Article Text

Abstract
Background Despite treatment advancements with immunotherapy, our understanding of response relies on tissue-based, static tumor features such as tumor mutation burden (TMB) and programmed death-ligand 1 (PD-L1) expression. These approaches are limited in capturing the plasticity of tumor–immune system interactions under selective pressure of immune checkpoint blockade and predicting therapeutic response and long-term outcomes. Here, we investigate the relationship between serial assessment of peripheral blood cell counts and tumor burden dynamics in the context of an evolving tumor ecosystem during immune checkpoint blockade.
Methods Using machine learning, we integrated dynamics in peripheral blood immune cell subsets, including neutrophil-lymphocyte ratio (NLR), from 239 patients with metastatic non-small cell lung cancer (NSCLC) and predicted clinical outcome with immune checkpoint blockade. We then sought to interpret NLR dynamics in the context of transcriptomic and T cell repertoire trajectories for 26 patients with early stage NSCLC who received neoadjuvant immune checkpoint blockade. We further determined the relationship between NLR dynamics, pathologic response and circulating tumor DNA (ctDNA) clearance.
Results Integrated dynamics of peripheral blood cell counts, predominantly NLR dynamics and changes in eosinophil levels, predicted clinical outcome, outperforming both TMB and PD-L1 expression. As early changes in NLR were a key predictor of response, we linked NLR dynamics with serial RNA sequencing deconvolution and T cell receptor sequencing to investigate differential tumor microenvironment reshaping during therapy for patients with reduction in peripheral NLR. Reductions in NLR were associated with induction of interferon-γ responses driving the expression of antigen presentation and proinflammatory gene sets coupled with reshaping of the intratumoral T cell repertoire. In addition, NLR dynamics reflected tumor regression assessed by pathological responses and complemented ctDNA kinetics in predicting long-term outcome. Elevated peripheral eosinophil levels during immune checkpoint blockade were correlated with therapeutic response in both metastatic and early stage cohorts.
Conclusions Our findings suggest that early dynamics in peripheral blood immune cell subsets reflect changes in the tumor microenvironment and capture antitumor immune responses, ultimately reflecting clinical outcomes with immune checkpoint blockade.
- immunotherapy
- tumor biomarkers
- translational medical research
Data availability statement
Data are available on reasonable request. Data are available in the supplementary tables of this manuscript and available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Snapshot tissue-based determinants of response to immune checkpoint blockade may not capture the plasticity of antitumor immune responses during therapy and are therefore not accurately reflecting clinical outcomes.
WHAT THIS STUDY ADDS
We studied dynamic changes in peripheral immune cell subsets in patients with both metastatic and early stage non-small cell lung cancer (NSCLC) receiving immune checkpoint inhibitors.
Longitudinal assessment of the neutrophil–lymphocyte ratio (NLR) and eosinophil levels captured induction of inflammatory responses in the tumor microenvironment, as well as intratumoral T cell repertoire reshaping in peripheral blood and ultimately reflected therapeutic response.
Non-invasive measurement of immune cell subsets during immune checkpoint blockade may complement circulating cell-free tumor DNA (ctDNA)-derived molecular responses and may be particularly informative in cases where ctDNA is undetectable.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Our findings provide insights into why NLR dynamics are correlated with clinical outcomes with immune checkpoint blockade and may allow for non-invasive early adaptive changes in therapeutic strategies.
Background
Advancements in immunotherapy have dramatically changed the clinical outcomes of patients with non-small cell lung cancer (NSCLC), and immune checkpoint inhibitors (ICIs) have significantly expanded the treatment landscape.1 2 Despite the significant progress, current approaches to predict therapeutic response with static, predominantly tissue-based biomarkers, are subject to limitations, related to technical challenges and poor reflection of the dynamic nature of antitumor immune responses with ICI. High tumor PD-L1 expression and tumor mutational burden (TMB) have been linked with responses to ICI, but not all patients with PD-L1 or TMB high tumors attain long-term responses.3 4 Tumor heterogeneity and purity impact the measurement of PD-L1 and TMB, rendering these features insufficient to accurately predict which tumors will respond to therapy.5 Importantly, the dynamic nature and plasticity of the tumor-immune system interplay in the context of immune checkpoint blockade cannot be adequately captured by static single feature analyses. Recent integrative analyses of tumor-intrinsic and immune cell-focused features,5–9 as well as multiomic meta-analyses,10 have identified nuanced characteristics of the tumor genomic landscape that together with proinflammatory signatures in the tumor microenvironment (TME) better distinguish responding from non-responding tumors.
The plasticity of tumor and immune cell dynamics during immunotherapy may be more accurately captured by circulating cell-free tumor DNA (ctDNA) kinetics via liquid biopsy analyses11 12 and by tracking the neutrophil-lymphocyte ratio (NLR) in peripheral blood.13 14 High NLR values may represent a higher density of tumor-associated neutrophils (TANs) in peripheral blood; these can promote tumor cell proliferation and ultimately immune escape.15 High pretreatment NLR has been found to be associated with worse prognosis in early and late stage lung cancer16 17 as well as with inferior outcomes with ICI.18–21 Despite the intuitive value of NLR as a prognostic or predictive biomarker, most prior studies have focused on pretreatment measurements of NLR without clear evidence that these reflect distinct immune surveillance states in the TME. NLR changes after immunotherapy initiation have been reported to correlate with treatment response in small cohorts of cancer patients receiving ICIs.13 14
Here, we used machine learning to model longitudinal dynamics of immune cell subsets in peripheral blood during ICI and predict therapeutic response for patients with metastatic NSCLC. We then linked peripheral blood immune cell dynamics and NLR changes with transcriptomic profiles of innate and adaptive immunity in the TME as well as with reshaping of the intratumoral T cell repertoire during ICI. Ultimately, we linked NLR dynamics with therapeutic responses at both a cellular level (evaluated by pathological response) and a molecular level (assessed by ctDNA kinetics).
Methods
Cohorts
Patients enrolled in the Thoracic Oncology Biorepository Protocol at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center were reviewed, and 239 serial patients were identified; these had a diagnosis of metastatic NSCLC and received ICI-containing regimens between February 2011 and March 2020 (online supplemental table S1). Clinical data including age, histology, smoking status, and peripheral complete blood counts (CBCs) with differential were retrieved from the medical records. Response to immunotherapy was evaluated by durable clinical benefit (DCB), which was defined as confirmed absence of progressive disease or death within 6 months from therapy initiation. An early stage cohort consisted of 26 patients with stage I to IIIA surgically resectable NSCLC that received neoadjuvant nivolumab 3 mg/kg every 2 weeks for two doses, or neoadjuvant nivolumab 3 mg/kg every 2 weeks for three doses with one dose of ipilumumab 1 mg/kg as part of a clinical trial between August 2015 and May 2019 (online supplemental table S2).22–24 Therapeutic response was assessed pathologically using immune-related pathologic response criteria25 and percent tumor regression and radiographically using RECIST V.1.1. Major pathologic response (MPR) was defined as ≤10% residual tumor (RT) and occurred in 11 patients (42%). Disease burden was calculated as the sum of the longest diameters of target lesions defined by RECIST V.1.1 criteria before and after neoadjuvant ICI. RECIST defined stable disease occurred in 21 patients (81%). Progression-free survival (PFS) and overall survival (OS) were defined as the time elapsed between the date of treatment initiation and the date of disease progression or death from disease, or the date of death, respectively.
