers and assess the implications of a preventive frameshift peptide neoantigen-based vaccine. We consider both preclinical and clinical experience to date. We discuss the feasibility of a cancer preventive vaccine for Lynch syndrome carriers and review current antigen selection and delivery strategies. Finally, we propose RNA vaccines as having robust potential for immunoprevention of Lynch syndrome cancers.
- CD8-Positive T-Lymphocytes
- Genetic Markers
- Immunogenicity, Vaccine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
At the dawn of the 20th century, infectious disease was the most common cause of death. Advances in medical sciences led to the elucidation of infectious disease mechanisms and clinical translation of potent vaccines and therapies. Today, we can leverage and build on the significant advances in our mechanistic understanding of the genomic architecture of cancer, cancer predisposition, and very recent rapid advances in vaccine technology. Such advances have reduced the death burden from common infections by polio, meningococcus, and many others.1–5 Taking these advances together with cancer immune prevention and interception vaccines, as well as improved therapies and early detection technologies, we now have the potential to substantially reduce cancer mortality in the 21st century.
Over the past two decades, cancer prevention strategies have undergone a major paradigm shift. Primary cancer prevention1 6 is thought as best targeted to individuals with increased cancer risk resulting from inherited cancer predisposition, a personal history of premalignant neoplasms, or carcinogen exposure.1 3 In parallel, another paradigm shift has been our understanding of the critical role of immune surveillance in primary cancer predisposition, prevention,2–4 7 and advanced cancer immunotherapy. Most cancer immunotherapy approaches exploit the killing capacity of CD8 +cytotoxic T cells. These cells can recognize tumor-specific antigens including neoantigens generated from mutant proteins, which are presented on the tumor cell surface via major histocompatibility complex (in humans, human leucocyte antigen, HLA) class I molecules. Vaccination strategies against tumor-specific neoantigens aim to enhance the specific recognition and killing of neoantigen-expressing tumor cells by T cells.8–10
Origins of tumor immunology
Physicians’ attempts to harness the immune system in the treatment of patients with cancer has been reported in sources from ancient times. In the early 20th century, Paul Ehrlich hypothesized that host immunity could prevent tumor formation.11 In parallel, William B. Coley observed tumor shrinkage in sarcoma patients following tumor site bacterial infection, local erysipelas and systemic inflammation. Based on this clinical observation Coley actively inoculated bacteria to induce immune responses at the tumor site and later developed Coley’s Toxin, a mixture of inactivated bacteria,12 as a therapy. This innovation established the paradigm of immunotherapy. However, the concept of harnessing the immune system for cancer prevention arose in parallel from observational epidemiology: a study in 1929 described a lower frequency of cancer in patients with tuberculosis (TB) and a higher frequency of TB among cancer survivors.13 In the following, Bacille Calmette-Guérin (BCG), a live strain of Mycobacterium bovis widely employed as an attenuated vaccine to prevent TB, BCG was used to augment cancer therapy in multiple clinical trials for different tumor types, such as non-muscle invasive bladder cancer or melanoma.14 Notably, intravesical BCG is still used clinically for immunotherapy of superficial bladder cancer and carcinoma in situ. A lower leukemia incidence was reported epidemiologically in children who had received neonatal BCG immunization, indicating a potential tumor-preventive effect.15 16
In parallel to these clinical observations, experimental evidence for immune surveillance and immune protection against tumors was increasing.
In the early years of the 20th century, studies on mouse sarcoma models17 indicated the possibility of systemic protection against tumor growth. The work of Gross18 on chemically induced sarcoma re-inoculation experiments provided evidence for tumor strain-specific immune protection in mice. However, it was not until a decade later that Foley19 and Prehn and Main20 confirmed the results of Gross, finding that each individual chemically induced tumor elicited tumor-specific immunity, suggesting for the first time that each individual tumor carries a unique set of antigens. These results provided proof-of-concept that immunization against defined tumor subtypes may 1 day become clinically feasible. Importantly, Klein et al21 could show that the immunological protection against chemically induced sarcoma is mainly mediated by lymphocytes.
With increasing knowledge about the mechanisms of anti-tumor immune responses, more refined strategies have been developed. On the one hand, sophisticated approaches to generally support the host’s antitumor response capacity have been developed, culminating in the clinical success of immune checkpoint blockade (ICB).22 23 On the other hand, the identification of antigens unique to or overexpressed in tumor cells has opened the perspective to cancer vaccines representing tailored interventions that specifically stimulate the expansion and activation of tumor-antigen specific immune cells.
