Article Text

Abstract
Background Immune checkpoint blockade (ICB) has achieved unprecedented success in treating multiple cancer types. However, clinical benefit remains modest for most patients with solid malignancies due to primary or acquired resistance. Tumor-intrinsic loss of major histocompatibility complex class I (MHC-I) and aberrations in antigen processing machinery (APM) and interferon gamma (IFN-γ) pathways have been shown to play an important role in ICB resistance. While a plethora of combination treatments are being investigated to overcome ICB resistance, there are few identified preclinical models of solid tumors harboring these deficiencies to explore therapeutic interventions that can bypass ICB resistance. Here, we investigated the combination of the epigenetic modulator entinostat and the tumor-targeted immunocytokine NHS-IL12 in three different murine tumor models resistant to αPD-1/αPD-L1 (anti-programmed cell death protein 1/anti-programmed death ligand 1) and harboring MHC-I, APM, and IFN-γ response deficiencies and differing tumor mutational burden (TMB).
Methods Entinostat and NHS-IL12 were administered to mice bearing TC-1/a9 (lung, HPV16 E6/E7+), CMT.64 lung, or RVP3 sarcoma tumors. Antitumor efficacy and survival were monitored. Comprehensive tumor microenvironment (TME) and spleen analysis of immune cells, cytokines, and chemokines was performed. Additionally, whole transcriptomic analysis was carried out on TC-1/a9 tumors. Cancer Genome Atlas (TCGA) datasets were analyzed for translational relevance.
Results We demonstrate that the combination of entinostat and NHS-IL12 therapy elicits potent antitumor activity and survival benefit through prolonged activation and tumor infiltration of cytotoxic CD8+ T cells, across αPD-1/αPD-L1 refractory tumors irrespective of TMB, including in the IFN-γ signaling-impaired RVP3 tumor model. The combination therapy promoted M1-like macrophages and activated antigen-presenting cells while decreasing M2-like macrophages and regulatory T cells in a tumor-dependent manner. This was associated with increased levels of IFN-γ, IL-12, chemokine (C-X-C motif) ligand 9 (CXCL9), and CXCL13 in the TME. Further, the combination therapy synergized to promote MHC-I and APM upregulation, and enrichment of JAK/STAT (janus kinase/signal transducers and activators of transcription), IFN-γ-response and antigen processing-associated pathways. A biomarker signature of the mechanism involved in these studies is associated with patients’ overall survival across multiple tumor types.
Conclusions Our findings provide a rationale for combining the tumor-targeting NHS-IL12 with the histone deacetylase inhibitor entinostat in the clinical setting for patients unresponsive to αPD-1/αPD-L1 and/or with innate deficiencies in tumor MHC-I, APM expression, and IFN-γ signaling.
- immunotherapy
- tumor microenvironment
- cytokines
- programmed cell death 1 receptor
- combined modality therapy
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- tumor microenvironment
- cytokines
- programmed cell death 1 receptor
- combined modality therapy
What is already known on this topic
Despite the success of immune checkpoint blockade (ICB) therapies for many cancers, most patients harbor innate or acquired resistance and do not respond to ICB. This has been shown in part to be due to either low/negative tumor PD-L1 (programmed death ligand 1) expression and/or defects in tumor antigen processing machinery (APM).
What this study adds
In this study, we demonstrate that a tumor-targeting immunocytokine (NHS-IL12) and a histone deacetylase (HDAC) inhibitor (entinostat) synergizes to produce potent antitumor effects in three anti-programmed cell death protein 1/anti-programmed death ligand 1 (αPD-1/αPD-L1) refractory models each harboring differences in major histocompatibility complex class I/APM deficiencies, interferon gamma responsiveness, and tumor mutational burden. Comprehensive transcriptome, proteome, and immune cell analyses were used to interrogate mechanism of action of the combination therapy.
How this study might affect research, practice, and/or policy
This study thus provides the rationale for the combined use of tumor-targeting IL-12 and an HDAC inhibitor such as entinostat for the therapy of patients whose tumors have innate or acquired αPD-1/αPD-L1 resistance and/or defects in APM.
Introduction
Despite unprecedented success of immune checkpoint blockade (ICB) therapies targeting the programmed cell death protein 1/programmed death ligand 1 (PD-1/PD-L1) axis across malignancies, most patients harbor innate or acquired resistance.1 In addition to low or negative tumor PD-L1 expression, tumor-intrinsic defects in major histocompatibility complex class I (MHC-I) and antigen processing machinery (APM), and alterations in genes associated with antigen presentation and interferon gamma (IFN-γ) signaling including B2M, JAK1/2, IFNGR, and NLRC5 are increasingly seen as hallmarks of ICB resistance resulting from loss of tumor immune recognition.2 3 In addition, whereas high tumor mutational burden (TMB-H) has been correlated with anti-programmed cell death protein 1/anti-programmed death ligand 1 (αPD-1/αPD-L1) response in certain tumor types,4 5 mounting evidence demonstrates that TMB-H does not always predict clinical responses, indicating alternative immune and tumor-intrinsic factors at play.6 7 Thus, there is a critical need to explore αPD-1/αPD-L1 refractory preclinical tumor models with varying TMB harboring these tumor-intrinsic defects as to identify therapies able to provide clinical benefit to patients refractory to PD-1/PD-L1 inhibition.
Interleukin 12 (IL-12), produced mainly by antigen presenting cells (APCs) including dendritic cells (DCs), macrophages, B cells and monocytes, can activate natural killer (NK) and T lymphocytes to produce IFN-γ.8 IL-12-mediated tumor suppression relies on high IFN-γ secretion, further activating APCs and upregulating tumor MHC-I/APM.8 However, early trials of systemic administration of recombinant IL-12 (rIL-12) demonstrated a narrow therapeutic window and high toxicity.8 NHS-IL12 is an immunocytokine encompassing the tumor-targeted NHS76 antibody fused with two IL-12 heterodimers. NHS76 binds to exposed DNA fragments in necrotic areas, allowing delivery of immune stimulatory IL-12 into the tumor microenvironment (TME).9 10 In the first in-human clinical study in patients with solid malignancies, NHS-IL12 was well tolerated with signs of clinical activity, although no objective responses, suggesting NHS-IL12 may achieve improved efficacy in a combinatorial setting.11
Overexpression of histone deacetylases (HDACs) is implicated in cancer initiation and progression.12 Entinostat, a class I HDAC inhibitor (HDACi), has been shown to increase tumor immune recognition by epigenetically upregulating MHC-I/APM.13 Increased PD-L1 expression, NK cell cytotoxicity, CD8+ T-cell tumor infiltration and neoantigen-specific immune responses have also been reported.14 15 Entinostat-mediated suppression of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) further supports its use in combination with immunotherapies.15 16 Preclinically, entinostat was shown to promote tumor necrosis9 15 and induce potent suppression of MHC-I+ tumors in combination with NHS-IL12.9 Entinostat has been proven safe and well tolerated in multiple clinical trials.17
Here we demonstrate that NHS-IL12 and entinostat synergize to produce potent antitumor efficacy in three αPD-1/αPD-L1 refractory tumor models harboring varying MHC-I/APM deficiencies, IFN-γ responsiveness, and TMB. Through comprehensive transcriptome, proteome, and immune cell analysis, we demonstrate that combination therapy can bypass αPD-1/αPD-L1 resistance by promoting cytolytic CD8+ T-cell infiltration and M1-like macrophages intratumorally, with robust increases in IFN-γ, antigen presentation and janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathways, resulting in survival benefit regardless of TMB status. Further tumor suppression is associated with NK and APC activation and dampening of M2-like macrophages and Tregs, in a tumor-dependent manner.
Methods
Reagents
Entinostat and recombinant NHS-murine IL12 were provided by Syndax and EMD Serono, respectively, under Cooperative Research and Development Agreements with the National Cancer Institute (NCI). Low-fat diet of 35% sucrose was enriched with entinostat for a target dose of 6 mg/kg/day (Research Diets). Anti-mouse PD-1 (clone RMP1-14, no. BE0146), PD-L1 (clone 10F.9G2, no. BE0101), rat IgG2a isotype (clone 2A3, no. BE0089), CD8α (clone 2.43, no. BE0061), and CD4 (clone GK1.5, no. BP0003-1) antibodies were purchased from BioXCell. Rabbit anti-asialo GM1 was purchased from Wako Chemicals USA.
Murine tumor cell lines
The MHC-class I+ HPV16 E6/E7+ lung cell line TC-1 was a kind gift from Dr T.-C. Wu (Johns Hopkins University, Maryland, USA) and cultured as described.18 The MHC-class I deficient TC-1 subline TC-1/a9 was a kind gift from Dr Michal Šmahel (Charles University, Czech Republic) and cultured as reported.19 The CMT.64 murine lung cancer cell line (Sigma-Aldrich, 10032301) was cultured as recommended. RVP3, a rous sarcoma virus-induced sarcoma cell line, was a kind gift from Dr Milan Reiniš (Institute of Molecular Genetics, Czech Academy of Sciences, Czech Republic) and cultured as described.20 All cell lines were used at low passage (<6) and tested negative for Mycoplasma (MycoAlert Kit; Lonza).
Mice
C57BL/6 and athymic nu/nu female mice aged 6–8 weeks were acquired from the NCI Frederick National Laboratory for Cancer Research Facility. Mice were co-housed under specific pathogen-free conditions, in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited National Institutes of Health (NIH) animal facility. All studies were reviewed and approved by the NIH Institutional Animal Care and Use Committee. Mice used for TC-1/a9 flow cytometry and RNA-seq were 4–6 months old.
Murine tumor studies
TC-1/a9 cells (5×104) were implanted subcutaneously into the right flank of C57BL/6 or nu/nu mice. CMT.64 cells (1×105 or 5×105) and RVP3 cells (2.5×104) were implanted subcutaneously into the right flank of C57BL/6 mice for respective studies. Mice randomization and treatment were initiated when tumors reached 50–80 mm3. In select studies, mice received αPD-L1 (200 µg) or phosphate-buffered saline (PBS) intraperitoneally on alternating days for a total of three doses. For entinostat+NHS-IL12 studies, mice were fed entinostat or control diet for 10 days. Two days after initiating entinostat diet, mice received NHS-IL12 (2 µg subcutaneously) or PBS ipsilaterally on alternate days for a total of three doses. Tumors were measured twice weekly using digital calipers, and volume determined as length2×width/2.