Supplemental material
Peripheral immune cell subset assessments
For the metastatic NSCLC cohort, peripheral blood values of absolute neutrophil count (ANC), absolute lymphocyte count (ALC), eosinophil fraction, neutrophil fraction and lymphocyte fraction were retrieved from the medical records at baseline (day of first treatment), at 4 weeks and at first radiographic follow-up. For the early-stage NSCLC cohort, blood cell counts, obtained as standard of care CBC with differential, were longitudinally measured at baseline (day of first treatment) and every 2 weeks until time of resection, as well as postoperatively at routine follow-up visits. NLR was calculated as the ratio of ANC to ALC, and relative change in NLR at subsequent time points was defined as the ratio of the NLR at the respective time point to the baseline NLR value. For the early stage NSCLC cohort, this was categorized into three groups: those with >10% decrease, unchanged (−10% to +10% change, inclusive), and >10% increase. Relative eosinophil percentages at 4 weeks were dichotomized as high or low using the median value at 4 weeks of 2.9%.
Machine learning
We employed XGBoost, a decision-tree based ensemble machine learning algorithm derived from a gradient-boosting framework,26 to integrate a total of 25 variables, including patient features, tumor characteristics, treatment history, peripheral blood cell values, and derived features of relative changes in immune cell subsets into a predictive model of DCB, defined as confirmed absence of progressive disease or death within 6 months. The cohort was split into two groups, 171 patients in a training set and 68 in an unseen testing set (online supplemental table S1). We employed XGBoost’s embedded feature selection within 10-fold cross validation loops for 100 iterations to reduce overfitting. The final model generated was an ensemble of 100 models. Performance was assessed using the area under the receiver operating characteristic (ROC) curve (AUC). A separate feature importance analysis through Shapley Additive Explanations (SHAP) was also performed to estimate each feature’s importance.27
RNA sequencing
Patients in the early-stage NSCLC cohort underwent a baseline tumor biopsy prior to therapy initiation and had tumor samples obtained at time of resection. Total RNA was extracted from fresh frozen tumors with the RNeasy Mini kit (Qiagen). The quality of total RNA was assessed using the RNA Integration Number measured with the RNA 6000 Nano Kit (Agilent Technologies). RNA-seq libraries for next-generation sequencing were generated by poly(A)-selection (NEBNext Poly(A) mRNA Magnetic Isolation Module) followed by reverse transcription into strand-specific cDNA libraries (NEBNext Ultra directional RNA library kit for Illumina). The generated libraries were sequenced at 100 bp paired-end on a HiSeq 2500 High Output instrument generating on average 200M total reads (online supplemental table S3). Sequence data per sample were inspected for outliers using principal component analysis (PCA); blinded variance stabilizing transformed values were used as inputs for PCA, and quality control metadata (sequencing batch and universal human reference RNA add-in batch) were visualized on the principal components. Outliers were removed based on all quality control metrics previously calculated.
Gene set enrichment analysis
DESeq2 was used to perform differential expression using a negative binomial model with a Wald test to determine significance.28 Default DESeq2 metrics were used to filter genes based on expression levels and calculate normalization factors. All visualizations of gene expression used values derived from a blind variance stabilizing transformation of raw counts. Volcano plots of results were generated using EnhancedVolcano29 with an adjusted p value threshold of 0.05 to indicate statistical significance and a fold change threshold of ±2. Results were subsequently processed by gene set enrichment analysis using fgseaMultilevel from the fgsea package30 with all Hallmark gene sets and a selection of gene sets relevant to cancer hallmarks and immune responses (online supplemental tables S4 and S5). Summary visualizations of gene set enrichment results were generated by unsupervised selection of the 10 most significant gene sets in the positive and negative direction. Gene sets fold changes were calculated in patients with samples at both baseline and post-therapy. After calculating fold changes per patient, genes were ranked by p value derived from a t-test between patients who were NLR responders, defined as those with decreased NLR of >10% at 4 weeks, and NLR non-responders, defined as those with unchanged or increased NLR >10% at 4 weeks. For a given gene set, the 30 most significant genes from the gene set were selected for inclusion in the heat map. Fold changes were thresholded to ±2.5 for visualization (online supplemental figure S1, tables S6–S8).
Supplemental material

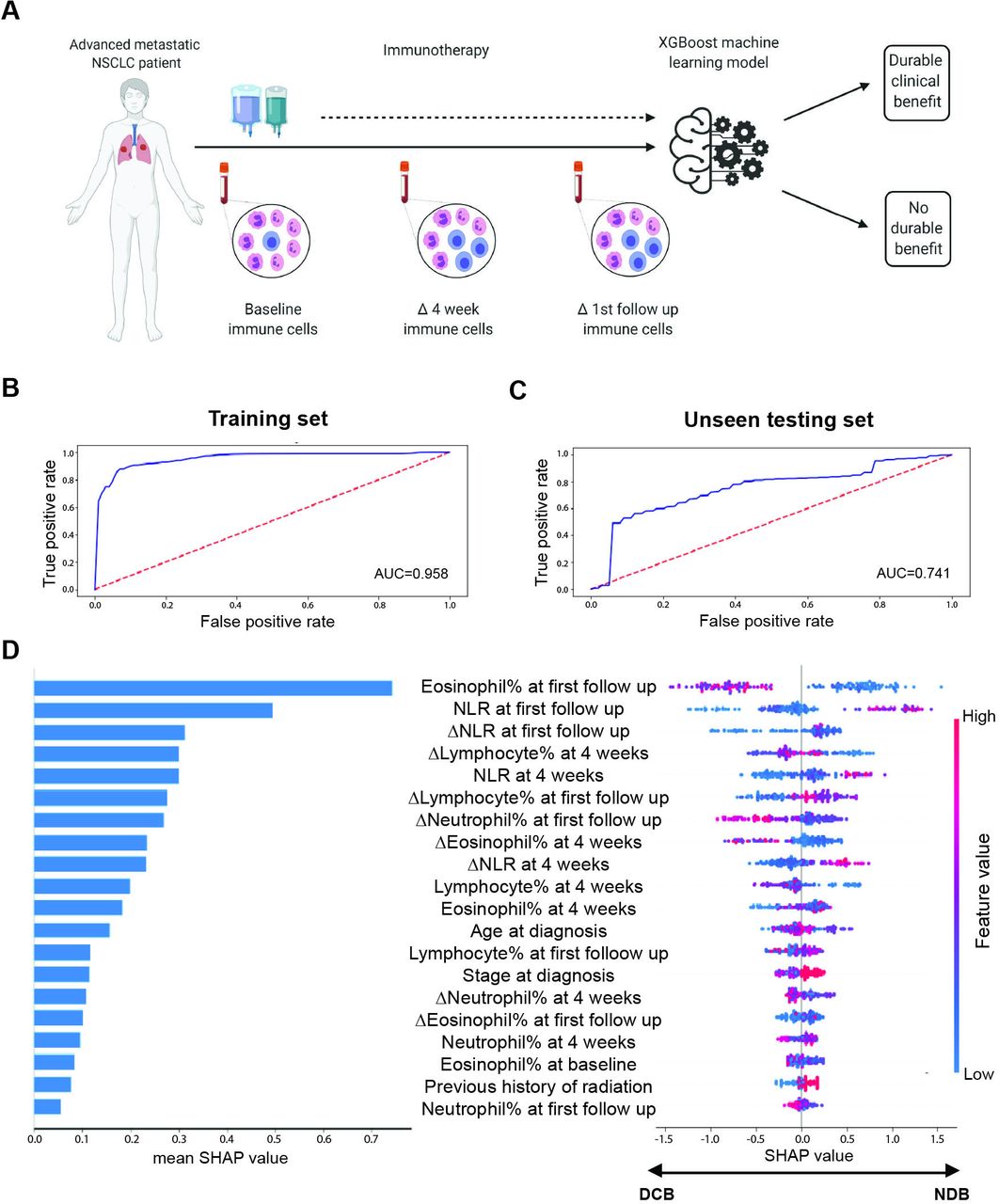
Machine learning integration of peripheral immune cell subsets predicts durable clinical benefit to ICI. (A) Sample collection schema for 239 patients with advanced metastatic NSCLC treated with ICI-containing regimens. We employed an XGBoost machine learning approach to integrate 25 variables, including clinical characteristics and peripheral immune cell dynamics to predict clinical outcome through training an ensemble of 100 models and incorporating feature selection within 10-fold cross validation loops. (B) Receiver operator curve (ROC) for model prediction of durable clinical benefit (DCB) in the training cohort of 171 patients. (C) ROC for model prediction of DCB in an unseen testing cohort of 68 patients. (D) Shapley feature importance analysis; on the left, feature importance is listed in descending order of mean absolute SHAP value, while on the right, the directional value of features are color-coded in association with SHAP value where negative SHAP value is associated with DCB and positive SHAP values are associated with no durable benefit (NDB). AUC, area under receiver operator curve; ICIs, immune checkpoint inhibitors; NSCLC, non-small cell lung cancer; SHAP, Shapley Additive Explanations; XGBoost, eXtreme gradient boosting.