Immune checkpoint blockade
Since the discovery of immune checkpoint molecules CTLA-4 and PD-1/PD-L1 as key inhibitory regulators of immune cell activity in the mid-1990s,24 25 antibody-based (and more recently small molecule inhibitor) strategies to selectively target these checkpoints have been developing rapidly. In the last decade, the clinical introduction of antibodies targeting immune checkpoints has fundamentally reshaped oncology. In 2011, ipilimumab, an antibody directed against the immune checkpoint CTLA-4, was approved by the US Food and Drug Administration, followed by the approval of two anti-PD-1 antibodies pembrolizumab and nivolumab in 2014. These new therapies, alone and in combination, showed remarkable success in patients with advanced melanoma, demonstrating high disease control rates and inducing full and durable remissions in a subset of patients.26 Clinical observations showed that efficacy of ICB was restricted to certain tumor types, and tumor mutational burden was identified as a predictor of therapy responsiveness.27 28
These observations indicated that the molecular pathogenesis pathways and the resulting mutational signatures in cancer cells were major determinants of their susceptibility toward immune therapy. Notably, cancers triggered by a deficiency of the DNA mismatch repair system (MMRd cancers) have an exceptionally high mutational burden.29 30 First clinical trials evaluating ICB in MMRd patients with cancer were highly successful,31 32 leading to the first tissue-agnostic approval of pembrolizumab for irresectable metastasized MMRd cancers irrespective of the organ site.33 However, due to a pronounced side effect profile, ICB is not suitable for immunoprevention of cancer.
Cancer vaccines
The hypothesis that cancer vaccines can support the immune system in its fight against cancer is based on the idea that cancer cells express antigens that can be detected as foreign by the immune system. Before the concept of somatic mutations in cancer cell genomes were available, the connection between the immune system and cancer was founded mainly on clinical observations.
However, several promising approaches to prevent cancer by specific stimulation of the immune system through vaccines have been developed and tested in preclinical and/or clinical studies: Vaccination with human papillomavirus (HPV)-derived antigens led to T helper 1 associated regression of HPV-associated premalignant lesions in a high proportion of patients.6 34–37 However, no regression of late stage HPV16-induced cancers was observed after HPV16 long peptide vaccination,38 highlighting the importance of vaccination before the local and systemic onset of immune suppression.36 39–41 Vaccination with HER-2 pulsed dendritic cells resulted in increased complete response of HER-2 positive ductal carcinoma in situ patients.42 43 For colorectal tumor prevention, a MUC1-derived peptide unmasked after tumor-associated carbohydrate changes has been proposed (NCT007773097, NCT02134925) as a preventive vaccine target for individuals with previous premalignant lesions in the colon. Over 40% of vaccinated participants showed long-lasting memory responses and MUC1-specific antibodies, and mouse model data showed promising results with regard to tumor prevention.44–48 Notably, MUC1 vaccination studies showed that high numbers of myeloid deriver suppressor cells (MDSCs) were observed in part of the vaccinated participants, suggesting that an immunosuppressive tumor microenvironment (TME) can already be established in the premalignant stage of advanced adenoma,45 underscoring the benefit of vaccinating before tumorigenesis and onset immune suppression also in the colorectum. MUC1 is additionally being tested as a preventive vaccine in smokers at high risk for lung cancer49 (NCT03300817).
In addition to MUC1, vaccination for colorectal cancer prevention has been evaluated using four candidate overexpressed tumor-associated antigens (TAAs). Two of the candidate TAAs induced peptide-specific T cell responses and protected 50% of the mice from tumor development.50 Overall, these data suggest that preventive vaccination with TAA can have a preventive effect, however, central tolerance remains an important consideration that reduces the pool of reactive T cells against self-antigens. Vaccination against neoantigens has the potential to overcome the tolerance problem; however, it requires the exact knowledge of the mutation events and resulting antigen structures occurring during tumor development. These latter prerequisites are given in cancers with DNA MMRd.
DNA mismatch repair deficiency and microsatellite instability
Genomic instability is a crucial enabling mechanism in human cancer. The majority of human cancers display chromosomal instability, which results in chromosomal aberrations. However, a significant subset of cancer with increasing clinical relevance is triggered by an inactivation of the DNA mismatch repair system.