Flow cytometry
Cells were stained using standard procedures (online supplemental methods). Gating strategies are shown in online supplemental table 2 and figure 7.
Supplemental material
Web databases
Correlation analysis of CESC immune infiltrates was completed using the Tumor Immune Estimation Resource (TIMER2.0; timer.cistrome.org) online platform.21 Survival analysis and differential contribution of signature genes were completed using the Gene Expression Profiling Interactive Analysis (GEPIA; gepia2.cancer-pku.cn) online platform.22
Statistics
Statistical analyses (two-tailed) were performed on GraphPad Prism V.9.3.0 (GraphPad software) and listed in figure legends. For volcano plots and Venn diagrams, a p value threshold of 0.05 and a log fold-change threshold of 2 and 0.5 were used to delineate points of interest, respectively. For GSEA, the t-statistic of differential expression was used for gene-rank scores. The Fisher’s exact test was used for gene pathway analysis with a p value cut-off of 0.001. Statistical significance was set at p<0.05 (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Additional materials and methods are described in the online supplemental methods.
Results
Entinostat and NHS-IL12 combination elicits potent antitumor efficacy against αPD-L1-resistant tumor models harboring APM and IFN-γ pathway deficiencies
To identify murine tumor models harboring MHC-I/APM deficiencies and/or IFN-γ pathway disruption, we examined MHC-I and PD-L1 expression in vitro in three distinct tumor cell lines, namely TC-1/a9 (HPV16 E6/E7+) and CMT.64 lung, and RVP3 sarcoma. The TC-1/a9 parental cell line TC-1 was used as MHC-I+ control. Baseline expression of H-2Db and H-2Kb MHC-I haplotypes, and PD-L1 was observed in TC-1 cells but not in TC-1/a9, CMT.64, and RVP3 cell lines (figure 1A). IFN-γ exposure increased H-2Kb and PD-L1 expression in TC-1/a9 and CMT.64 cells. However, no changes were observed in the RVP3 cell line previously reported as IFN-γ resistant (figure 1A).23

Entinostat and NHS-IL12 combination elicits potent CD8 T and NK cell-dependent antitumor efficacy in αPD-L1‒resistant tumor models harboring APM deficiencies. (A) Cell surface expression of MHC-I haplotypes and PD-L1 on untreated cells were compared with isotype controls or IFN-γ treated cells for TC-1, TC-1/a9, CMT.64, and RVP3 cell lines. (B) Oncoplot of gene mutations in the IFN-γ response pathway for TC1a9, CMT.64, and RVP3 cell lines; top bar plot shows tumor mutation burden per cell line. (C–E) Graphs show tumor growth for (C) TC-1/a9, (D) CMT.64, and (E) RVP3 tumor-bearing mice treated with PBS or αPD-L1 as per respective schematic insets (blue squares denote αPD-L1 dosing), n=4–5 mice/group. (F–H) Treatment schedule and tumor growth curves of (F) TC-1/a9, (G) CMT.64, and (H) RVP3 tumor-bearing C57Bl/6 mice treated with PBS or entinostat+NHS-IL12, n=4–5 mice/group. (I) Treatment schedule and tumor growth curves of TC-1/a9 tumor-bearing mice treated with PBS, entinostat, and/or NHS-IL12 as per schematic, n=8–9 mice/group. (J) Frequency of tumor non-immune (CD45neg) cells expressing H-2Db and H-2Kb on day 21 post-tumor implant (7 days after last dose of NHS-IL12) as in figure 1I, n=6 mice/group. (K) Treatment schedule and tumor growth curves for TC-1/a9 tumor-bearing nu/nu mice treated with PBS, entinostat, and/or NHS-IL12 as per schematic, n=9–10 mice/group. (L) Treatment schedule, tumor mean and individual tumor growth curves, and survival for TC-1/a9 tumor-bearing mice treated with PBS or entinostat+NHS-IL12 as per schematic with CD8 or NK cell (asialo GM1) depletion, n=4–9 mice/group; ns=not significant. Insets denote number of cured mice/treatment group, and mOS in days (d). All graphs show mean±SEM; data are representative of 1 (B, D–E, J–L), 2 (A, I), 3 (C, G–H) or 4 (F) independent experiments yielding similar results. Tumor volumes: two-way ANOVA; Survival: Mantel-Cox; Bar graphs: one-way ANOVA with Tukey’s multiple comparisons test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. In vivo schemas were created with BioRender.com. αPD-L1, anti-programmed death ligand 1; ANOVA, analysis of variance; APM, antigen processing machinery; IFN-γ, interferon gamma; MHC-I, major histocompatibility complex class I; mOS, median overall survival; NK, natural killer; PBS, phosphate-buffered saline.
Next, we investigated the effect of IFN-γ on B2m, Lmp2, and Nlrc5 APM gene expression in vitro. NLRC5 (NOD-like receptor family CARD domain containing 5) is a key transactivator of MHC-I-associated APM genes such as beta-2-microglobulin (B2M) and the proteasome subunit LMP2 (PSMB9).24 IFN-γ significantly enhanced B2m, Nlrc5, and Lmp2 gene and protein expression in TC-1/a9 and CMT.64 cell lines without impacting RVP3 gene expression (online supplemental figure 1). Whole-exome sequencing (WES) of each cell line revealed varying somatic missense mutations in the IFN-γ response pathway attributing to varying responses to IFN-γ (figure 1B; online supplemental figure 1). TMB-H, clinically defined as >10 mutations/megabase (Mb), generally correlates with clinical response to ICB in non-small cell lung cancer and other solid tumors.4 5 TC-1/a9 harbored the lowest number of mutations (<5 mutations/Mb), followed by CMT.64 (10–20 mutations/Mb), with RVP3 displaying the highest number of mutated genes (>30 mutations/Mb). Of note, no mutations were identified in the PD-L1 gene (Pdcd1lg1).
Next, we interrogated the response of TC-1/a9, CMT.64, and RVP3 tumors in vivo to αPD-1 and/or αPD-L1 antibodies with known antitumor activity against ICB-responsive tumor models.25–27αPD-L1 induced minimal antitumor activity without survival benefit in TC-1/a9 tumor-bearing mice (figure 1C, online supplemental figure 1). CMT.64 and RVP3 tumors were unresponsive to αPD-L1 despite their TMB-H status (figure 1D,E). TC-1/a9 and CMT.64 tumors were also unresponsive to αPD-1 therapy (online supplemental figure 1).
Next, we investigated the effects of entinostat and IFN-γ combination on APM in TC-1/a9, CMT.64, and RVP3 cells in vitro. IFN-γ treatment alone or combined with entinostat significantly increased Nlrc5, B2m, and Lmp2 gene expression in TC-1/a9 and CMT.64 cells (online supplemental figure 1). In RVP3 cells, only combination treatment increased Nlrc5 gene expression, with B2m remaining unchanged (online supplemental figure 1). Protein expression of LMP2 further corroborated gene findings (online supplemental figure 1). These data suggest that while IFN-γ is the major potentiator of MHC-I/APM, entinostat and IFN-γ-inducing NHS-IL12 therapy could potentially synergize to further promote antitumor efficacy.
Thus, we examined the antitumor efficacy of one cycle of entinostat+NHS-IL12 combination against the MHC-I-deficient/IFN-γ-responsive TC-1/a9 and CMT.64 tumor models. Combination therapy elicited significant control of TC-1/a9 tumors, extending median overall survival (mOS) by more than twofold with a 40% effective cure rate (no relapses) (figure 1F, online supplemental figure 1), while resolving all CMT.64 tumors (100% cures; no relapses) (figure 1G, online supplemental figure 1). Next, we examined this combination against MHC-I-deficient/IFN-γ-resistant RVP3 tumors. We observed significant tumor growth control and increased mOS with a 20% effective cure rate (no relapses; figure 1H, online supplemental figure 1). Collectively, these data demonstrate that combination therapy with entinostat and NHS-IL12 has significant antitumor efficacy against various αPD-1/αPD-L1‒resistant solid tumor models.
Combination therapy requires T cells to elicit potent antitumor activity
Next, and for subsequent studies, we dissected the tumor response to combination therapy and its mode of action versus PBS or single therapy controls in all three tumor models. Combination therapy elicited significant TC-1/a9 tumor growth inhibition compared with PBS-treated or monotherapy-treated mice, respectively (figure 1I). Tumor examination 7 days after final NHS-IL12 dosing showed a significant enhancement in cell-surface expression of H-2Kb and H-2Kd on non-immune (CD45neg) cells only in combination-treated mice (figure 1J). To probe if T cells were determinant for antitumor efficacy, TC-1/a9 tumor-bearing athymic mice received entinostat+NHS-IL12. In these mice, the antitumor efficacy of combination therapy was similar to that of NHS-IL12 monotherapy (figure 1K). Further, tumor growth inhibition elicited by combination therapy was 75% less than that attained in syngeneic mice, completely negating survival benefit (figure 1K, online supplemental figure 1). These findings suggested that T cells largely but not singularly mediated the antitumor effects of combination therapy. Depletion of CD8+ T and NK cells led to 77% and 70% decrease in antitumor efficacy, respectively. Depletion of either immune subset completely abrogated the survival benefit invoked by combination therapy, decreasing mOS from 35 days to 25 days (figure 1L). CD4 depletion increased antitumor efficacy of combination therapy (online supplemental figure 1).