TCR sequencing
T cell receptor (TCR) sequencing data from tumor tissue and peripheral blood of patients in the early-stage NSCLC cohort was retrieved from an earlier publication.31 For each patient, T cell clones found in that individual’s post-treatment tumor sample were identified in the matched pretreatment and post-treatment blood samples and deemed to be tumor-specific clones for that patient. Productive TCR frequencies, calculated as the normalized count of unique productive TCR rearrangements among the total number of productive TCR rearrangements detected in any given sample, were further analyzed and are shown in online supplemental table S9. For each sample, a clonality metric was estimated to quantitate the extent of monoclonal or oligoclonal expansion by measuring the shape of the clone frequency distribution. Clonality values range from 0 to 1, where values approaching 1 indicate a nearly monoclonal population. An intersection analysis of all TCR clonotypes identified in tumor and peripheral blood among patients was performed to identify public versus private clonotypes. For each patient, differential abundance analysis using Fisher’s exact test with false discovery rate (FDR) correction was performed to determine significant changes in TCR clone relative abundance during treatment. Aggregation of significantly increasing or decreasing clone counts per patient, including tumor-specific clones, was performed for comparisons of patients according to NLR dynamics (online supplemental table S9). To examine variability in signal for clone dynamics, clones were characterized as increasing or decreasing during therapy by: (1) maximum relative abundance pre-ICI, (2) minimum relative abundance in post-ICI, and (3) maximum FDR adjusted p value, with or without normalization to the total unique productive clones. Statistical comparisons of the aggregated TCR clone metrics across patients were performed using the Mann-Whitney U test with FDR correction.
Next-generation sequencing analyses
Tumor samples were processed for whole exome sequencing as previously described.5 Serial blood samples were collected prior to treatment, every 2 weeks until the time of resection and postoperatively at routine follow-up visits and processed for targeted error-correction sequencing (TEC-Seq) as previously described (online supplemental tables S10–S12).11 Briefly, cell-free DNA (cfDNA) was isolated from plasma using the Qiagen Circulating Nucleic Acids Kit (Qiagen GmbH). TEC-Seq cell-free DNA libraries were prepared, followed by targeted capture using a custom set of hybridization probes (online supplemental table S10) and sequenced using 100 bp paired end runs on the Illumina HiSeq 2000/2500 (Personal Genome Diagnostics, Baltimore, Maryland, USA).32 For a subset of samples, matched white blood cell sequencing was also performed to filter out variants related to clonal hematopoiesis (online supplemental table S12). TEC-Seq characteristics are described in online supplemental table S13. Genomic alterations identified in plasma cfDNA were cross-referenced against tumor and white blood cell sequencing to determine variant origin (online supplemental table S14). Molecular response was defined as clearance of tumor-derived variants prior to surgery. Patients with no tumor-derived variants at any time point were considered to have non-detectable ctDNA.
Statistical analyses
Features associated with major pathological response were compared using the χ2 exact test for categorical variables. Spearman rank-order correlation coefficient was used to assess correlations between continuous variables and residual tumor at the time of resection. The Mann-Whitney U-test was used to assess differences in residual tumor. Fisher’s exact test was used to assess differences in NLR and ctDNA dynamics in patients with differential pathological responses. Survival analysis was completed through log-rank test. ROC calculations were performed using the pROC package in R.33 Statistical analyses were performed using STATA (release 16) and R (V.4.0.2).
Results
Machine learning links early immune cell subset dynamics with response to immunotherapy
We investigated the value of peripheral immune cell dynamics in predicting DCB (see Methods) for a cohort of 239 patients with metastatic NSCLC treated with ICI containing regimens (figure 1A, online supplemental table S1). We employed XGBoost, a decision-tree-based ensemble machine learning algorithm, to integrate baseline and longitudinal values, as well as relative changes of immune cell subsets with clinical characteristics and predict clinical benefit from ICI (see Methods). Following splitting of the cohort into a training (n=171) and unseen testing set (n=68), we trained an ensemble of 100 models, incorporating feature selection within 10-fold cross validation loops. The resulting model predicted DCB with an area under the ROC curve (AUC) of 0.96 for the training (95% CI 0.96 to 0.96), 0.72 for cross-validation testing (95% CI 0.68 to 0.75) and 0.74 for the unseen dataset (95% CI 0.74 to 0.74; figure 1B,C). In contrast, the AUCs for PD-L1 and TMB were 0.54 (95% CI 0.44 to 0.64) and 0.61 (95% CI 0.50 to 0.73), respectively, for the training dataset, and 0.61 (95% CI 0.45 to 0.76) and 0.49 (95% CI 0.25 to 0.73), respectively, for the unseen dataset (online supplemental figure S2A–D). Feature importance analysis revealed that peripheral blood NLR and eosinophil fraction at first radiographic follow-up, NLR at 4 weeks, as well as relative change in NLR at these time points were the strongest predictors of clinical benefit (figure 1D, online supplemental figure S2E). Specifically, a low NLR value, decrease in NLR at first radiographic follow-up, or high eosinophil percentage all favored clinical benefit from ICI (figure 1D).