MMRd cancers develop in the colorectum (about 15% of all colorectal cancers), the endometrium (up to 30%), and other organ sites at lower frequency.51
Due to insufficient DNA repair, MMRd cancers accumulate numerous mutations during their evolution. The mismatch repair system physiologically is mainly responsible for detecting and correcting base mismatches resulting from polymerase slippage during DNA replication.52 These mismatches typically affect repetitive sequence stretches termed microsatellites. In MMRd cells, uncorrected mismatches become manifest as insertion/deletion (indel) mutations. Indels of coding microsatellites (cMS) located in tumor suppressor genes, such as TGFBR2 or ACVR2 represent key driver mutations responsible for malignant transformation and progression of MMRd cells into manifest cancers.53 54 Due to the functional relevance of cMS indels, their distribution in manifest MMRd cancers is non-random and follows Darwinian principles of selection (figure 1). Importantly, recurrent cMS indels are well known and have been documented by many independent studies for different MMRd tumor types.53–56

Schematic illustration of mismatch repair deficient (MMRd) cancer evolution. Lynch syndrome is caused by mono-allelic germline DNA mismatch repair gene variants. Second hit inactivation of the remaining functional allele gives rise to DNA mismatch repair deficiency (flash). Insertion and deletion (indel) mutations that occur during DNA replication cannot be repaired, leading to the accumulation of mutations as the affected cell clone divides. Indel mutations affecting coding microsatellites (cMS) result in the generation of potentially immunogenic frameshift peptide (FSP) neoantigens. Due to Darwinian evolution, CMS mutations that favor cell survival and tumor growth will accumulate. As a result, manifest cancers share growth-promoting indels at CMS and a pool of FSP neoantigens. FSP-derived epitopes are presented on HLA class I molecules on the surface of the tumor cells, enabling potential recognition by CD8 +T cells. HLA, human leucocyte antigen.
Such mutations not only inactivate tumor-suppressive pathways, but also trigger shifts of the translational reading frame, thereby giving rise to the generation of novel frameshift peptide (FSP) sequences.57 In contrast to point mutations, which lead to the change of single amino acids, indel-mediated frameshifts can lead to long stretches consisting of amino acid sequences entirely foreign to the host’s immune system.57 Hence, the immunogenicity of MMRd cancers is not only caused by the sheer number of somatic mutations, but also the high number potential epitopes in FSPs triggered by indel mutations.
Lynch syndrome and sporadic MSI
The pathogenesis of MMRd cancers is heterogeneous. They can develop sporadically, typically in the elderly female patients. Sporadic MMRd cancers are either caused by promoter methylation silencing the MMR gene MLH1, often associated with the CpG island methylator phenotype (CIMP),58 or by biallelic somatic MMR gene mutations.59 However, a significant subset of MMRd cancers represent hallmark tumors of the most common inherited cancer predisposition syndrome, Lynch syndrome.60 Lynch syndrome is caused by monoallelic germline variants affecting one of the four mismatch repair genes MLH1, MSH2, MSH6 or PMS2. In about 90% of Lynch syndrome-associated cancers, expression loss of the MMR proteins MLH1 or MSH2 is observed, resulting from second hit inactivation of the remaining functional allele.61 62
Lynch syndrome carriers have a lifetime risk of up to 80% of developing MMRd cancers, mainly affecting the colon and the endometrium, similar to the typical presentations of MMRd cancers in general.63 Regular colonoscopy is therefore recommended to carriers to detect and remove precancerous lesions or to diagnose incident cancers early.64 Epidemiology data indicate that colonoscopy surveillance of Lynch carriers has limited effectiveness for colorectal cancer prevention, possibly because part of Lynch colorectal cancers develop too rapidly or from non-polypous lesions that escape detection by colonoscopy.65 66 Such non-polyp precursor lesions may be associated with MLH1 rather than MSH2 germline variants; however, additional studies are required to validate this association.66 The fact that colonoscopy as a prevention measure does not eliminate the colorectal cancer burden in Lynch carriers underlines the need for novel primary prevention approaches, such as vaccination-based prevention.
Several studies reported elevated immune cell recruitment and infiltration in Lynch-related compared with sporadic MMRd colorectal cancers.67–69 This observation potentially reflects lifelong ‘self-education’ of Lynch patients’ immune systems, which recurrently are confronted with MMRd cells expressing FSPs.70–72 So far, however, no significant difference in immune therapy response rates between Lynch syndrome carriers and sporadic MMRd patients with cancer have been reported.73 This counterintuitive observation may be explained by the fact that Lynch-associated MMRd cancer cells have evolved particularly effective strategies of evading attacks by the host’s immune cells. This hypothesis is supported by several studies reporting an elevated frequency of immune evasion mechanisms in Lynch-associated compared with sporadic MMRd colorectal cancers.67
Immune evasion and immunoediting in MMRd cancers
Immune evasion of MMRd cancers according to current knowledge can take place on three levels: complete loss of HLA class I antigen presentation, partial or selective loss of HLA class I, or counterselection of mutations that induce particularly immunogenic neoantigens.