Combination therapy elicits a proinflammatory TME conducive to immune engraftment, activation, and proliferation
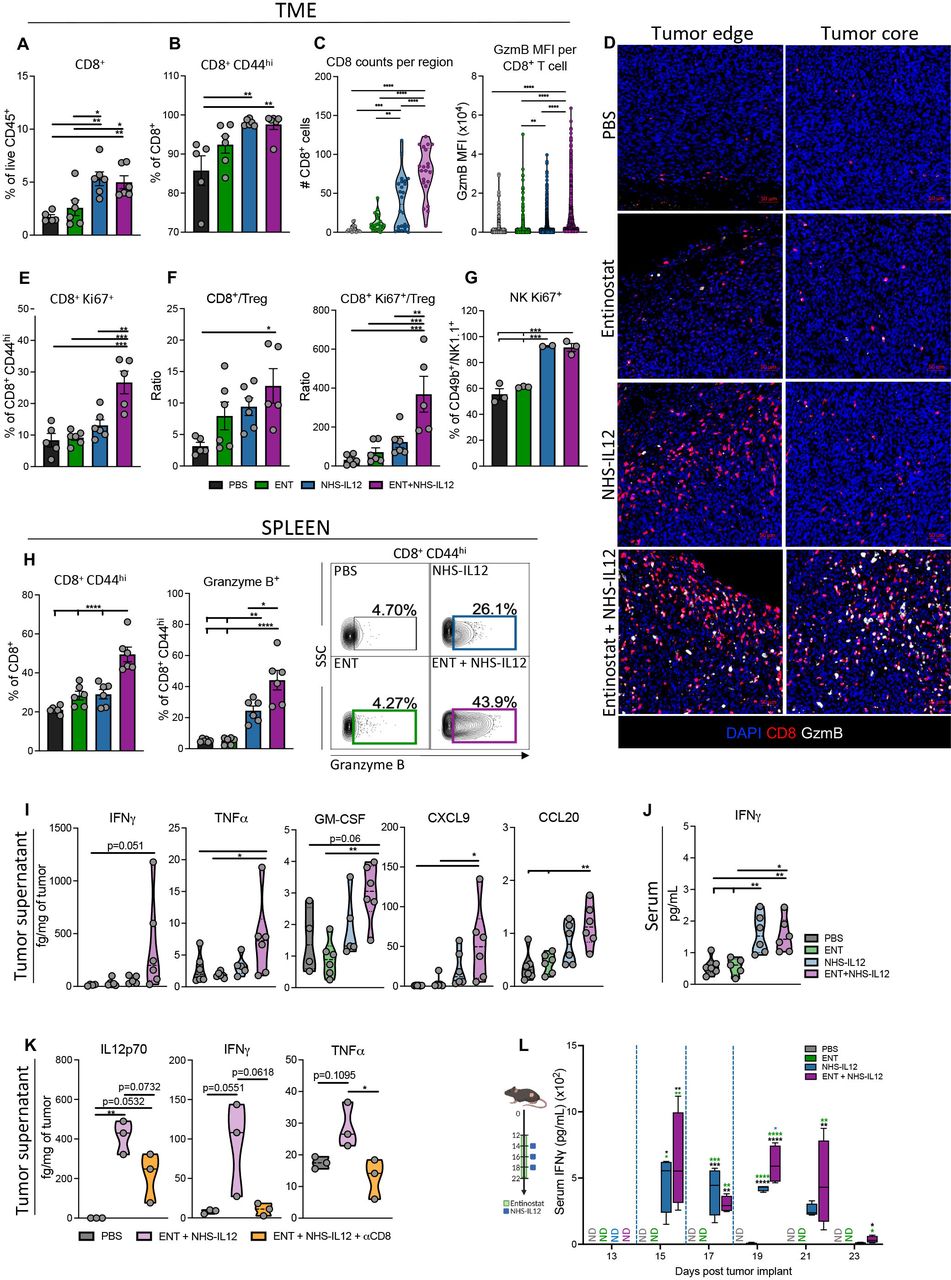
To dissect the contribution of CD8+ T cells and NK cells to tumor suppression invoked by combination therapy, we examined TC-1/a9 tumor-infiltrated and splenic immune cells 7 days after final NHS-IL12 dosing. Tumor analysis by flow cytometry demonstrated significantly increased CD8+ T-cell frequency and activation (CD44hi) in NHS-IL12-treated and combination-treated mice, although no significant effect on CD8+ tumor-infiltrating lymphocyte (TIL) numbers (figure 2A,B, online supplemental figure 1). However, spatial analysis of tumor sections revealed greater CD8+ and granzyme B (GzmB) expression with a stark increase in CD8 counts in the tumor core of combination-treated compared with NHS-IL12-treated mice (figure 2C,D, online supplemental figure 2). Additionally, only combination therapy enhanced Ki67 expression on activated CD8+ TILs indicating increased proliferative capacity (figure 2E). Combination therapy did not alter Treg tumor infiltration, significantly increasing tumor CD8+ and proliferative CD8+Ki67+ T cell-to-Treg ratios versus PBS controls (figure 2F, online supplemental figure 2). Increased Ki67 expression was also observed in NK TILs from NHS-IL12- and combination-treated mice 2 days after final NHS-IL12 dosing (figure 2G). Moreover, combination therapy significantly increased activated splenic CD8+ T cells and their GzmB expression (figure 2H).
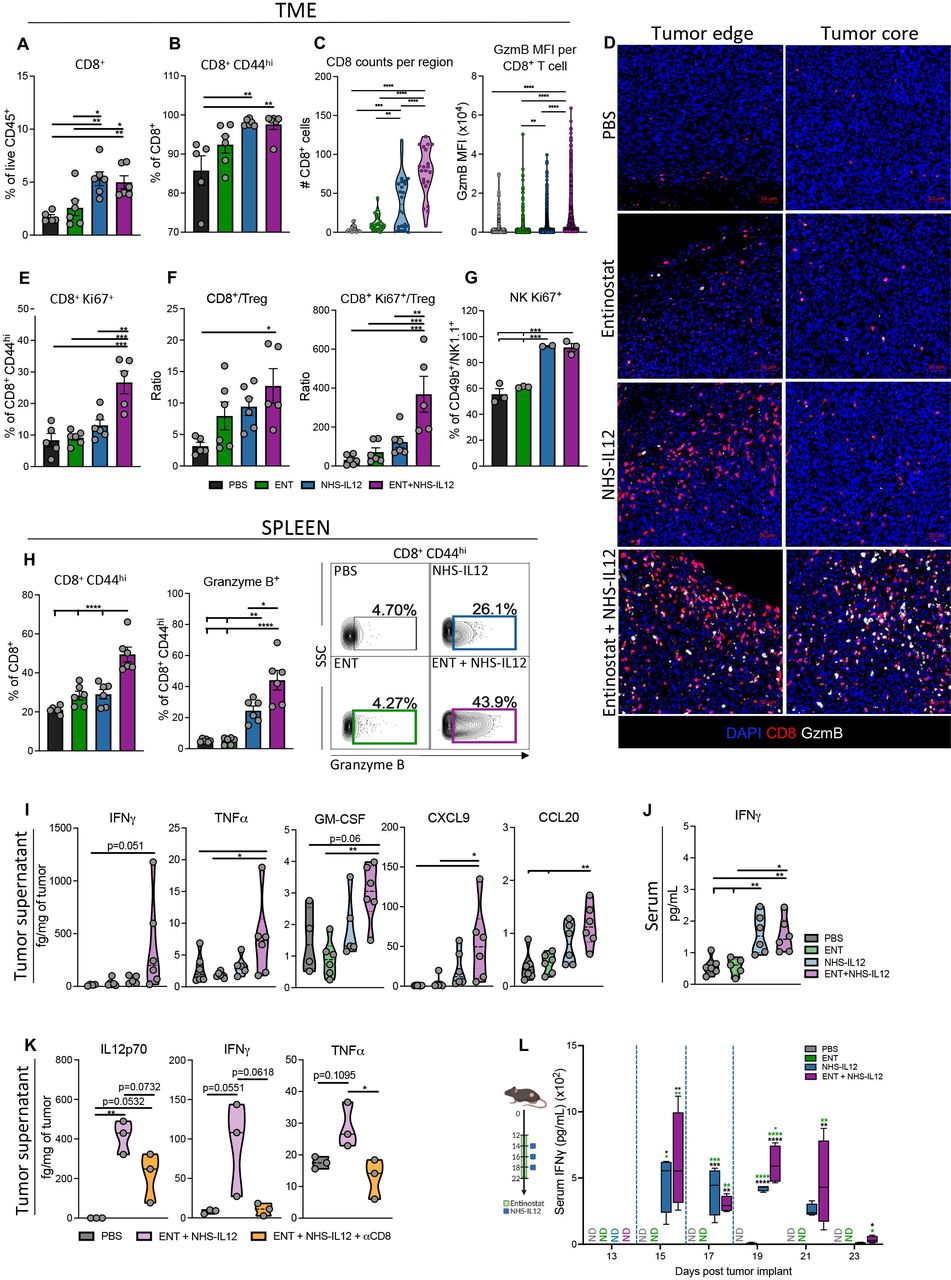
Combination therapy increases proliferative CD8+ T and NK cells with an antitumorigenic cytokine and chemokine profile in MHC-I deficient TC-1/a9 tumors. TC-1/a9 tumor-bearing mice treated with PBS, entinostat, NHS-IL12 or entinostat+NHS-IL12 were sacrificed 7 days after last dose of NHS-IL12 as in figure 1I, n=5–6 mice/group (A and B, E and F, H and J) or 2 days after last dose of NHS-IL12, n=3–4 mice/group (C and D, G, K, L). CD8+ T and NK cells were analyzed by flow cytometry. (A) CD8+ TILs as frequency of CD45+ cells. (B) Frequency of CD8+CD44hi TILs. (C–D) CD8+ T cells and granzyme B were examined by immunofluorescence (IF). (C) IF quantification of CD8+ T cells per region of interest (six equal sized regions per tumor; 1 dot=1 region) and mean fluorescence intensity (MFI) of granzyme B per CD8+ T cell in tumor core. (D) Representative images of immune fluorescent staining for DAPI (blue), CD8 (red), and granzyme B (white); scale bar, 50 µm. (E) Frequency of CD8+CD44hi cells expressing Ki67. (F) Tumor CD8+ (CD44hi) TIL-to-Treg and CD8+Ki67+ (CD44hi) TIL-to-Treg ratios. (G) Frequency of CD49b+/NK1.1+ NK cells expressing Ki67. (H) Frequency of splenic CD8+ T cells expressing CD44hi and CD8+CD44hi T cells expressing granzyme B with representative contour plots. (I) Levels of designated cytokines and chemokines in the TME. (J) Levels of serum IFN-γ. (K) Levels of designated cytokines in the TME. (L) Serum IFN-γ kinetics in TC-1/a9 tumor-bearing mice as treated in schematic (blue dashed line indicates timing of NHS-IL12 dosing), n=4 mice/group. Bar graphs show mean±SEM. Data shown are representative of 1 (C,D, F–L) or 2 (A,B, E) independent experiments yielding similar results. Truncated violin plots show values from individual mice with contours denoting distribution density, dashed line denoting median, and dotted lines denoting quartiles. Bar graphs and violin plots: One-way ANOVA with Tukey’s multiple comparisons test; Box plot: Two-way ANOVA with Tukey’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; IFN-γ, interferon gamma; MHC-I, major histocompatibility complex class I; NK, natural killer; PBS, phosphate-buffered saline; TIL, tumor-infiltrating lymphocyte; TME, tumor microenvironment; Treg, regulatory T cell.