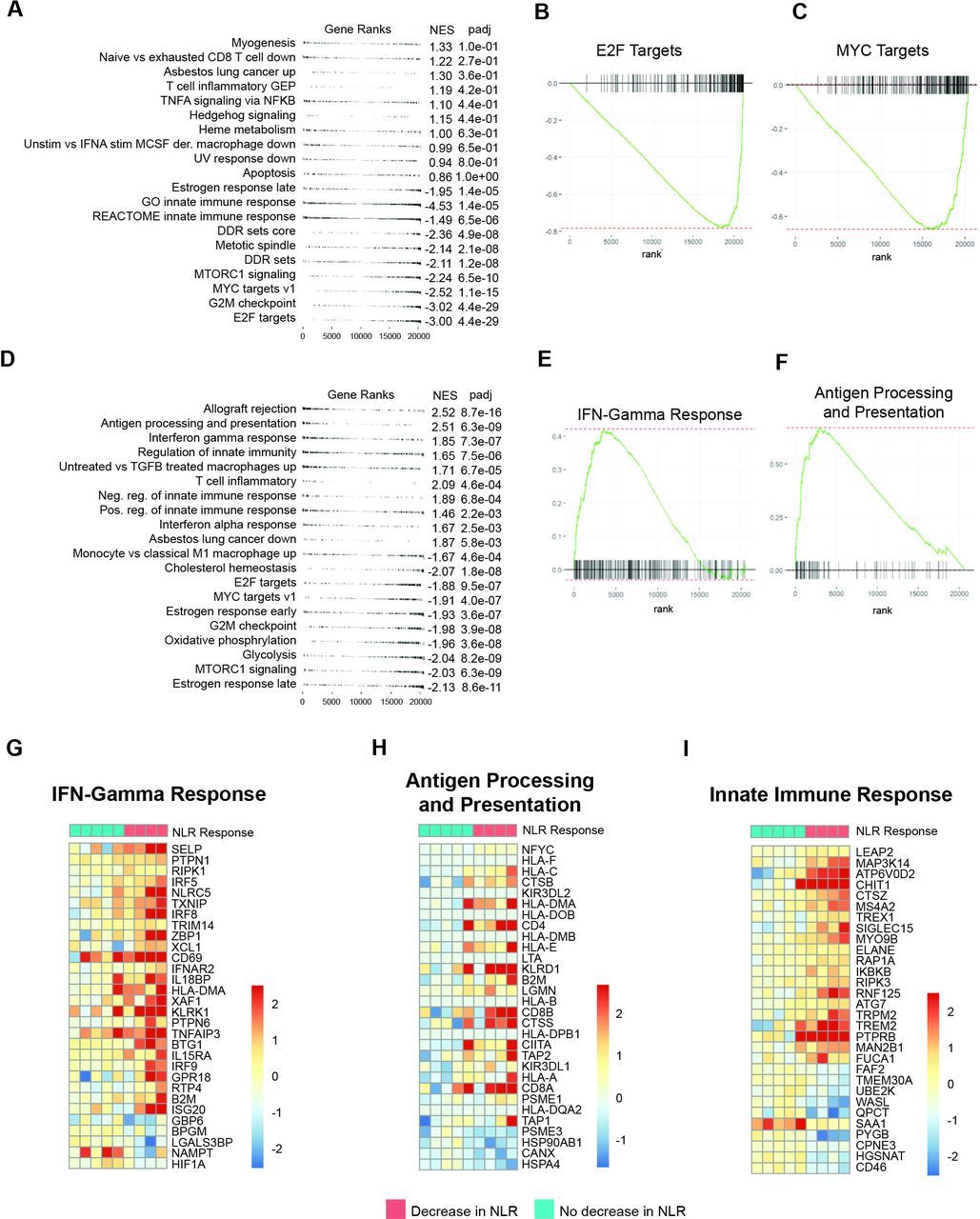
NLR dynamics point to distinct transcriptomic profiles in the tumor microenvironment. (A–C) Gene set enrichment analysis in baseline (pre-ICI treatment) tumor samples with (A) gene sets ranked by p value. Leading edge analyses are shown for (B) E2F targets and (C) MYC targets, which are enriched in patients who do not manifest a decrease in NLR. (D–F) Gene set enrichment analysis in post-ICI tumor samples with (D) gene sets ranked by p value. Leading edge analysis are shown for (E) IFN-γ and (F) antigen processing and presentation gene expression programs, which are significantly upregulated in the TME of patients with a peripheral blood decrease in NLR. (G–I) Fold change paired analysis between pre-ICI and post-ICI tumors for (G) IFN-γ, (H) antigen presentation, and (I) innate immune response gene signatures, suggesting peripheral NLR dynamics reflect an ICI-associated antitumor adaptive immune response. Dn, Down; ICI, immune checkpoint inhibition; IFN, interferon; NES, normalized enrichment score; NLR, neutrophil–lymphocyte ratio; TME, tumor microenvironment.
Peripheral blood NLR dynamics reflect distinct transcriptional profiles in the TME
As TANs can suppress innate and adaptive immune responses,34 we next sought to investigate whether peripheral NLR dynamics were associated with distinct transcriptional signatures in the context of ICI therapy. We evaluated 41 serial tumor samples from an independent cohort of 26 patients with early-stage NSCLC treated with neoadjuvant ICI prior to definitive surgical resection (Methods; online supplemental figure S3 and table S2) and performed serial RNA sequencing of baseline and post-ICI tumors coupled with sequence data deconvolution (online supplemental table S3). Dichotomizing based on a decrease of >10% in NLR at 4 weeks, which was identified in the metastatic NSCLC cohort as associated with clinical benefit from immunotherapy, we performed differential enrichment analyses for pre-ICI and post-ICI treated tumors and further evaluated relative changes in proinflammatory profiles during therapy (online supplemental figure S1A,B and tables S4,S5). These analyses revealed an enrichment in E2F targets (adjusted p=4.4e-29) and G2M checkpoint cell cycle progression (adjusted p=4.4e-29) as well as MYC targets (adjusted p=1.1e-15) in pre-ICI tumors from patients without a decrease in NLR (figure 2A–C), suggesting a proliferation advantage of these tumors in an immunosuppressive TME.35 Importantly, after induction with ICI, an upregulation of IFN-γ and antigen processing and presentation expression programs (adjusted p=7.3e-07 and p=6.3e-09, respectively) alongside with activation of conserved immune rejection signatures (adjusted p=8.7e-16), differentiated the TME of patients with a peripheral blood decrease in NLR (figure 2D–F). We then focused on paired analyses of pre-ICI and post-ICI tumors, which similarly showed an induction of IFN-γ and antigen presentation programs for patients with a decrease in NLR, suggesting an effective adaptive immune response and subsequent T cell cytotoxicity (figure 2G–H) in conjunction with upregulation of genes involved in innate immune responses (figure 2I; online supplemental tables S6–S8).