Most commonly, immune evasion of MMRd colorectal cancer cells is caused by mutations of the Beta-2-microglobulin (B2M) gene, which have been reported in up to 30% of cases.67 74 B2M mutations lead to a breakdown of the HLA class I antigen presentation machinery of tumor cells.75 Whereas somatic B2M mutations are the most common alteration contributing to immune evasion in MMRd colorectal cancer and also gastric cancer, MMRd endometrial cancers very rarely display B2M mutations. In contrast, JAK1 mutations have been reported as an alternative route of immune evasion prevalent in MMRd endometrial cancers and other MMRd cancer types.76 77 MMRd colorectal cancers devoid of B2M mutations show other alterations that may explain their survival in a hostile immune milieu. In about half of these B2M-wild type cancers, other components involved in antigen presentation are affected by alterations, including mutations of HLA class I heavy chain encoding genes HLA-A, HLA-B, and HLA-C, as well as the transporter of antigen presentation TAP1/TAP2 and the HLA class I transactivator NLRC5.78 Alternatively, MMRd cancers with no detectable alterations in the antigen processing and presentation machinery display an interesting pattern of FSP-generating indel mutations: mutations that are predicted to result in highly immunogenic HLA class I epitopes occur at a significantly lower frequency compared with mutations resulting in peptides that are poor HLA class I binders.7
Counterintuitively, immune evasion phenomena in MMRd cancer, such as loss of B2M and HLA class I on the cell surface, do not result in a worse prognosis, but apparently in an improved outcome and less aggressive clinical behavior. Patients with B2M-mutant MMRd colorectal cancer show lack of disease recurrence and prolonged survival, as has been demonstrated by independent studies,79 80 suggesting that B2M-mutant cancers in most cases represent a surgically manageable disease. B2M mutations are also associated with a lack of distant hematogeneous metastasis in MMRd colorectal cancer and in completely different tumor types, such as uveal melanoma, indicating a general biological mechanism.74 81–83 Although the mechanistic basis still remains partly elusive, it has been suspected that natural killer (NK) cells may restrict metastasis formation of B2M-mutant cancers, because they lack the NK cell-inhibitory effect of HLA class I antigens.82 84 85
From therapy to immunoprevention in Lynch syndrome
The occurrence of frequent immune evasion phenomena in Lynch syndrome-associated cancers supports the concept that they evolve under continuous immune surveillance. It also implies that immune interventions to support this surveillance and to increase the likelihood of immune-mediated elimination of cancer cells or precancerous cells should be applied timely, before immune evasion occurs.
Conceptually, primary prevention strategies that can be applied before the emergence of initiated precancer cell clones develop would be ideal to reduce the probability of cancer development in Lynch carriers. However, available potent interventions, such as ICB are often associated with severe adverse reactions, which hamper their application as preventive measures in healthy, tumor-free individuals.32 75 Therefore, alternative approaches such as vaccines are required to support natural immune surveillance in Lynch carriers.
Evidence for endogenous immune surveillance in Lynch syndrome
Besides the observation of immune evasion phenomena, which indirectly imply the existence of immune surveillance in Lynch syndrome, there are more direct clues. First, the study by Schwitalle et al reported FSP-specific T cell immune responses in MMRd patients with colorectal cancer, but notably also in Lynch carriers who never had developed any cancer or adenoma.86 These immune responses indicate the clinically unnoticed exposure of the immune system toward FSPs generated by MMRd cells during life. Non-neoplastic or early dysplastic MMRd cells have been shown to be highly prevalent in the intestines and other organs of Lynch carriers,71 87 and mutations generating FSPs have been proven in colonic MMRd crypts.88 Moreover, pronounced local immune responses in Lynch syndrome have been demonstrated in Lynch colorectal cancers, adenomas, MMRd crypts and even entirely normal-appearing colonic mucosa.89–92
All these observations together suggest that active surveillance by the adaptive immune system plays a major role in controlling and suppressing the outgrowth of MMRd cancers in individuals affected by Lynch syndrome. Immune surveillance thus likely contributes to the limited penetrance of the disease60 63 through the elimination of precancerous MMRd cells.65 This hypothesis is further supported by the clinical observation that immunosuppression can unmask Lynch syndrome, as Lynch carriers under immunosuppressants following organ transplantation can develop a multitude of rapidly growing cutaneous tumors.93
The concept of cancer-preventive vaccines in Lynch syndrome
Based on the strong evidence of naturally existing immune surveillance as a major tumor-suppressive factor in Lynch syndrome, further reducing tumor risk by stimulating the immune system is a highly promising perspective for Lynch carriers.