These effects were paralleled with significant elevation in tumor and serum IFN-γ protein levels in addition to other inflammatory cytokines such as tumor necrosis factor alpha (TNFα) and granulocyte-macrophage colony-stimulating factor in the TME of combination-treated mice, with limited impact on IL-10 levels (figure 2I,J, online supplemental figure 2). These data suggested that combination therapy promoted increased TME inflammation, supportive of T cell, NK cell, and macrophage activation and migration. Combination therapy significantly elevated CXCL9, CCL5, and CCL20 chemokines in the TC-1/a9 TME (figure 2I, online supplemental figure 2), known to drive tumor infiltration of CD8+ T, NK, and dendritic cells.28 Examining tumor and sera from TC-1/a9 tumor-bearing mice at earlier timepoints, we observed elevations in total IL-12p70, IFN-γ, and TNFα cytokine levels as well as significant increases in NHS-IL12 levels (figure 2K,L, online supplemental figure 3). These cytokine increases were lost on CD8+ T-cell depletion specifically within the TME despite the presence of elevated NHS-IL12 levels, corroborating the role of CD8+ T-cell trafficking into the TME for antitumor efficacy (figure 2K, online supplemental figure 3). Other chemokines such as CCL5, CCL20, CXCL9, CXCL10, CXCL13, important in T cell, NK cell, DC, and B-cell recruitment into the TME, were significantly increased in combination-treated mice but not in CD8+ T-cell depleted mice with the exception of CXCL9 (online supplemental figure 3).28
Combination therapy promotes antigen presentation in the TME
Analysis of APCs infiltrating TC-1/a9 tumors revealed that combination therapy significantly increased M1-like (CD11b+F4/80+CD38+CD206neg) tumor-associated macro-phage (TAM) frequency while dampening M2-like (CD11b+F4/80+CD38negCD206+) TAMs in frequency and numbers (figure 3A–C), resulting in significant elevation of M1-like to M2-like ratio versus PBS controls (figure 3D).

Combination therapy shifts macrophage M1-to-M2 ratio while increasing genes associated with antigen presentation, CD8+ T cells, and monocytic/macrophage lineage. TC-1/a9 tumor bearing mice treated as in figure 1I were sacrificed on day 21 post-tumor implant (7 days after last dose of NHS-IL12), n=6 mice/group. M1-like and M2-like macrophages and DCs in TME and/or spleen were analyzed by flow cytometry. (A) Representative flow cytometry contour plots of M1-like (F4/80+/CD38+/CD206neg) and M2-like (F4/80+/CD38neg/CD206+) TAMs in mice treated with PBS or combination therapy. M1- and M2-like TAMs as (B) frequency of CD45+ cells and (C) number of cells per milligram of tumor. (D) Ratio of M1-to-M2 TAMs. (E) Numbers of DCs per milligram of tumor and PD-L1 expression (gMFI) on DCs. (F) Frequency of CD45+ splenic DCs plus PD-L1 and MHC-II expression (gMFI) on DCs. (G–K) TC-1/a9 tumor-bearing mice treated as in figure 1I were sacrificed on day 25 post-tumor implant and tumor whole transcriptome analysis was performed, n=3–4 mice/group. Volcano plots show differentially expressed genes (p<0.05) relative to PBS control for (G) entinostat, (H) NHS-IL12, and (I) combination therapy, and combination therapy relative to (J) entinostat and (K) NHS-IL12. Right upper quadrant shows genes upregulated. Horizontal lines indicate threshold for significant changes in gene expression. Gene names highlighted are as follows: green, genes associated with antigen presentation; blue, genes associated with T-cell activation; black, genes involved in migration and differentiation of monocytic/macrophage lineages. Bar graphs show mean±SEM. Data are representative of 1 (F–K) to 2 (A–E) independent experiments yielding similar results. Bar graphs: one-way ANOVA with Tukey’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; DC, dendritic cell; gMFI, geometric mean fluorescence intensity; MHC-II, major histocompatibility complex class II; PBS, phosphate-buffered saline; PD-L1, programmed death ligand 1; TAM, tumor-associated macrophage; TME, tumor microenvironment.
While combination therapy only expanded splenic DCs with a significant increase in MHC-II expression, it significantly increased PD-L1 expression in both TC-1/a9 tumor-infiltrating and splenic DCs (figure 3E,F), collectively suggesting active antigen uptake and presentation. These observations are consistent with findings in tumor-draining lymph nodes (dLNs) 2 days after last NHS-IL12 dose demonstrating that combination therapy significantly increased activated antigen-specific HPV16 E7 tetramer+ CD8+ T cells, associated with significantly higher frequency of cross-presenting conventional type 1 DCs (cDC1) but not type 2 (cDC2). These results were abated with CD8+ T-cell depletion (online supplemental figure 3).
TC-1/a9 tumor rechallenge revealed 60% protective memory elicited by combination therapy (online supplemental figure 3). To probe protective antigen-specific memory, splenocytes from combination-treated mice that evaded primary and secondary tumor challenges were examined by ELISpot for antigen specificity against an HPV16 E7 15-mer peptide. A significant increase in IFN-γ production was observed in combination-treated mice but not in naive tumor-bearing mice (online supplemental figure 3).
To further dissect the immune-mediated mechanism elicited by combination therapy, we performed tumor bulk RNA-seq 11 days after last NHS-IL12 dosing. Combination therapy revealed an expansion of upregulated genes associated with antigen presentation (H2-Aa, H2-Ab1, H2-Eb1, H2-Q7, Cd86, Ciita, Nlrc5, Tap2, Psmb8, Psmb9), CD8+ T-cell activation and chemotaxis (Cd8a, Cxcl9, Cd3d, Cd3e), and drivers of monocytic/macrophage and dendritic cell compartments (Csf1r, Csf2ra, Csf2rb, Csf2rb2, Csf3r, Ccl6, Ccl9) versus PBS (figure 3G–I) or individual therapies (figure 3J,K).24 29–32 Additionally, corroborating our findings of DC activity, combination therapy demonstrated increased expression of genes previously identified as cross-presenting cDC1s, IL-12-secreting DCs, and markers of DC activation and maturation (Cd74, Cd86, H2-Aa, Fabp5, Ccr1, Itgax, Ccl8, Cd8a, Ccr5, Cxcl12, Cxcl9, Psmb8, Ciita, Irf8, Tlr7, Tap2, Cd40, Psmb9, Nlrc5, Fcgr3, Ctsc) (online supplemental figure 3).33
Combination-treated tumors display significant enrichment in IFN-γ, antigen processing, and effector T-cell pathways
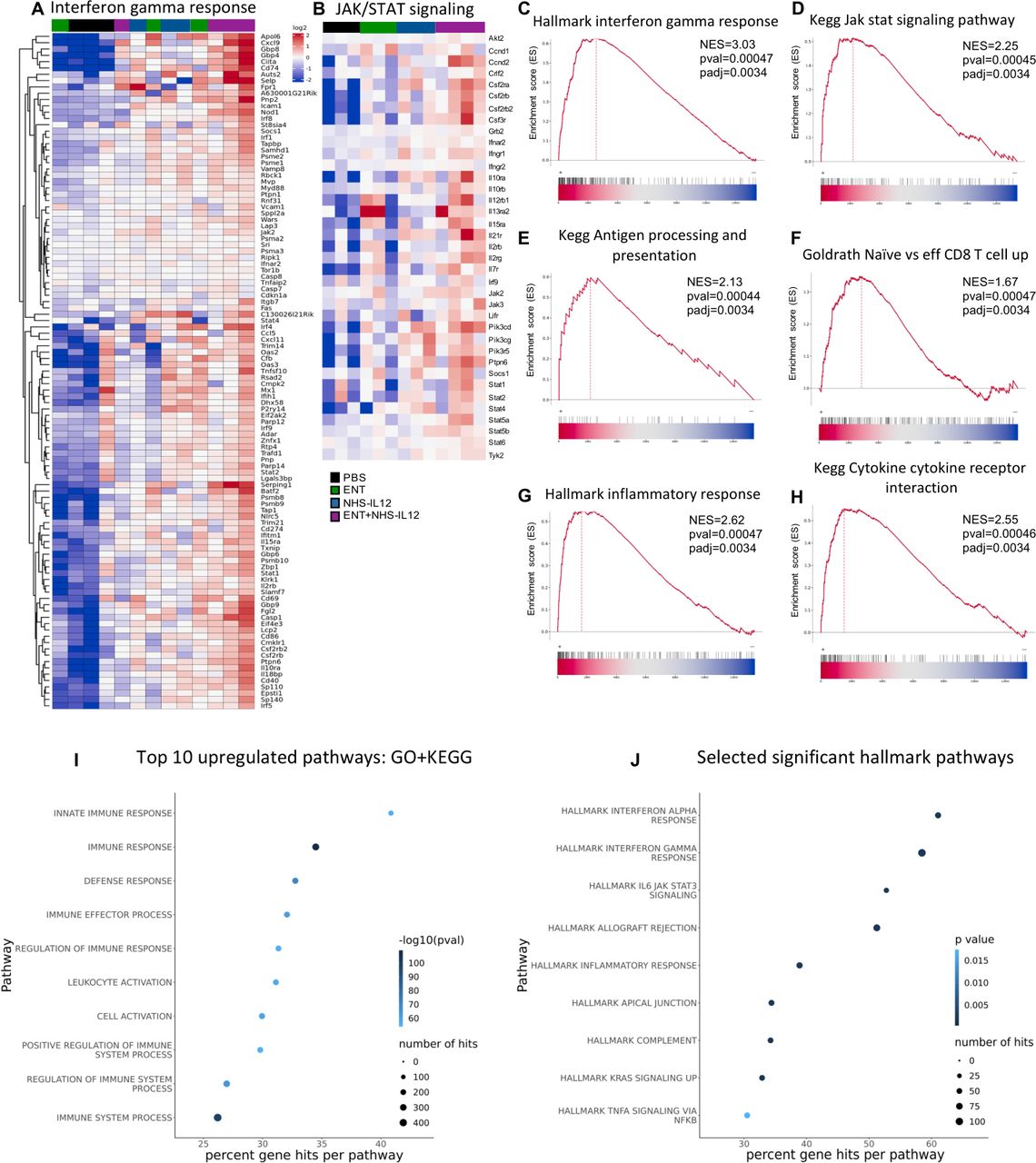
In addition to altered tumor MHC and APM signaling pathways, disruption in IFN-γ-driven JAK/STAT signaling pathway is often observed in patients harboring ICB resistance.2 Gene set enrichment analysis (GSEA) of combination therapy-treated tumors demonstrated significant enrichment of genes involved in IFN-γ response and JAK/STAT signaling pathways (figure 4A–D), in addition to antigen processing/presentation, effector CD8+ T-cell, inflammatory and cytokine/cytokine receptor interaction pathways (figure 4E–H, online supplemental figure 4). Analysis of top upregulated pathways from gene ontology (GO) plus Kyoto Encyclopedia of Genes and Genomes (KEGG), and Hallmark gene set databases further demonstrated significant increases in IFN-α, IFN-γ, inflammatory and immune responses in tumors from combination-treated mice (figure 4I,J). Analysis of the top 10 GO pathways with the highest normalized enrichment scores showed entinostat contributing to high positive regulation of cytokine secretion and immune responses in addition to leukocyte migration and leukocyte chemotaxis (online supplemental figure 5). NHS-IL12 showed enrichment of multiple pathways associated with innate and adaptive immune responses. Multiple comparison analysis indicated NHS-IL12 to drive the upregulation of antigen processing and presentation pathway, also observed with combination therapy (online supplemental figure 5). The combination of entinostat and NHS-IL12 demonstrated positive synergy for multiple pathways including cytokine–cytokine receptor interaction, chemokine signaling, NK cell-mediated cytotoxicity, and T-cell receptor signaling. In addition, combination therapy demonstrated downregulation of multiple pathways involved in cancer stemness and tumor progression, including Hippo signaling, signaling pathways regulating pluripotency of stem cells, Wingless/integrated (Wnt) and mitogen-activated protein kinase (MAPK) signaling (online supplemental figure 5).