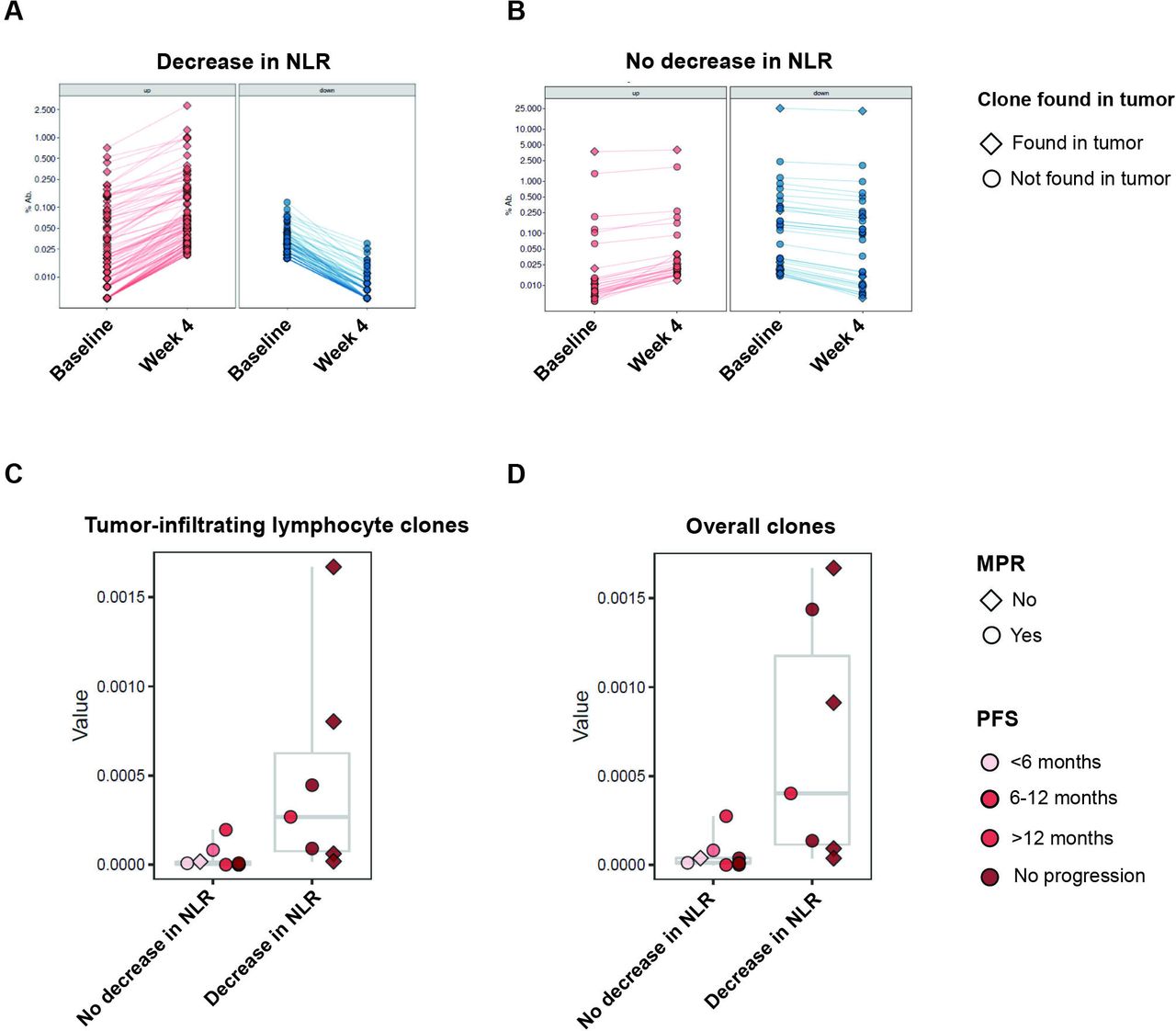
T cell repertoire dynamics are reflected in peripheral blood NLR dynamics. (A and B) Representative examples of TCR reshaping, signified by clonotypic expansions (red), notably of TCR clones also found in the tumor (diamond), or retractions (blue), notably of TCR clones not found in the tumor (circle), in peripheral blood, for (A) a patient with a decrease in NLR, compared with limited TCR repertoire changes in (B) a patient without a decrease in NLR, highlighting more clonotypic expansion in patients with a decrease in NLR in both overall clones and tumor-infiltrating clones. (C and D) Proportion of clonotypic expansion relative to baseline, defined as a statistically significant increase in clonotypic abundance in the on-therapy samples compared with pretreatment abundance. Compared with patients with unchanged or increased NLR, patients with decreased NLR had greater amounts of clonotypic expansion in both (C) clones identified in tumor and (D) overall clones. MPR, major pathologic response; NLR, neutrophil–lymphocyte ratio; PFS, progression-free survival; TCR, T cell receptor.
NLR dynamics in peripheral blood capture T cell repertoire reshaping post-ICI
Building on the differential gene expression data and under the premise that NLR dynamics may identify patients with effective T cell adaptive immune responses resulting in clinical responses with ICI, we sought to further investigate the relationship between changes in NLR and intratumoral T cell clonal dynamics in the peripheral blood during immune checkpoint therapy. We analyzed TCR Vb sequencing data in paired pre-ICI and post-ICI PBMC samples and post-ICI tumor samples from 16 patients with NSCLC treated with neoadjuvant ICI (online supplemental table S9).31 Examining the intersection of intratumoral repertoires, we found that the majority of productive intratumoral TCR clonotypes were private (96.7%, online supplemental figure S4). In looking at pretreatment peripheral T cell repertoires, with a focus on clones also found in the tumor (see Methods), we did not identify any statistically significant differences in aggregate T cell repertoire features among patients with differential NLR dynamics. However, patients with a decrease in NLR showed significant expansions in peripheral blood of matched intratumoral TCR clones 4 weeks after ICI initiation (Mann-Whitney FDR-adjusted p=0.018; figure 3A–D). The majority of intratumoral clones with significant clonotypic reshaping in peripheral blood during ICI were largely private (92.2%; online supplemental figure S4). Taken together, these findings may suggest that decreases in the peripheral neutrophil populations during immunotherapy may be linked with emergent T cell responses that may mediate DCB.