Although vaccines have significantly contributed to the reduction of serious, life-threatening infectious diseases, early generation cancer vaccines largely failed to fulfill the high initial expectations.94 We now understand that vaccination-triggered stimulation of the activation of antigen-specific immune responses are often insufficient to control and eliminate advanced cancer, mainly because cancer cells have many opportunities to evolve and successfully dodge the host’s immune attacks. Several critical steps need to be completed for a successful antitumor response: release of cancer antigens through cancer cell death or vaccination, cancer antigen presentation on antigen presenting cells, priming and activation of T cells, migration of activated T cells to the tumor site, infiltration of T cells into the tumor, recognition of the tumor cells by the T cells via the TCR-peptide-HLA complex, and finally killing of cancer cells.95 96 The complex set of host, tumor and environmental factors that influence the success of an immune response had not been taken into account before.97 Advanced cancer cells may have already intervened in many of the critical steps, such as by establishing an immunosuppressive TME that inhibits any effector T cells previously activated by vaccination, or by dysregulating their own antigen presentation machinery, among others.95 As such, cancer vaccines have not performed like vaccines against infectious diseases, which by nature are foreign to the hosts immune system and allows for a robust immune system activation by vaccines.
On the other hand, preventive vaccination allows targeting and elimination of arising tumor cells before the onset of an immunosuppressive TME or evolution of other immune escape mechanisms. However, vaccination against cancer requires the possibility to predict antigens that are expected to occur in the tumors, which may be prevented by the vaccine. In most tumor types, mutations are either highly variable and therefore not predictable or associated with low immunogenicity of the resulting peptides.98 99 Lynch syndrome, in contrast, represents an ideal target for evaluating the feasibility and effectiveness of cancer prevention by vaccination.57 This is due to the following reasons: (1) predictable MMRd-induced somatic mutation landscape in Lynch tumors leading to (2) a predictable set of recurrent FSPs as a neoantigen pool, (3) the existence of a well-defined high-risk population and (4) a high lifetime tumor risk allowing monitoring the preventive effectiveness of vaccination. Vaccines to enhance the immune system’s capacity to detect and attack emerging pre-cancer cell clones hold high potential for reducing tumor incidence in Lynch carriers. In the following, we will discuss previous pioneering studies, the current status and future directions.
FSPs as vaccine targets
Recurrent frameshift neoantigens are a major reason why Lynch syndrome represents an ideal scenario for cancer-preventive vaccines. Frameshifted peptides result in the generation of peptides with completely new amino acid sequences starting from the site of the frameshift. This new stretch of amino acids is foreign to the immune system and can potentially encompass multiple immunogenic non-self HLA class I and class II epitopes,100 whose length typically ranges from 8–10101 to 13–19102 amino acids, respectively. A study that compared somatic mutation load with predicted immunogenic peptides found that frameshift indels result in the generation of more neoantigens than point mutations.103 Therefore, frameshifted peptides are potentially better vaccine targets than proteins derived from point mutations that result in a single amino acid change.100 103 104 T cell activation requires binding of the T cell receptor to the peptide-HLA complex on the surface of professional antigen presenting cells.105 Thereafter, the activated effector T cell can recognize the same peptide-HLA complex on the surface of target cancer cells to be eliminated. MMRd cancer cells carry a high number cMS mutations that result in the generation of multiple FSPs neoantigens that are potentially presented on the surface of the cell in complex with HLA molecules, rendering them detectable by the immune system and as such, prime targets for an FSP-based vaccine.7
The immunogenicity of certain neoantigens derived from the MMRd-induced FSP pool had already been demonstrated more than 20 years ago.106 107 The detection of FSP-specific T cell and humoral immune responses in the peripheral blood of Lynch patients confirmed processing and presentation of these antigens indirectly.86 Additionally, a study of two unrelated LS families identified three immunogenic neoantigens that elicit CD8 +specific T cell responses.108 More recently, an immunopeptidomics study for the first time formally confirmed HLA-mediated presentation of FSP-derived epitopes on the cell surface.109
Notably, transcriptional errors of cMS and mis-splicing of exons have also been proposed as mechanisms by which FSP neoantigens are generated in MMR proficient tumor cells,110–112 as RNA transcription, splicing and quality control systems are disturbed in tumor cells.113 A preventive vaccine composed of such FSP neoantigens is being tested in a canine clinical trial.114
Preclinical and clinical experience
The first-in-human clinical trial published in 2020 was based of a combination of three recurrent FSPs TAF1B(−1), HT001/ASTE1(−1), and AIM2(−1), all derived from cMS indels occurring with a high frequency in MMRd colorectal and endometrial cancer.115 The study with primary endpoints safety and immunological effectiveness included 22 participants with history or current MMRd colorectal cancer. All patients receiving the full cycle of vaccinations developed pronounced humoral and T cell responses against the vaccine peptides. T cell responses were mainly CD4 +responses, CD8 +responses were detected in 9 of the 22 patients, probably indicating that, according to in silico predictions, certain HLA class I genotypes were required to develop CD8 +responses. This finding underlines the need to account for HLA class I diversity of target populations when selecting FSPs as vaccine targets. No systemic vaccine-related side effects occurred in vaccinated patients, demonstrating the general feasibility of the approach.