Transcriptomic analysis of combination-treated tumors shows significant enrichment of IFN-γ, antigen processing, and effector T-cell pathways. TC-1/a9 tumor-bearing mice treated as in figure 1I were sacrificed on day 25 post-tumor implant and tumor whole transcriptome analysis was performed. Gene set enrichment analysis was carried out for entinostat+NHS-IL12 combination-treated tumors compared with PBS. Heatmap of significantly altered genes in the (A) Hallmark IFN-γ response and (B) KEGG JAK/STAT signaling pathways. Enrichment plots show NES by ranked genes for (C) Hallmark IFN-γ response, (D) KEGG JAK/STAT signaling, (E) KEGG antigen processing and presentation, (F) GOLDRATH naive versus effector CD8 T-cell response, (G) Hallmark inflammatory response, and (H) KEGG cytokine–cytokine receptor interaction pathways. (I) Top 10 GO/KEGG and (J) selected significant Hallmark pathways. Data represents one experiment, n=3–4 mice/group. Fisher’s test for top pathways applied top 10% genes with p value cut-off of p=0.001. GO, gene ontology; IFN-γ, interferon gamma; JAK, janus kinase; KEGG, Kyoto Encyclopedia of Genes and Genomes; NES, normalized enrichment score; PBS, phosphate-buffered saline; STAT, signal transducers and activators of transcription.
Combination therapy elicited significant control of MHC-I-deficient CMT.64 tumors by promoting a dynamic proinflammatory milieu in the TME and periphery supporting cytolytic T-cell activation.
Next, we examined antitumor efficacy and mode of action of combination therapy against CMT.64, which harbors multiple missense mutations in IFN-γ response genes (figure 1B) and is reported to have MHC-I and TAP (transporter associated with antigen processing) deficiencies inducible by IFN-γ.34 35 One cycle of combination therapy elicited significant tumor growth inhibition versus control (93%), entinostat (69%), or NHS-IL12 (28%) monotherapies, and 20% effective tumor resolution given two out of five mice relapsing after 20 days post-tumor cure. Collectively, combination therapy induced a significant 77% increase in mOS (62 days) versus PBS controls (35 days) (figure 5A). To better capture on-treatment immune responses, comprehensive immune analysis was performed 2 days after last NHS-IL12 dosing when antitumor effects were first emerging (online supplemental figure 6). In the TME, both NHS-IL12 alone and combination treatment enhanced cell-surface expression of H-2Kb on CD45neg cells and elevated CD8+ TIL frequency with trending Ki67+ elevation on activated (CD44hi) CD8+ TILs compared with PBS controls, suggesting tumor cell recognition by CD8+ TILs (figure 5B,C, online supplemental figure 6). However, only combination therapy significantly increased GzmB expression on activated (CD44hi) CD8+ TILs (figure 5D). Concomitant dampening of Treg TILs contributed to a significant increase (12.5-fold) in CD8+ TIL-to-Treg ratio (figure 5E, online supplemental figure 6). These effects were paralleled by significantly increased tumor deposition of NHS-IL12 in combination-treated mice (figure 5F). TME immune spatial analysis corroborated previous findings in TC-1/a9 tumors with significantly higher CD8+ T-cell infiltration in the core of combination-treated CMT.64 tumors relative to PBS-treated or monotherapy-treated mice (figure 5G). While total IFN-γ and TNFα levels were increased in tumors from NHS-IL12-treated and combination-treated mice, only combination therapy elicited greater IL12p70 and CXCL9 levels (figure 5H). Further, other chemokines (CCL5, CXCL10, and CXCL13) associated with T cell and other immune cell recruitment were significantly elevated in NHS-IL12- and combination-treated mice (online supplemental figure 6).

Combination therapy elicited significant tumor control in the MHC-I-deficient CMT.64 lung tumor model, associated with increased T-cell activity and reduced immune regulatory cells, while potentiating peripheral NK and APCs. (A) Treatment schedule, tumor growth curves and survival for CMT.64 tumor-bearing mice treated with PBS, entinostat and/or NHS-IL12. Green area represents time course of entinostat treatment, whereas blue squares denote NHS-IL12 administration, n=5–6 mice/group. Insets denote number of cured mice/group and mOS (days). (B–M) CMT.64 tumor-bearing mice were treated as in online supplemental figure 6 and sacrificed 2 days after last NHS-IL12 administration (day 15), n=3–4 mice/group. Immune and non-immune cells in the TME and spleen were analyzed by flow cytometry. Tumor CD8+ cells were examined by immunofluorescence (IF). (B) Cell surface expression of H-2Kb on tumor non-immune (CD45neg) cells. (C) CD8+ TILs as frequency of CD45+ cells and frequency of CD8+ CD44hi TILs expressing Ki67 and (D) granzyme B. (E) Tumor-infiltrating Tregs as frequency of CD45+ cells and CD8+ TILs-to-Treg ratio. (F) Levels of NHS-IL12 in TME and (G) quantification of CD8+ T cells per region of interest (six equal sized regions per tumor; 1 dot=1 region) in tumor core and representative images for DAPI (blue) and CD8 (red) IF staining; scale bar, 50 µm. (H) Designated cytokines and chemokines in the TME. (I) M1- and M2-like TAMs as frequency of CD45+ cells. (J) Tumor-infiltrating DCs as frequency of CD45+ cells and PD-L1 expression (gMFI) on DCs. (K) Frequency of splenic CD8+ CD44hi T cells expressing granzyme B and Ki67 proteins. (L) Frequency of splenic CD49b+/NK1.1+ NK cells expressing Ki67. (M) DCs as frequency of CD45+ cells, PD-L1 expression (gMFI) of DCs, cDC1 as frequency of DCs and CD45+ splenocytes. Bar graphs show mean±SEM. Data are representative of 1 (B–H, K–M) to 2 (A, I–J) independent experiments yielding similar results. Truncated violin plots show values from individual mice with contours denoting distribution density, dashed line denoting median, and dotted lines denoting quartiles. Tumor volume graphs: two-way ANOVA; Survival: Mantel-Cox; Bar graphs and violin plots: One-way ANOVA with Tukey’s multiple comparisons test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. In vivo schemas were created with BioRender.com. ANOVA, analysis of variance; APC, antigen presenting cell; cDC, conventional type 1 dendritic cell; DC, dendritic cell; gMFI, geometric mean fluorescence intensity; MHC-I, major histocompatibility complex class I; mOS, median overall survival; NK, natural killer; PBS, phosphate-buffered saline; PD-L1, programmed death ligand 1; TAMs, tumor-associated macrophages; TILs, tumor-infiltrating lymphocytes; TME, tumor microenvironment; Tregs, regulatory T cells.
This prompted us to dissect the effects of combination therapy on APCs in CMT.64 tumors. M1-like and M2-like TAMs demonstrated similar findings as with the TC-1/a9 model, with frequency of M1-like but not M2-like TAMs significantly increased with NHS-IL12 alone and combination therapy (figure 5I, online supplemental figure 6). Increased frequency of tumor-infiltrating DCs with elevated expression of MHC-II and PD-L1 was also observed, implicating activation (figure 5J, online supplemental figure 6).
To better capture combination therapy-induced immunity, we examined splenic immune cells. Activated CD8+ T cells showed significantly increased cytolytic and proliferative capacity (figure 5K), the latter also observed in NK cells (figure 5L). Whereas the frequency of activated splenic DCs remained unchanged, these displayed significant PD-L1 upregulation in response to combination therapy. Significantly increased cross-presenting cDC1 in both NHS-IL12 and combination-treated mice suggested activation of antitumor immunity (figure 5M).36 This was further supported by significantly elevated levels of NHS-IL12 as well as total IL12p70, IFN-γ, IL-10, TNFα, and IL-6 observed in sera from combination-treated mice (online supplemental figure 6).