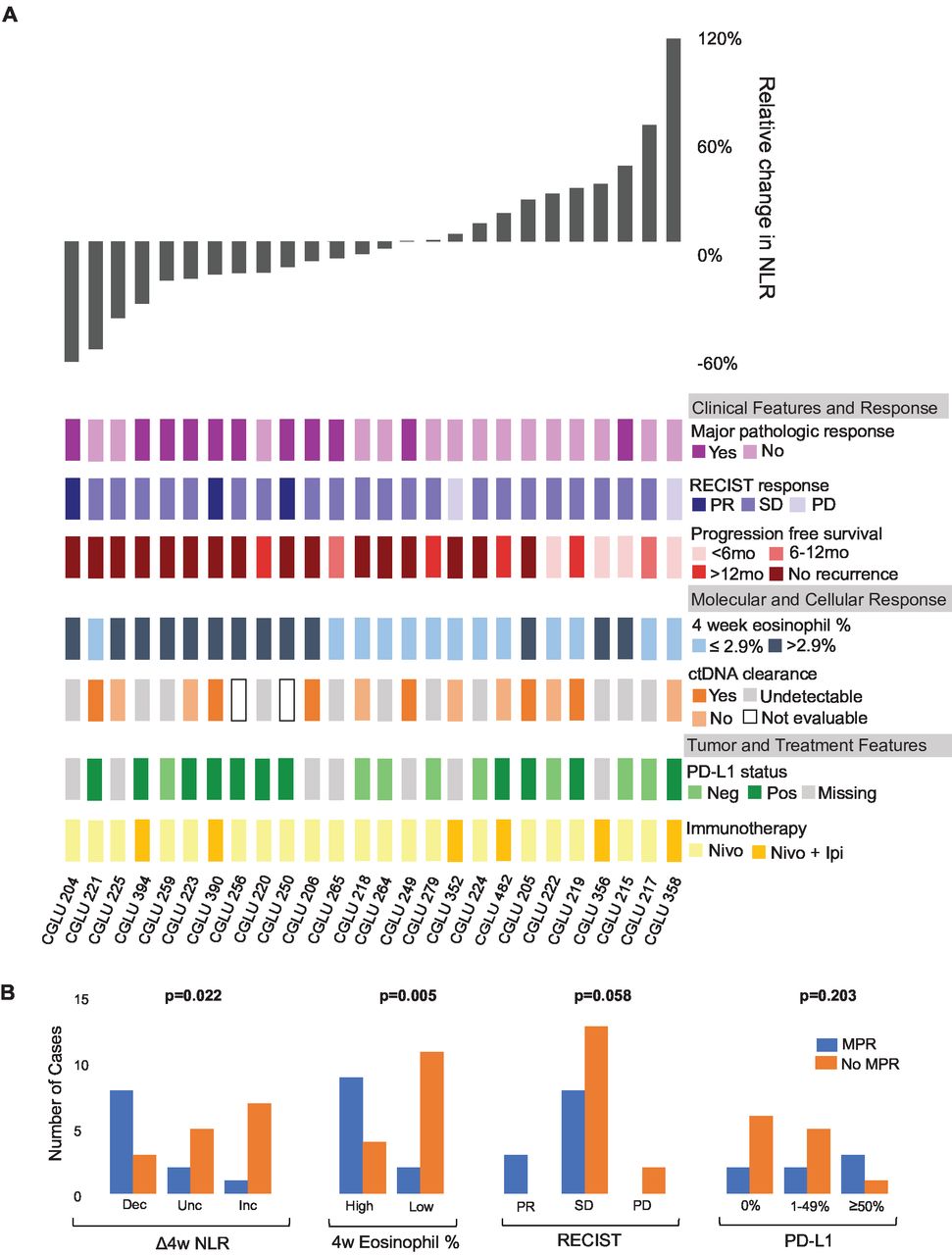
Peripheral immune cell subset dynamics are associated with response to ICI. (A) Representation of each patient in the early-stage NSCLC as a column, showing change in NLR along with clinical features, molecular and cellular features, and tumor and treatment features. Decreases in NLR and elevated on treatment eosinophil fractions were associated with major pathologic response and longer progression free survival. NLR dynamics did not appear to be associated with radiographic response, PD-L1 status, or type of immunotherapy received. Majority of patients showed stable disease by RECIST, highlighting difficulties of traditional radiography in capturing response to immunotherapy. (B) Early changes in NLR and on treatment eosinophil fraction are statistically associated with MPR, while PD-L1 and RECIST are not. χ2 test are based on two-sided testing for on treatment eosinophil fraction, RECIST, and PD-L1. Early changes in NLR was based on one-sided χ2 testing. ICI, immune checkpoint inhibitor; MPR, major pathologic response; Nivo, nivolumab; Nivo+Ipi, nivolumab+ipilimumab; NLR, neutrophil–lymphocyte ratio; NR, no response; NSCLC, non-small cell lung cancer; PD, progressive disease; PR, partial response; R, response; SD, stable disease.
Peripheral blood immune cell dynamics are reflective of pathologic response to ICI
Ultimately, we hypothesized that peripheral immune cell dynamics, being reflective of tumor rejection, would predict the depth of response to ICI. Using the cohort of patients with NSCLC treated with neoadjuvant ICI, we determined tumor regression post-ICI at a cellular level by immune-related pathological response criteria (see Methods; online supplemental figure S2 and table S2).25 Consistent with our hypothesis, we found that peripheral blood immune cell dynamics during immunotherapy, determined by NLR dynamics but not based on differences in the absolute number of neutrophils, were predictive of pathologic response. An early decrease of >10% in NLR at 4 weeks after ICI initiation was associated with tumor regression (Mann-Whitney test, p=0.0032) and MPR (χ2, p=0.007) at time of resection. Notably, an elevated eosinophil count at 4 weeks (see Methods) was also predictive of tumor regression (Mann-Whitney test, p=0.0086) and MPR (χ2 p=0.005), which was consistent with our findings in the metastatic NSCLC cohort. In line with previous studies highlighting the challenges with radiographic imaging and PD-L1 expression to capture therapeutic responses,36 baseline PD-L1 status and radiographic RECIST response failed to predict pathological response in this cohort (χ2 p=0.21 and p=0.06 respectively; figure 4). Patients with a decrease in NLR at 4 weeks had a significantly longer recurrence-free (log-rank p=0.0097) and a trend towards longer OS (log-rank p=0.070; figure 5A,B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
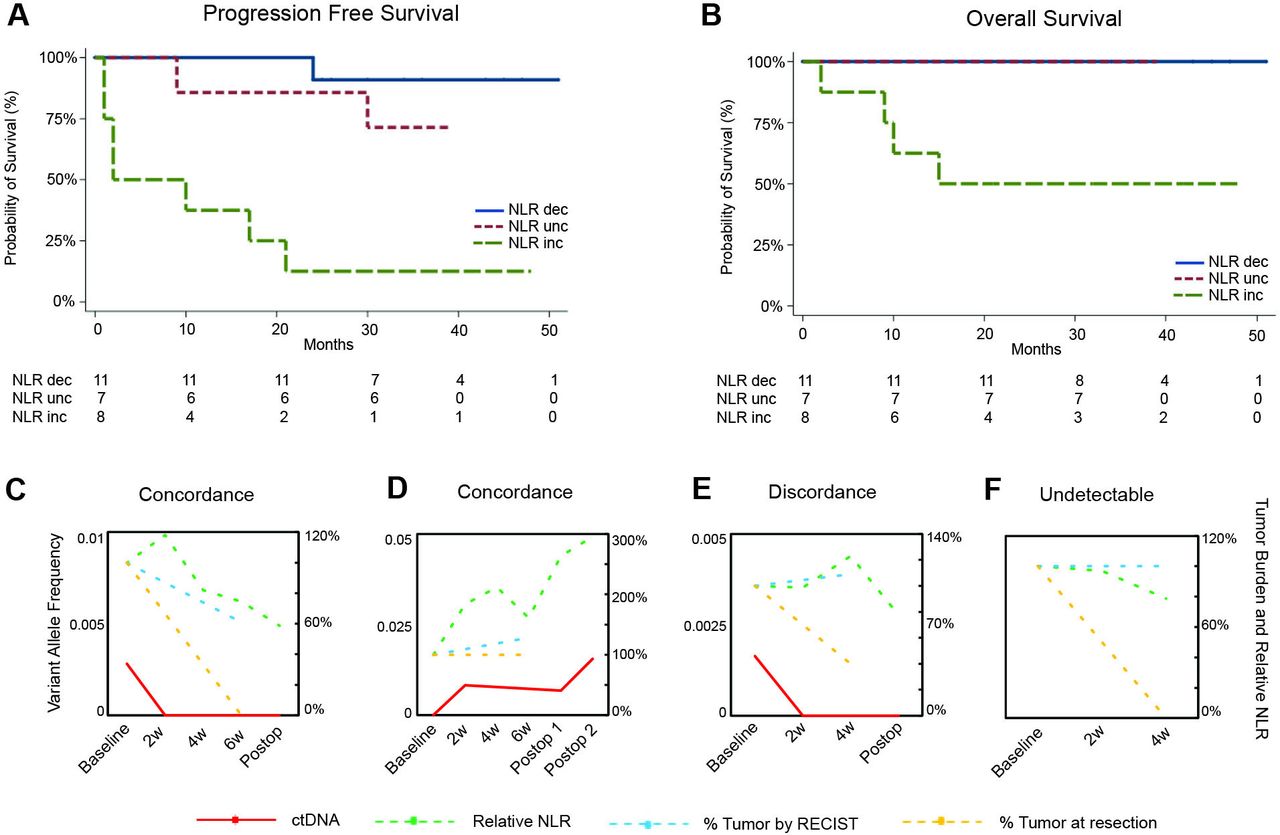
NLR dynamics predict survival and complement ctDNA molecular responses. (A and B) Progression-free survival (A) and overall survival (B) stratified by NLR dynamics in the early-stage NSCLC cohort, demonstrating that a decreased NLR was significantly associated with longer PFS (log-rank p=0.0097) and OS (log-rank p=0.07). Survival curves were compared by using non-parametric log-rank test. (C–F) Comparison of ctDNA and NLR dynamics with tumor evaluation by RECIST and pathologic response at resection in representative examples. Variant allele frequency is shown on the left axis for variants confirmed to be tumor derived. Percent tumor burden and NLR value relative to baseline are shown on the right axis. For a patient with MPR (C), an early decrease in NLR captured therapeutic effect and was consistent ctDNA molecular clearance (KRAS G12C mutation) compared with RECIST tumor burden, which showed partial response. In contrast, for a patient with no tumor regression post-ICI (D), an early increase in NLR was consistent with ctDNA molecular persistence (KRAS Q61H mutation) and radiographic progressive disease. (E) In a patient where an early increase in NLR was discordant with ctDNA molecular clearance (TP53 R248L mutation), pathologic evaluation of his primary tumor and a satellite nodule revealed two separate histologies, suggesting ctDNA molecular clearance reflecting one tumor’s response and increasing NLR reflecting the other tumor’s lack of response. In a patient with undetectable ctDNA (F), early decrease in NLR accurately captured the therapeutic effect and MPR compared with RECIST tumor burden. ctDNA, circulating tumor DNA; Dec, decreased; Inc, increased; MPR, major pathologic response; NLR, neutrophil–lymphocyte ratio; NSCLC, non-small cell lung cancer; OS, overall survival; PFS, progression free survival; Unc, unchanged.