A viral-vectored vaccine encoding 209 shared FSPs against MSI tumors, referred to as Nous-209, was recently developed by Nouscom S.R.L.116 The FSPs were selected based on analysis of frameshift mutations overlapping with cMSs in the cancer genome atlas (TCGA) for MSI colorectal cancer, gastric cancer, and endometrial cancer tumor samples versus matched normal samples. Further selection criteria for the 209 FSPs included prevalence of ≥5% for each tumor type, in ≤25% of alleles of matched healthy tissues, in <2% of alleles of healthy tissues samples,≥8 amino acid length and mapping to protein coding regions. Interferon gamma (IFNg) Enzyme-Linked Immunosorbent Spot assay (ELISpot) performed with splenocytes from vaccinated mice restimulated with pools of peptides showed induction of specific T cell responses. However, it is not clear how neoantigen peptide T cell responses were influenced and restricted by presentation on mouse host HLA, rather than human HLA and consequently these results cannot be directly translated to immunogenicity in humans. Also, due to a sizeable number of FSP neoantigens and possible multiple epitopes, immunological interference is of concern as this could dilute immune response to the strongest neoantigens. To test this, Leoni et al vaccinated a group of mice only with FSP 1–45 and another with all 209 FSPs. While IFNg ELISpot showed no significant difference, there was a reduction of approximately 20% in the intensity of the specific response to the peptide pool 1–45 in the mice vaccinated with the 209 FSPs. Again, these data are in the context of xeno-presentation by mouse HLA that makes interpretation complex.
A phase I clinical trial combining the Nous-209 vaccine with anti-PD-1 antibody pembrolizumab is currently underway in patients with metastatic colorectal cancer, gastric and gastro-esophageal cancer (NCT04041310).116 Interim results show that out of 12 evaluable subjects, 7 had partial responses, 2 stable disease, and 3 progressive disease. Importantly, the combination treatment was reported to be safe, tolerable and immunogenic for T cells as shown by ex-vivo IFNg ELISpot.117 However, because the study used combination therapy, how much of the response was attributable to pembrolizumab and how much to the Nous-209 vaccine is unclear at present.
Future clinical trials for Lynch syndrome vaccines need to address safety and efficacy to reduce the burden of cancers and advanced neoplasia. Evaluation of the immunological effectiveness should encompass T cell activation, humoral immune responses and analysis of local immune environments.44 To that end, assays can be applied that have been commonly used in previous cancer vaccine trials, such as standardized ELISpot, ELISA and flow cytometric analysis of immune cell subtypes including MDSCs. In addition, Lynch syndrome-specific markers could assess microsatellite instability in the non-tumoral colonic mucosa or immune cell infiltration to monitor MMR-deficient crypt foci and local mucosal immune surveillance.88 92
Current research questions and perspectives
Antigen selection
In contrast to most other tumor types, Lynch syndrome-associated MMRd cancers present with high numbers of mutational frameshift neoantigens, which constitute a large pool of potential vaccine targets. One of the key questions for designing the optimal cancer-preventive vaccine is therefore the selection of the ideal antigen composition. Previous approaches have followed different rationales, either focusing on recurrent FSPs derived from functionally relevant driver mutations or combining large numbers of more than 200 frameshift antigens in one vaccine.115 116 118
Common to both approaches is that a major criterion for the selection of antigens is the occurrence of the respective indel mutations in MMRd cancers or precancerous lesions. Here, different sources of tumor mutation typing data provide divergent information, as somatic mutation profiling of Lynch cancers has technically been limited by the fact that short-read next-generation sequencing approaches have a limited sensitivity for detecting indels affecting repetitive sequence stretches.119
Another aspect that needs to be considered is that diversity of the HLA class I and class II gene loci will lead to different antigen spectra visible for the immune system in different patients. A recent study suggested that MMRd cancer patients’ HLA genotype is reflected in the somatic tumor mutation landscape.7
Delivery strategies
Vaccination with peptide FSPs has been shown to be safe and feasible without inducing severe systemic adverse reactions. However, a subset of Lynch patients vaccinated with FSP peptides plus the adjuvant Montanide developed pronounced injection site reactions, which has been previously associated with Montanide.115 Thus, vaccine delivery strategy is an important consideration. Clinical and pre-clinical trials evaluating FSP vaccination for Lynch and MMRd patients with cancer have used peptide vaccination,115 120 dendritic cell vaccination,118 121 or vector-based vaccination.116
For dendritic cell vaccines, protein-adjuvant mix is loaded ex vivo onto autologous dendritic cells derived from patient monocytes, which are reinfused into the patient. Advantages of this delivery system include no extraneous non-specific antigens, so on-target immunodominance is maintained and no vector immune interference, such as from viral vectors. However, since antigens are endocytosed by professional APC that are manipulated in vitro, antigens can have inefficient entry into the HLA-I pathway and poor presentation to CD8+ T cells in vivo. Additionally, antigens bypass endogenous post-translational modifications and proteasomal processing. Loading of ex vivo autologous DCs is also laborious and costly. DC vaccines are often designed with infectious diseases in mind, and as such it’s common for Th2- or Tfh-polarizing adjuvants to be used; however, the ideal adjuvants for skewing toward Th1 and CD8 immunity are less understood and require more study.