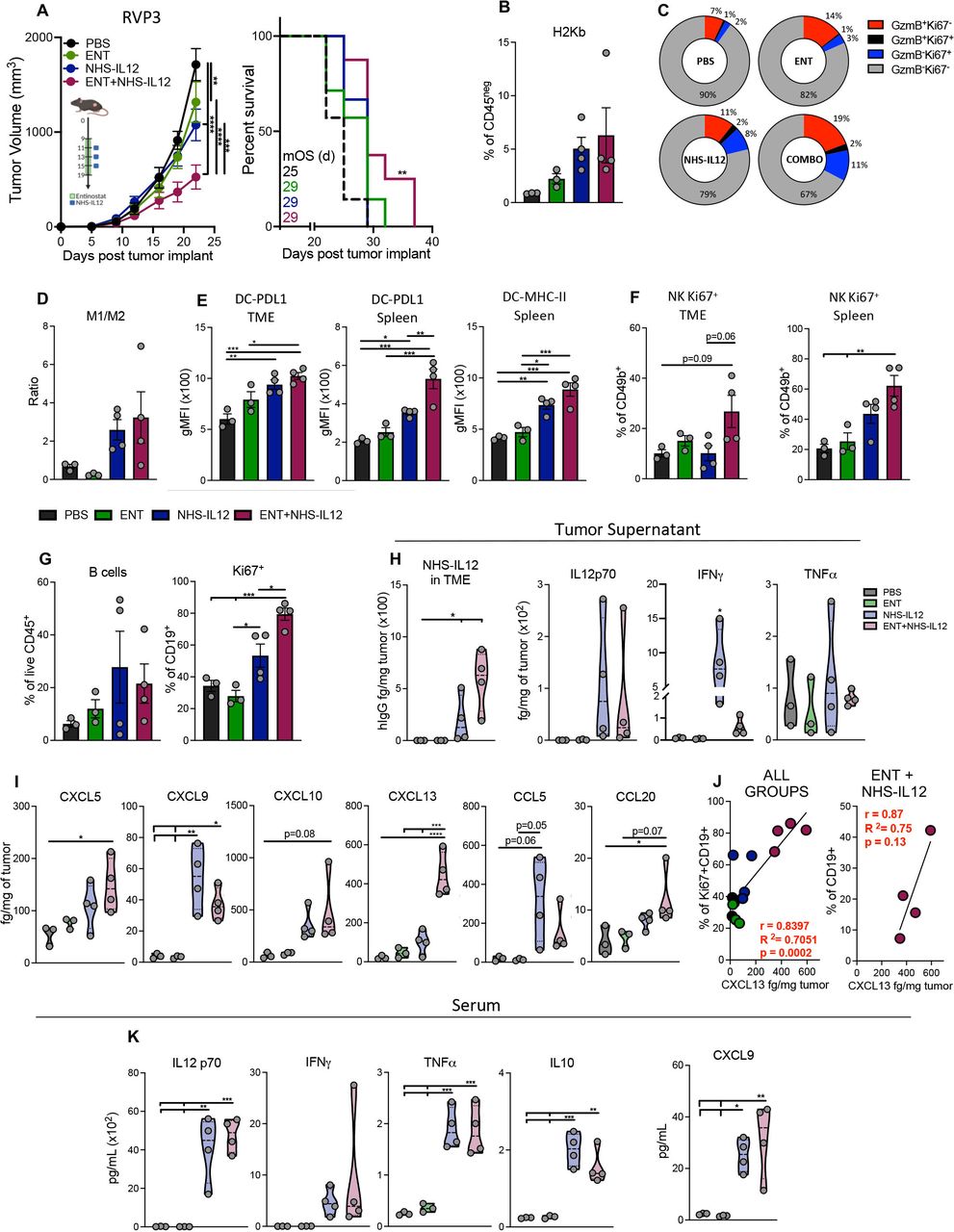
Combination therapy maintained moderate control of IFN-γ-resistant RVP3 tumors, associated with increased CD8+ T, M1-like macrophages, and B-cell activity
Next, we examined the efficacy of combination therapy against the IFN-γ-resistant RVP3 sarcoma to investigate potential clinical translation to αPD-1/αPD-L1-resistant tumors harboring loss of IFN-γ sensitivity. Comprehensive analysis of immune subsets was performed 2 days after last NHS-IL12 dosing. Combination therapy elicited significant antitumor efficacy relative to either entinostat or NHS-IL12 monotherapies, resulting in increased survival (figure 6A). Consistent with previous findings in TC-1/a9 and CMT.64 tumors, combination therapy upregulated H2Kb in non-immune cells and augmented cytolytic and proliferative CD8+ TILs, although to a milder degree (figure 6B,C). In parallel, NHS-IL12 alone and combination therapy promoted M1-like while decreasing M2-like TAMS, resulting in a mild increase of M1- to M2-like macrophage ratio (figure 6D, online supplemental figure 6). Increased PD-L1 expression on DCs in both tumor-infiltrating and splenic DCs was also observed with significantly elevated MHC-II expression and a trending increase in splenic DC frequency although milder than in previous tumor models (figure 6E, online supplemental figure 6). Given these findings, we examined the possible role of NK cells within the TME. While the number of NK TILs remained unchanged, combination therapy significantly increased the frequency of proliferative splenic and tumor-infiltrating NK cells (figure 6F).

Combination therapy elicited moderate control of IFN-γ-resistant RVP3 tumors, associated with increased B cell and CD8+ TIL activation, and augmented M1-like TAM infiltration. (A) Treatment schedule, tumor growth curves and survival for RVP3 tumor-bearing mice treated with PBS, entinostat, and/or NHS-IL12. Green area represents time course of entinostat treatment while blue squares denote NHS-IL12 administration, n=6–8 mice/group. (B–J) Two days after last NHS-IL12 administration (day 17), n=3–4 mice/group, TME cells were analyzed by flow cytometry. (B) Cell surface expression of H-2Kb as frequency of tumor non-immune (CD45neg) cells. (C) Expression frequencies of granzyme B+ and/or Ki67+ in CD8+CD44hi TILs. (D) Ratio of M1- to M2-like TAMs. (E) PD-L1 and MHC-II expression (gMFI) on tumor infiltrating and splenic DCs. (F) Frequency of CD49+ NK cells expressing Ki67 in TME and spleen. (G) B cells (CD19+) as frequency of CD45+ cells and frequency of B cells expressing Ki67. (H) Levels of NHS-IL12 and designated cytokines and (I) chemokines in TME. (J) Pearson correlation of CXCL13 tumor levels from individual combination-treated mice with the frequency of Ki67+ B cells and with frequency of B cells (CD19+) in the TME. (K) Serum levels of designated cytokines and chemokines. Bar graphs show mean±SEM from one experiment. Truncated violin plots show values from individual mice with contours denoting distribution density, dashed line denoting median, and dotted lines denoting quartiles. Tumor volume graphs: two-way ANOVA; Survival: Mantel-Cox; Bar graphs and violin plots: One-way ANOVA with Tukey’s multiple comparisons test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. In vivo schemas were created with BioRender.com. ANOVA, analysis of variance; DCs, dendritic cells; IFN-γ, interferon gamma; gMFI, geometric mean fluorescence intensity; MHC-II, major histocompatibility complex class II; NK, natural killer; PBS, phosphate-buffered saline; PD-L1, programmed death ligand 1; TAM, tumor-associated macrophage; TIL, tumor-infiltrating lymphocyte; TME, tumor microenvironment.
Given prior increases in CXCL13 and its role in B-cell recruitment, we examined pan B cells within the TME. While their overall frequency remained unchanged, combination-treated mice had significantly higher frequency of proliferative B cells (figure 6G). We further examined tumor cytokines and chemokines to fully assess immune infiltration into the TME. Despite NHS-IL12 levels being significantly increased in combination-treated mice, no significant changes were observed in total IL-12, IFN-γ, or TNFα cytokines (figure 6H). Notably, chemokines involved in the recruitment of T cells and APCs including CXCL9, CXCL13, CCL20, and CXCL5 were all significantly increased within the TME of combination-treated mice (figure 6I). Furthermore, CXCL13 tumor levels correlated to the relative frequency of proliferative and overall tumor-infiltrating B cells, implicating their role as APCs in antitumor immunity (figure 6J, online supplemental figure 6). Significant increases in total serum IL-12, TNFα, IL-10, and CXCL9 but not IFN-γ were observed, suggesting that even in tumors with loss of IFN-γ signaling, combination therapy is able to activate and recruit CD8+ T cells, NK cells, and APCs to achieve antitumor efficacy, although to a lesser degree (figure 6K).