Peripheral NLR dynamics complement ctDNA molecular responses
We further hypothesized that dynamic assessment of NLR would be complementary to ctDNA-based measurements of circulating tumor burden and molecular response. To evaluate the association between NLR dynamics and clinical responses by an orthogonal molecular method, we performed longitudinal targeted error-correction sequencing of 82 serial plasma and matched leukocyte DNA samples as well as whole exome sequencing for 24 tumor samples from patients in the neoadjuvant ICI cohort (online supplemental tables S10–S14). As illustrative examples, graphical representation of change over time in NLR, ctDNA variant allele frequency, RECIST assessment of tumor, and pathologic assessment of tumor for four patients are shown (figure 5C–F). Non-invasive assessment by either NLR change at 4 weeks or ctDNA kinetics were associated with lack of clinical progression or recurrence (Fisher’s exact p=0.014). For the subgroup of patients with detectable ctDNA (n=13 of 24, 54%), response by ctDNA clearance or decrease in NLR was associated with long-term outcome (seven of eight patients with a decreased NLR or ctDNA response had no disease progression or recurrence, while three of five patients without a decrease in NLR or ctDNA response had disease progression or recurrence). For the subgroup of patients that did not have detectable ctDNA (n=11 of 24, 46%), NLR dynamics were concordant with MPR (three of four patients with a decrease in NLR had MPR, while five of seven patients without a decrease in NLR did not have MPR) and clinical progression or recurrence (three of four patients with a decrease in NLR did not have progression or recurrence, while five of seven patients without a decreased NLR did). Despite the small sample size, these findings suggest that NLR dynamics complement ctDNA dynamics in reflecting long-term clinical response to immunotherapy treatment and may be informative when ctDNA is undetectable.
Discussion
Immunotherapy has revolutionized the treatment options for patients with NSCLC, but our current static, pretreatment tissue-based predictive biomarkers, such as PD-L1 and TMB, do not reflect the plasticity of antitumor immune responses during ICI therapy. Here we have examined dynamic changes in peripheral immune cell subsets in patients with both metastatic and early-stage NSCLC receiving ICIs. Using transcriptomic analysis along with T cell repertoire trajectories, we showed that decreases in NLR are associated with an induction of innate and adaptive immune responses in the TME, as well as expansion of individual intratumoral T cell clones in the periphery. Taken together, this suggests that NLR dynamics may be an indirect peripheral measurement of an emerging antitumor immune response during immunotherapy and ultimately, favorable clinical outcomes. Furthermore, we showed that NLR dynamics may be complementary to ctDNA kinetics and informative in cases where ctDNA is undetectable.
NLR reflects a broad interaction between systemic inflammation and overall immune function, and in the context of cancer, may serve as a proxy for the equilibrium between tumor-mediated inflammation and antitumor immunity. An elevated pretreatment NLR has been associated with poor outcomes and has also been associated with lack of immunotherapy response.20 These observations have been attributed to both circulating and TANs and their roles in tumorigenesis, angiogenesis, and moderation of the tumor immune microenvironment through T cell suppression, contributing to immunotherapy resistance.6–8 10 15 Tumors produce IL-8, a chemokine that attracts neutrophils and is strongly associated with a tolerogenic TME and poor response to ICI.37 Neutrophils, in turn, secrete chemokines that promote tumor proliferation, invasion, and angiogenesis, such as neutrophil elastase, matrix metallopeptidase-9 and vascular endothelial growth factor.34 Within circulation, neutrophil extracellular traps are believed to play a key role facilitating the concentration of cancer effectors resulting in metastasis formation.34 Additionally, there is evidence that neutrophils can associate with circulating tumor cells, enabling establishment of metastatic sites of disease.38 In contrast, lymphocytes, particularly CD8+ T cells, play a key role in antitumor response by inhibiting tumor cell proliferation and migration as well as inducing cytotoxic cell death.39 40 Notably, lymphocytopenia has been associated with poor survival in numerous settings, as tumors may induce lymphocyte apoptosis both within the TME and in peripheral circulation as a means of avoiding immune recognition.41–43 In line with this notion, we showed that peripheral expansion of T cell clones found in the TME may be reflected in NLR dynamics and in particular a decrease in NLR values during immunotherapy.