Viral-vector vaccines are well studied. Important advantages of viral delivery are viral “aduvanticity“ to drive immune response, efficient antigen delivery and endogenous post-translational modification such as phosphorylation or acetylation, that may be important for T cell recognition. Disadvantages can include viral vector protein cytotoxicity, host viral infection response to downregulate HLA-I antigen presentation and dampened costimulatory ligand expression to inhibiti DC maturation. Viral vectors can also cause immune interference by shifting immunodominance away from the vaccine antigen cargo and toward viral antigens that elicit strong memory responses.
A novel delivery system is mRNA-lipid nanoparticles (mRNA-LNP). This can efficiently deliver vaccine antigen cargo into the cytosol (by facilitating endosomal escape) without any extraneous antigens to drive immune interference. mRNA-LNP also has the advantage of enabling antigen post-translational modifications and larger protein cargo capacity This platform can also incorporate potential advances such as cell type-specific antibody targeting or vaccine payloads that include checkpoint inhibitors or Treg inhibitors to prolong immune response duration. However, compared with viral vector or peptide vaccines, there are practical challenges with large-scale manufacturing and long-term cold supply chain storage—as observed with COVID-19 mRNA vaccines. Also, because mRNA-LNP vaccines drive extremely robust immune response, its intensity may provoke T and B cell exhaustion that reduce duration of immunity.
Recently, for advanced melanoma patient immunotherapy, BioNTech reported clinical data for FixVac, an intravenously administered liposomal RNA vaccine encoding four TAA prevalent in melanoma-in a first-in-human phase I trial (Lipo-MERIT trial, ClinicalTrials.gov identifier NCT02410733). This study showed early data that FixVac, alone or in combination with IV anti-PD-1 therapy, caused durable objective responses in checkpoint-inhibitor (CPI)-experienced patients with unresectable melanoma.122 Clinical responses were associated with the induction of strong CD4 +and CD8+T cell immunity against the vaccine antigens and safety studies did not show adverse events unexpected from immunotherapy. Thus, LNP RNA vaccination has strong potential as a technology to be repurposed for immunoprevention. The unmet medical need and challenges for cancer-preventive vaccines in Lynch syndrome are summarized in figure 2.

{kind=link}
{kind=link}
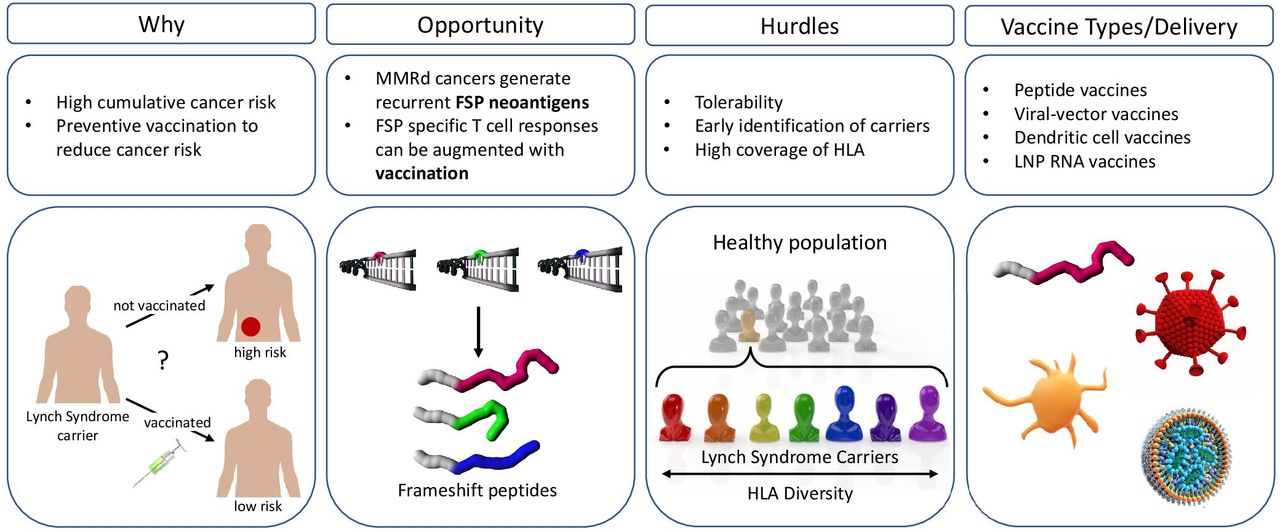
Opportunities and hurdles for the development of a frameshift peptide (FSP) vaccine for the prevention of mismatch repair deficient (MMRd) cancers. Why: Lynch syndrome (LS) carriers have a 50% to 80% lifetime risk to develop cancer. therefore, there is a big unmet medical need for preventive approaches to reduce LS cancer incidence. Opportunity: errors that occur during DNA replication, such as slippage events (marked with an arrow) that lead to insertion and deletion (indel) mutations, cannot be repaired. Indel mutations in coding microsatellites result in the generation of immunogenic FSP neoantigens that can be targeted by FSP-specific cytotoxic T cells augmented with vaccination. Hurdles: for an effective administration of a preventive cancer vaccine, Lynch syndrome carriers should be identified early. Preventive vaccine strategies should consider coverage to prevent different tumor types, tolerability, and account for HLA diversity. Vaccine delivery: options for vaccine delivery methods comprise among others peptide vaccines, viral-vector vaccines, loaded dendritic cells or lipid nanoparticle RNA vaccines. HLA, human leucocyte antigen.