Combination therapy-associated signature shows translational relevance in HPV+ and HPVneg malignancies associated with high frequencies of MHC-I and APM deficiencies
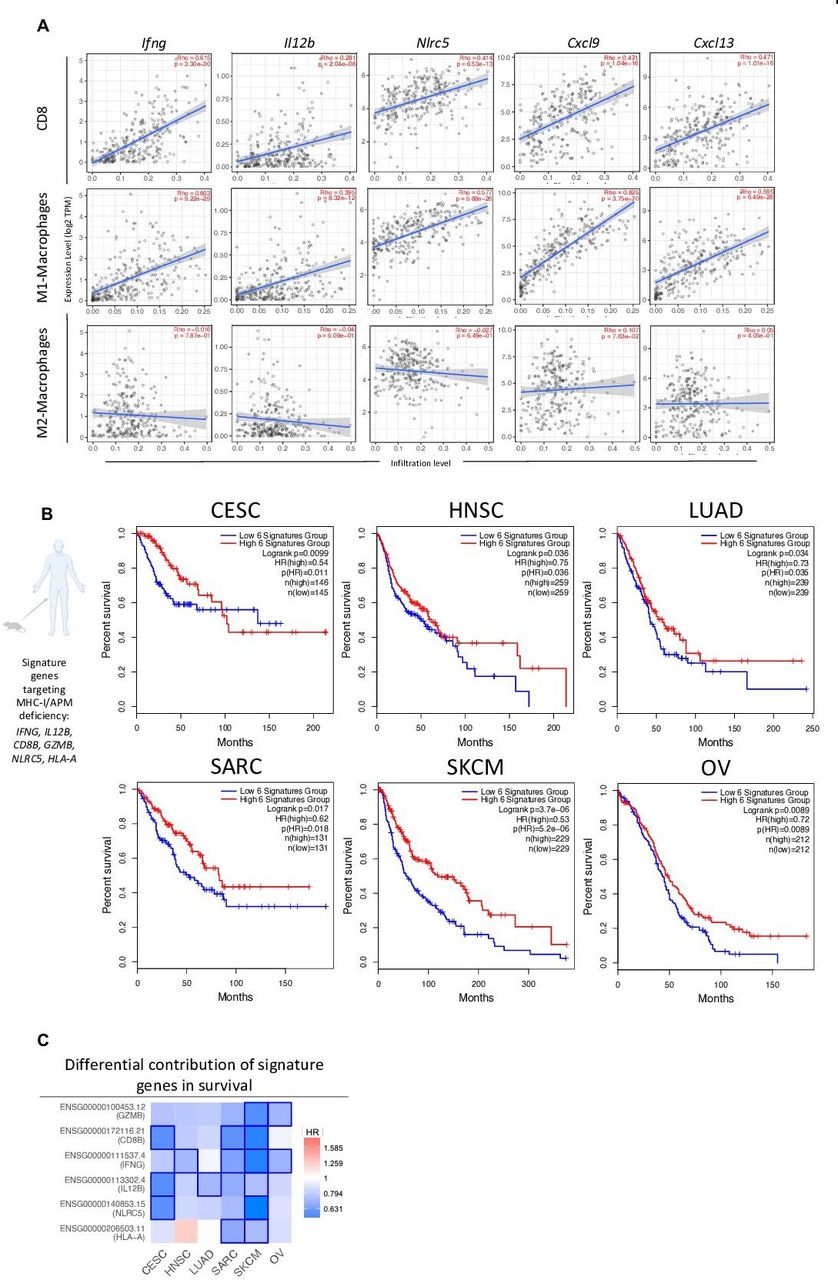
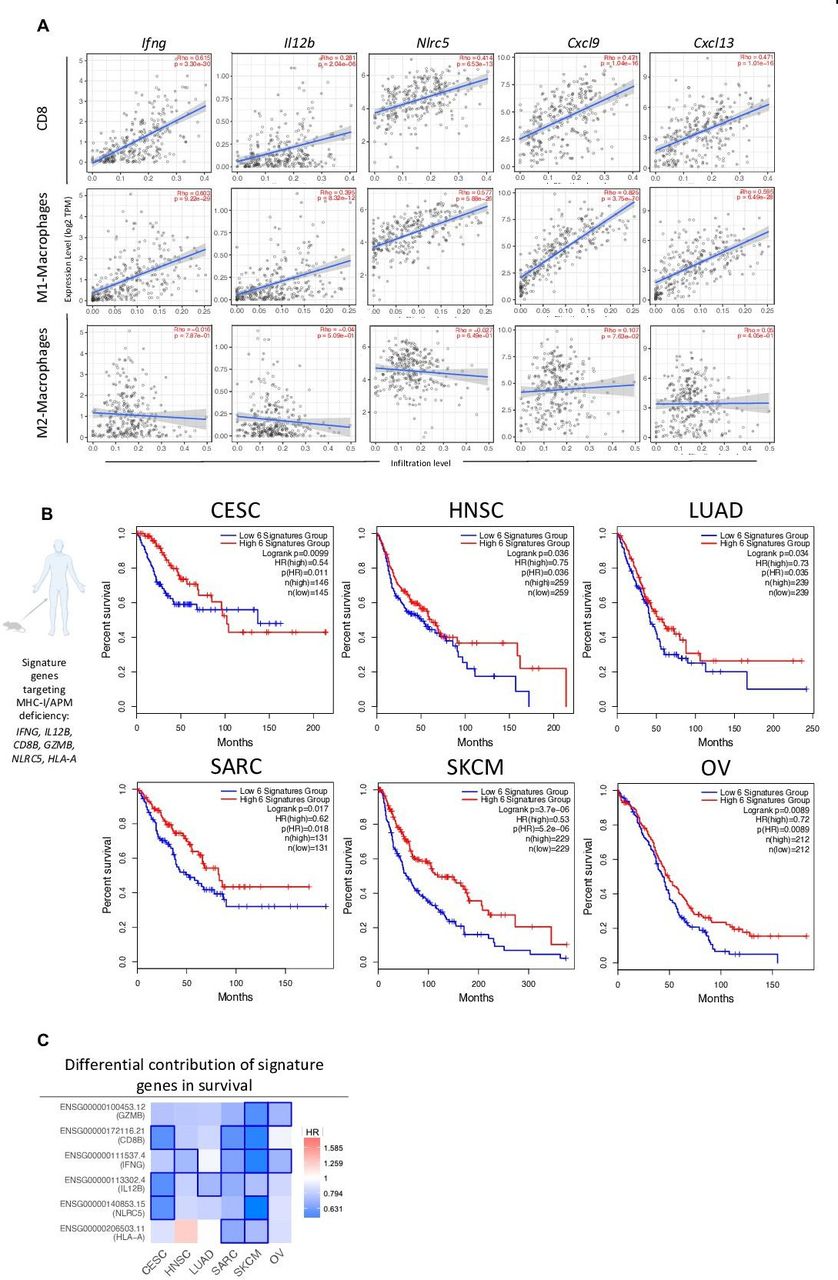
To investigate the potential of combination therapy for clinical translation, we analyzed available molecular data categorized by The Cancer Genome Atlas (TCGA). Since most of our mechanistic studies targeted the HPV16+ TC-1/a9 murine tumor model, we first examined immune signatures of combination therapy in human papilloma virus (HPV)-linked malignancy, such as cervical cancer. Recent TCGA data analysis of cervical cancers identified >50% of cases with decreased human leukocyte antigen (HLA) expression and up to 20% gene alterations in MHC-I, APM, or MHC-I signaling.37 We performed a correlative analysis between genes (IFNG, IL12B, NLRC5, CXCL9, and CXCL13) associated with TC-1/a9 tumor response to combination therapy and infiltration of CD8+ T cells, M1 and M2 macrophages in human cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC). Notably, all genes examined correlated with increased CESC infiltration of CD8+ T cells and M1 but not M2 macrophages (figure 7A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combination therapy-associated signature shows translational relevance in HPV+ and HPVneg malignancies. TIMER (A) and GEPIA (B–C) TCGA data analysis platforms were used for producing immune correlations and Kaplan-Meier survival curves across multiple cancers. (A) Dot plots of two-tailed Spearman’s correlation of CD8+ T cell, M1- and M2-macrophage tumor infiltration to genes (IFNG, IL12B, NLRC5, CXCL9, CXCL13) encoding combination therapy-induced key immune markers from TCGA CESC dataset (n=291 patients). Spearman’s r >0 indicates positive correlation. Spearman’s r <0 indicates negative correlation. Line represents best-fitting regression line with 95% CIs (gray shading). (B) Correlation between clinical overall survival for designated cancers with key immune signature (IFNG, IL12B, CD8B, GZMB, NLRC5, and HLA-A) observed in murine models of MHC deficiency. (C) HR of key immune signature genes to survival across malignancies. Kaplan-Meier curves were generated with a median high/low 50% cut-off. Mouse and human icons created with BioRender.com. CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CI, confidence interval; GEPIA, Gene Expression Profiling Interactive Analysis; HNSC, head and neck squamous cell carcinoma; HPV, human papilloma virus; HR, hazard ratio; LUAD, lung adenocarcinoma; MHC, major histocompatibility complex; SARC, sarcoma; SKCM, skin cutaneous melanoma; TCGA, The Cancer Genome Atlas; TIMER, Tumor Immune Estimation Resource; OV, ovarian serous cystadenocarcinoma.
Next, we created a human gene signature (IFNG, IL12B, CD8B, GZMB, NLRC5, and HLA-A) resembling key response markers from TC-1/a9, CMT.64, and RVP3 tumor models to combination therapy. This signature was used to predict survival in CESC, head and neck squamous cell carcinoma (HNSC), lung adenocarcinoma (LUAD), sarcoma (SARC), skin cutaneous melanoma (SKCM), and ovarian serous cystadenocarcinoma (OV) from TCGA datasets. CESC, HNSC, LUAD, and SARC represent HPV+-linked, lung, and sarcoma malignancies, respectively, similar to our ICB-resistant MHC-I/APM-deficient preclinical models. SKCM and OV also harbor high rates of gene alterations in MHC-I/APM.37 Analysis predicted significantly higher overall survival in patients with a high signature in all six tumor types examined. In patients with CESC, this was most clearly observed in the first 100 months of follow-up (figure 7B). Analysis of the differential contribution of signature genes to survival across tumor types identified SKCM as the tumor type with the greatest significance of each gene examined (figure 7C). This finding is supported by a recent report indicating SKCM to have the highest frequency of gene alterations in MHC-I and MHC-I signaling among TCGA tumor types.37 Altogether, we show that entinostat and NHS-IL12 combination therapy has potent antitumor activity in MHC-I/APM-deficient tumors refractory to ICB through modulation of CD8+ T cells, NK cells, macrophages, and APCs. These data provide the rationale to support the clinical application of entinostat and NHS-IL12 as a potential therapy for patients refractory to ICB therapy harboring MHC-I/APM deficiencies.
Discussion
Here, we identified three tumor models refractory to αPD-L1 and/or αPD-1, each with varying MHC-I and APM deficiencies, responses to IFN-γ, TMB and IFN-γ response pathway gene mutations (RPGMs). TC-1/a9 harbors low TMB and the lowest number of IFN-γ RPGM. CMT.64 is TMB-H and displays several IFN-γ RPGM. Both TC-1/a9 and CMT.64 are deficient in MHC-I/APM expression but respond to IFN-γ. However, RVP3 harbors the highest TMB and IFN-γ RPGM. RVP3 lacks response to IFN-γ, in concordance with reported, silenced, and IFN-γ-unresponsive APM, although inducible by HDACi.23
High TMB is a promising biomarker of ICB response in lung cancer and other TMB-H malignancies.4 5 However, its predictive value across solid malignancies remains unclear.6 Here, the lack of response to αPD-L1 and/or αPD-1 in TMB-H tumor models underscores alternative determinants of αPD-1/αPD-L1 response, in concordance with clinical findings across malignancies.6
NHS-IL12 in combination with entinostat has been shown to elicit robust antitumor activity against murine PD-L1+/MHC-I+ breast and colorectal tumors through IFN-γ-driven innate and adaptive immune cross-talk.9 Here we examined the antitumor effects of NHS-IL12 plus entinostat as a potential combination therapy for patients harboring resistance to αPD-1/αPD-L1 therapies associated with defects in antigen presentation and IFN-γ signaling. Our data suggest that the antitumor effects in αPD-1/αPD-L1‒resistant tumor models were attained via epigenetic and IFN-γ-mediated induction of MHC-I/APM, CD8+ T, and NK cell-mediated antitumor immunity, engaging M1-like TAMs and other APCs, and decreasing suppressive cells in the TME. Despite the presence of defects in antigen processing/presentation and IFN-γ signaling in these tumor models, one cycle of combination therapy elicited significant tumor control and increased survival, although less pronounced than previously observed in αPD-L1-sensitive/MHC-I+ MC38 and EMT6 tumor models.9
Notable increases in NHS-IL12 tumor deposition were observed in combination-treated mice. While IFN-γ-inducing NHS-IL12 seemed to drive CD8+ T-cell responses in IFN-γ-responsive TC-1/a9 and CMT.64 tumors, combination therapy achieved significant and prolonged tumor growth control in all tumor models, including the IFN-γ-resistant RVP3. These effects specific to combination therapy may be attributed to sustained IFN-γ and IL-12 levels as well as T-cell homing chemokines within the TME and/or systemically, converting immune-excluded (TC-1/a9) or immune-poor (CMT.64) into an immune-inflamed TME in an IL-12/IFN-γ-mediated loop conducive to tumor rejection.