Furthermore, we identified a correlation between increased peripheral eosinophils during ICI and clinical response. Previous studies have demonstrated that elevated blood eosinophil content correlates with intratumoral eosinophil concentration and activation and may predict response to ICI.44 Eosinophils are known to be present in the TME, but their role is not fully understood, and there is conflicting evidence regarding the prognostic impact of tumor eosinophilia in different tumor types.44 45 Immune cell subsets such as lymphoid cells, natural killer T cells, and mast cells, as well as cancer cells, can release interleukin-5, an important cytokine for eosinophil growth, differentiation, and activation, resulting in eosinophilic infiltration of tumor.46 Eosinophils in turn may exert direct cytotoxic effect against cancer cells, particularly when activated by IFN-γ, and can promote recruitment of CD8+ T cells through secretion of chemokines CCL5, CCL9, and CXCL10, which combined with alteration of the TME vasculature may lead to tumor regression.47 48 The interaction of intratumoral CD8+ T cells and eosinophils has been shown to induce M1-like activated macrophage polarization.48 Furthermore, histamine may induce the immunosuppressive M2 macrophage phenotype and addition of H1-antihistamine can enhance immunotherapy response.49 Eosinophils have bidirectional interaction with mast cells50; therefore, this may represent another pathway through which eosinophils influence ICI response.
Our study has several limitations; given its retrospective nature, there may be uncaptured sources of bias or confounding factors. Additionally, the number of patients included in the early-stage NSCLC cohort was limited, as was necessitated by the large number of samples analyzed from each patient. However, our findings are strengthened by the concordance with the larger metastatic NSCLC cohort. Furthermore, though we did not assess tumor-associated and mutation-associated epitope TCR reactivity, our analyses identified largely private intratumoral TCR clonotypic expansions in peripheral blood during immune checkpoint blockade, suggesting that these may be involved in the antitumor immune responses in the context of therapy.
In summary, our findings support the notion that NLR dynamics are associated with underlying changes in the quality of the antitumor immune response in the TME as well as with reshaping of the T cell repertoire in the periphery, providing the biological basis as to why NLR dynamics are associated with therapeutic response with immune checkpoint blockade. As an indirect reflection of immunologic changes in the TME with immunotherapy, NLR dynamics complement assessment of molecular response with ctDNA, particularly for early stage patients with undetectable ctDNA, a finding that has not been previously reported, to the best of our knowledge. Moving forward, we envision integrative predictive models of response that incorporate these non-invasive, readily available, and dynamic biomarkers. Capturing the tumor-immune cell equilibrium may allow for early identification of patients less likely to attain a long-term survival on immunotherapy, allowing for rapid adaptive changes in therapy.
Data availability statement
Data are available on reasonable request. Data are available in the supplementary tables of this manuscript and available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Patients provided written informed consent; the study protocol was approved by the institutional review boards of JHU and MSKCC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @SmithImmunology, @ValsamoA
MH, JVC and SR contributed equally.
Contributors MH, JVC, SR, TS and VA conceived the study, developed, and implemented the methodology, performed data analysis, interpreted the data and wrote the manuscript. GZ, JRW, CC, AB, NN performed data analyses. ZB, GP, AC, JZ, KNS, LS, JER, KM, JCM, QKL, VL, BPL, CH, VEV, JRB, and PMF interpreted the data and wrote the manuscript. VA was responsible for the the study's oversight and is the study's guarantor. All authors have read and approved the final version of the manuscript.
Funding This work was supported in part by Bristol Myers Squibb through its International Immuno-Oncology Network, the US National Institutes of Health grant CA121113 (VA and VEV) and CA 9071-40 (MH and JVC), the Maryland Department of Health and Mental Hygiene (Cigarette Restitution Fund Program; MH), the ECOG-ACRIN Thoracic Malignancies Integrated Translational Science Center grant UG1CA233259 (VA and VEV), the Bloomberg-Kimmel Institute for Cancer Immunotherapy (VA and PMF), the V Foundation (VA and VEV), Swim Across America (VA), the LUNGevity Foundation (VA, VEV), the Emerson Collective (VA), the Johns Hopkins Research Program in Quantitative Sciences (JVC), and the Pearl M. Stetler Research Fund (JVC).
Competing interests VA receives research funding to her institution from Bristol-Myers Squibb and Astra Zeneca. PMF has received research funding to his institution from AstraZeneca, Bristol-Myers Squibb, Novartis, Corvus, Kyowa. He has also served as a consultant for Amgen, AstraZeneca, Bristol-Myers Squibb, Daiichi Sankyo, Iteos, Janssen, Mirati, Novartis, Sanofi and as a DSMB member for Polaris and Flame Therapeutics. KNS receives research funding to her institution from Bristol-Myers Squibb, Astra Zeneca, and Enara Bio, and holds founder’s equity in manaT Bio. VEV is a founder of Delfi Diagnostics and Personal Genome Diagnostics, serves on the Board of Directors and as a consultant for both organizations, and owns Delfi Diagnostics and Personal Genome Diagnostics stock, which are subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics and Personal Genome Diagnostics. VEV is an inventor of multiple licensed patents related to technologies from Johns Hopkins University. Some of these licenses and relationships are associated with equity or royalty payments directly to Johns Hopkins and VEV. VEV is an advisor to Bristol-Myers Squibb, Danaher, Genentech, and Takeda Pharmaceuticals. Within the last five years, VEV has been an advisor to Merck and Ignyta. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. JW is a consultant for Personal Genome Diagnostics, is the founder and owner of Resphera Biosciences and holds patents, royalties or other intellectual property from Personal Genomic Diagnostics. JER is in the advisory board/consultant of Oncocyte, receives speaking fees for Astrazeneca, and has received research funding to his institution from Genetech/Roche, and Verastem. JB is in the advisory board/consultant of Amgen, AstraZeneca, BMS, Genentech/Roche, Eli Lilly, GlaxoSmithKline, Merck, Sanofi and Regeneron, receives grant research funding from AstraZeneca, BMS, Genentech/Roche, Merck, RAPT Therapeutics, Inc and Revolution Medicines and is in the Data and Safety Monitoring Board/Committees of GlaxoSmithKline, Sanofi and Janssen. TS is in the advisory board/consultant of Cue Biopharma, Dracen, Innate, Nanobiotix, Merck, Sanofi, Synthekine, receives grant research funding from AstraZeneca, BMS, Cue Biopharma, Genentech/Roche, Merck, Nanobiotix, Synthekine, and is in the Data and Safety Monitoring Board/Committees of Astra Zeneca, and Nektar. VL has received research funding to his institution from AstraZeneca, Bristol-Myers Squibb, Merck, SeaGen. He has also served as a consultant for Takeda, SeaGen, Bristol-Myers Squibb, AstraZeneca, and Guardant Health.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.