Combination of vaccines with chemoprevention
Non-steroidal anti-inflammatory drugs (NSAIDs), particularly aspirin (ASA), have been intensively studied for gastrointestinal (GI) cancer prevention. NSAIDs reduce cyclooxygenase 1 and 2 production of prostaglandin E2 (PGE2), which binds to EP1-4 receptors.123 PGE2 drives intestinal tumorigenesis by both promoting protumorigenic EP2/4 driven intestinal epithelial and stem cell proliferation and inhibiting immune surveillance and immune-interception of tumor neoantigens.124 125 ASA reduces Lynch syndrome colorectal cancer penetrance and is widely used for Lynch syndrome GI cancer prevention.123 126 However, a recent large-scale (approximately 20 000 participants) cancer chemoprevention randomized clinical trial in community-dwelling older men and women has raised questions whether ASA may actually increase overall pan-cancer rates and mortality.127 Recently, Naproxen (NAP), a propionic acid NSAID derivative, has shown greater cancer-preventive activity compared with ASA in Lynch syndrome mouse models, and increased immune surveillance in Lynch syndrome patients128 129 suggesting potential chemopreventive benefits of NAP regimen in targeted cohorts, such as Lynch syndrome. NAP treatment enhanced activation of dendritic cells, macrophages, T and B cells without increasing the number of lymphocytes in the colorectal mucosa, suggesting immune cell activation over recruitment or proliferation.128 The combination of vaccination to elicit neoantigen-specific T cells with NSAIDs that enhance local immune surveillance is therefore promising, as NSAIDs might prevent the establishment of an immunosuppressive TME.
We recently showed in pre-clinical studies that the combination of NAP and recurrent FSP vaccine was more effective to reduce tumor burden in Lynch syndrome mouse models than either NAP chemoprevention or recurrent FSP vaccination immunoprevention alone.120 The observation that only FSP vaccination, but not NAP treatment, enhanced CD4 +and CD8+TIL counts in Lynch syndrome mice support the hypothesis that FSP vaccination and NAP work by different mechanisms. That NAP in Lynch syndrome mice seem primarily to act as anti-inflammatory further indicates that the combination of FSP vaccine and NSAIDs achieves beneficial effects through complementary anti-inflammatory (NSAIDs) and enhanced adaptive immunity (FSP vaccine) mechanisms, rather than by together both driving increased immune surveillance. Our study for the first time addressed the important question as to whether FSP vaccination and NSAID chemoprevention are synergistic or antagonistic in Lynch syndrome cancer prevention. The increased survival and reduced tumor burden of mice receiving both FSP vaccination and NAP treatment is consistent with a role for combined chemo- and immunoprevention to reduce tumor burden in a practical manner.
Conclusions
At the beginning of the 20th century, advances in vaccine science led to a significant reduction in morbidity and mortality from infectious diseases, such as polio and meningococcus. Today, because of recent advances in the understanding of inherited cancer, cancer genomics and tumor immunology, we have similar potential, this time to substantially reduce cancer mortality in the 21st century. Lynch syndrome is a paradigmatic disease for a cancer immunoprevention vaccine. Present and future immunoprevention vaccine studies for Lynch syndrome will guide technology development that can then be applied more broadly to help individuals at increased cancer risk from other etiologies.
Summary of clinical and preclinical studies assessing the prevention of Lynch syndrome
Ethics statements
Patient consent for publication
References
Footnotes
Contributors AH-S, MG, KY, SSS, MK and SL wrote this review.
Funding This study was funded by Division of Cancer Prevention, National Cancer Institute (U01 CA233056).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.