Within the TME of all three models, combination therapy led to increases in cytolytic and/or proliferative CD8+ TILs, noted as determinant for antitumor activity. In the HPV16+ TC-1/a9 tumor model, these changes were associated with antigen specificity. On combination therapy, all three tumor models converged in significant upregulation of CXCL9, CXCL10, and CCL5 chemokines identified as potent drivers of CD8+ T-cell trafficking.28 Proinflammatory cytokines IFN-γ, TNFα, and IL-12, indicative of T helper 1 cell (Th1) immunity, were significantly increased in combination-treated TC-1/a9 tumors. However, this elevation was abrogated on CD8+ T-cell depletion, implicating CD8+ TILs as determinant for robust antitumor activity. CD4+ T-cell depletion improved antitumor activity of combination therapy, possibly due to early depletion of immune-suppressive CD4+ Tregs and/or expansion of tumor-reactive CD8+ TILs.38
The IFN-γ-resistant RVP3 model was the least responsive to combination therapy, associated with the lack of significant increases in tumor IFN-γ, TNFα, and total IL-12, and minimal elevation in CD8+ TILs. However, the increased presence of cytolytic CD8+ T cells may have contributed to the augmented survival.
NK cells are contender effector cells for ICB resistance due to their capacity to kill low/null MHC-I expressing tumor variants.39 Here, NK cells were determinant for combination therapy efficacy against TC-1/a9, with increased presence of proliferative NK TILs, also observed in CMT.64 and RVP3 tumor models peripherally. As NK cells have been implicated in an antitumor role at earlier stages of tumor progression,40 it is possible that NK cells play an early role, warranting further investigation. This is further supported by our tumor RNA-seq findings demonstrating combination therapy to upregulate the NK cell-mediated cytotoxicity pathway.
In accordance with increases in Th1-inflammatory cytokines, we observed a shift from M2-like to M1-like TAMs in TC-1/a9 tumors and overall increases in frequency and/or numbers of M1-like TAMs in all three models. However, in a stark contrast from previous findings in MHC-I+ tumor models treated with combination therapy, the overall increase in M1/M2 ratios was milder and driven mostly by NHS-IL12.9 Due to the IFN-γ-inducible nature of MHC-I/APM deficiencies in TC-1/a9 and CMT.64 tumors, this difference may have been due to simultaneous activation of DCs and antigen-specific immunity. In addition to significant increases in cDC1 in tumor dLNs and/or spleens, TC-1/a9 and CMT.64 models also had significant increases in CCL5 and CCL20 chemokines, both implicated in the recruitment of antitumor cross-presenting cDC1s.28 Bulk RNA-seq analysis of TC-1/a9 tumors further demonstrated enrichment of antigen presentation pathways and genes associated with DC activation including Cd11c (Itgax), Cd40, and Cd86. These findings are consistent with the observed upregulation of IFN-α response pathway elicited by combination therapy, known to stimulate proinflammatory macrophages, DC maturation, migration to lymph nodes, and cross-priming.41
IL-12 can exert distinct immune responses in a context-dependent manner.8 IL-12 has been shown to have cascading effects in B cells leading to Th1 immunity and IFN-γ production by NK cells.42 43 Interestingly, in IFN-γ-resistant RVP3 tumors, while tumor IL-12 and DC activity were unchanged, combination therapy elicited a significant NHS-IL12-driven increase in the frequency of proliferative B cells. These were highly correlated with the B- (and T-) cell trafficking chemokine CXCL13, also significantly upregulated in combination-treated TC-1/a9 and CMT.64 tumors. CXCL13 and CXCL9 have recently been shown to predict response to ICB therapy, suggesting a similar role here.7
A key observation that corroborated our findings of synergistic antitumor immunity with combination therapy was the increase in number of differentially expressed genes from whole transcriptome analysis of TC-1/a9 tumors. Most genes involved in antigen presentation, T-cell activation and migration, and differentiation of monocytic/macrophage lineages were driven by NHS-IL12 alone. However, upregulation of Cd3d, Cd3e, Cd8, and Cxcl9 genes was unique to combination-treated tumors, implicating ongoing activation of the T-cell receptor complex, important in antigen recognition as well as tumor CXCL9-driven CD8+ T-cell engraftment.44 45 Notably, upregulation of APM genes including Tap2, Psmb9, and Nlrc5 was exclusive to combination therapy. Interestingly, entinostat induced upregulation of colony stimulating factor (CSF) receptors, as a monotherapy and with combination treatment. This is concordant with reports indicating that CSFs promote commitment of myeloid precursor cells and entinostat can promote the direct conversion of M-MDSCs to TAMs.16 46 Combination therapy upregulated Ccl6 and Ccl9 chemokines, shown to recruit macrophages and DCs, further supporting their role in antitumor immunity.29 47 In TC-1/a9 tumors, combination therapy induced significant upregulation of IFN-γ response and JAK/STAT signaling pathways, whose deficiency results in ICB resistance. Moreover, the downregulation induced by combination therapy of multiple pathways involved in cancer stemness and tumor progression, such as signaling regulating pluripotency of stem cells, Hippo, Wnt and MAPK signaling, may have further contributed to tumor suppression, warranting future investigation.48 49 Collectively, these findings demonstrate that combination therapy promotes a dynamic inflammatory milieu in both the periphery and TME, conducive to activation and tumor recruitment of CD8+ T cells, NK cells, and APCs such as DCs, B cells, and macrophages, ultimately synergizing to potentiate IFN-γ and JAK/STAT pathways to circumvent tumor-intrinsic mechanisms of αPD-1/αPD-L1 resistance.
Future studies need to be conducted to further elucidate the mechanism in the TME by which combination therapy drives tumor suppression, including the precise contribution of macrophages, cDCs, and IFN-α response, as well as the functional effects on immunosuppressive entities such as MDSCs and CD25hi Tregs. Previously, entinostat has been shown to induce neoantigen-specific immune responses.15 In future studies, it will be important to further explore antigen-specific immune responses elicited by combination therapy, including to neoepitopes, across these TMB-differing tumor models.
To date, αPD-L1 is approved for patients with cervical cancer with recurrent or metastatic PD-L1+ tumors. However, of the 79% of these patients, only 14% are seen with overall response rates demonstrating a critical need for therapies that can bypass this resistance.50 Here we show that expression of IFNG, IL12B, NLRC5, CXCL9, and CXCL13 genes, associated with antitumor efficacy in murine tumor models, correlated with increased CD8+ T-cell and M1-macrophage infiltration but not M2 macrophages in CESC patient biopsies. We further identified a biomarker gene signature associated with favorable overall survival across tumor types including CESC, HNSC, LUAD, SARC, SKCM, and OV, all of which reported to have significant decreases in HLA expression and increased defects in antigen presentation signaling.37
While the vast majority of clinical studies and FDA-approved treatments involving ICB for the treatment of solid tumors use αPD-1/αPD-L1 monotherapies, anti-cytotoxic T-lymphocyte associated protein 4 (αCTLA-4) alone or in combination with PD-1 inhibition is approved for various advanced malignancies and shares common mechanisms of intrinsic resistance.2 Future preclinical studies examining the potential of entinostat/NHS-IL12 combination therapy to reverse innate and acquired resistance to ICB, including to αCTLA-4 and/or αPD-1, will further our understanding of the potential clinical translation of this therapy. In addition, whether combinations involving other IFN-γ-inducing cytokines and/or alternative HDACis can elicit significant suppression of tumors harboring intrinsic defects in antigen processing and IFN-γ signaling remains to be investigated.
Currently, the combination of entinostat and NHS-IL12 is being evaluated in a phase I/II study (NCT04708470) in combination with bintrafusp alfa (αPD-L1/TGFβRII bifunctional agent) for patients with advanced colorectal and HPV-associated malignancies.
To conclude, our findings provide a rationale for combining NHS-IL12 with entinostat in the clinical setting and underscore the potential of this combination to bypass αPD-1/αPD-L1 resistance stemming from tumor-intrinsic defects in antigen processing and IFN-γ signaling.
Supplemental material
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank Curtis Randolph for his excellent technical assistance and Debra Weingarten for her excellent editorial assistance in the preparation of this manuscript. We acknowledge Dr Maggie Cam, Dr Mayank Tendon, and the rest of the NCI CCR Collaborative Bioinformatics Resource (CCBR) group for their excellent assistance with bulk RNA-seq and WES analyses. Lastly, we thank Yongmei Zhao and the rest of the Sequencing Facility at the Office of Science and Technology Resources at NCI for performing WES.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JS and SRG contributed equally.
Contributors SRG, CMM, and JS conceptualized the study; CMM and SRG performed data curation; CMM performed analysis of data and generated figures; CMM, SRG, and JS drafted the manuscript; CMM performed all in vitro and in vivo animal experiments, flow cytometry experiments, and RNA-seq analysis; PLC and CMM quantified proteins in the TME. LAH performed and quantified immunofluorescent staining. CP supervised and curated immunofluorescent data analysis. PLC and KCH contributed to in vivo immune studies. CMM and SRG conducted the human TCGA correlation studies. SRG and JS co-supervised the project and associated experiments. All authors reviewed and approved the manuscript. JS acted as guarantor.
Funding This research was supported by the Intramural Research Program of the Center for Cancer Research, NCI, National Institutes of Health, and Cooperative Research and Development Agreements between the NCI and Syndax Pharmaceuticals, and the NCI and EMD Serono.
Competing interests The Laboratory of Tumor Immunology and Biology/Center for Immuno-Oncology, NCI, has a Cooperative Research and Development Agreement with Syndax Pharmaceuticals and EMD Serono. KCH is now an AstraZeneca employee.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.