Article Text

Abstract
Background Gasdermin D (GSDMD) is well known as a downstream of inflammasomes. However, the roles of GSDMD itself in hepatocellular carcinoma (HCC) remain unclear.
Methods Two independent cohorts of patients with HCC were analyzed to evaluate the pathological relevance of GSDMD/pTBK1/PD-L1. GSDMD knockout (GSDMD-/-) mice, Alb-Cre mice administered with an adeno-associated virus (AAV) vector that expressed the gasdermin-N domain (AAV9-FLEX-GSDMD-N) and their wild-type littermates were used to induce hepatocarcinogenesis or metastatic HCC. Combination of anti-programmed cell death protein-1 (PD-1) and GSDMD inhibitor dimethyl fumarate (DMF) was used to test for improved therapeutic efficacy. RNA sequencing was used to explore the mechanisms how GSDMD promoted HCC progression.
Results The expression of GSDMD and GSDMD-N was upregulated in HCC tissues or metastatic HCC tissues and positive GSDMD expression indicated grim prognosis. Diethylnitrosamine/carbon tetrachloride or thioacetamide-treated GSDMD-/- mice exhibited decreased liver tumors. In contrast, AAV9-FLEX-GSDMD-N promoted hepatocarcinogenesis. RNA sequencing manifested that knockout of GSDMD impacted the cyclic GMP-AMP synthase (cGAS) pathway and immune-associated pathway. GSDMD damped cGAS activation by promoting autophagy via outputting potassium (K+) and promoted programmed death ligand-1 (PD-L1) expression by histone deacetylases/signal transducer and activator of transcription 1 (STAT1)-induced transactivation of PD-L1 via importing calcium (Ca2+). High Mobility Group Box 1/toll-like receptor 4 (TLR4)/caspase-1 pathway contributed to the overexpression and cleavage of GSDMD. Anti-PD-1 combining with DMF largely impaired HCC progression and metastasis.
Conclusions Targeting GSDMD could promote expression of interferons through inactivation of cGAS pathway and downregulated the PD-L1 expression. Therefore, combined anti-PD-1 and GSDMD inhibitor might serve as an effective treatment option for patients with HCC with GSDMD upregulation.
- Immunotherapy
- Inflammation
- Liver Neoplasms
- CD8-Positive T-Lymphocytes
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Gasdermin D (GSDMD) has been known as a downstream of inflammasomes.
Checkpoint blockade immunotherapy has limited efficacy in patients with hepatocellular carcinoma (HCC).
High Mobility Group Box 1 (HMGB1) could cleave pro-caspase-1 and promotes HCC metastasis.
WHAT THIS STUDY ADDS
Upregulated-GSDMD contributes to hepatocarcinogenesis and indicates poor prognosis.
GSDMD deficiency ameliorates hepatic tumorigenesis and gasdermin-N domain upregulation promotes hepatic tumorigenesis.
GSDMD damped cyclic GMP-AMP synthase activation through autophagy by outputting potassium (K+) and promoted programmed death ligand-1 (PD-L1) expression by histone deacetylases/signal transducer and activator of transcription 1 (STAT1)-induced transactivation of PD-L1 via importing calcium (Ca2+).
HMGB1/toll-like receptor 4 (TLR4)/caspase-1 pathway contributed to the overexpression and cleavage of GSDMD.
Combined anti-programmed cell death protein-1 (PD-1) and GSDMD inhibitor might serve as an effective treatment option for patients with HCC with GSDMD upregulation.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Combined anti-PD-1 and GSDMD inhibitor might serve as an effective treatment option for patients with HCC with GSDMD upregulation.
Introduction
Hepatocellular carcinoma (HCC) has been one of the most aggressive malignancies with an increased incidence in the world, and the mortality ranks top two among all the cancer-related deaths worldwide.1 Although advanced therapeutic options have witnessed substantial progress, the 5-year survival rate of HCC remains low.2 Recent years, immune-checkpoint inhibitors (ICIs) have made great success in treating cancers including HCC. However, single-agent ICIs provide clinical benefits only in 15%–20% of patients with HCC.3 Therefore, exploring the driving events involving in HCC progression and microenvironment is of paramount importance in identifying therapeutic targets and improving the prognosis.
Gasdermin D (GSDMD) is identified as the pyroptosis executioner and could be cleaved to release its gasdermin-N domain (GSDMD-N) via caspase-1 or caspase-11/4/5.4 GSDMD-N locates in plasma membrane leading to pore formation, cell swelling, massive release of the pro-inflammatory cellular contents, and ion fluxion like potassium efflux and calcium influx.5–7 GSDMD-induced pyroptosis strengthens the immune defense function.8 Accumulating evidences have illustrated that GSDMD-induced pyroptosis participates in human diseases like liver fibrosis, non-alcoholic steatohepatitis,9 10 spinal cord injury11 and colitis.12 However, no evidence is currently available on whether GSDMD itself may contribute to HCC progression. Our previous work has reported that inflammation and glutamate exchange facilitate HCC progression.13 Considering the roles of GSDMD in inflammation and species exchange, we hypothesized that GSDMD could be a new avenue for therapeutic intervention of HCC.
Cyclic GMP-AMP synthase (cGAS) has been discovered as a cytosolic DNA sensor to mount innate immunity and viral defense.14 Once the activation of cGAS by double-stranded DNA (dsDNA), type I interferons (IFNs) are produced through STING-TBK1-IRF3 pathway.14 Activation of cGAS-STING in cancer cells functions in restricting tumorigenesis, attenuating tumor growth, and recruiting immune cells for tumor clearance by upregulating inflammatory genes, such as type I IFNs.15 Therefore, exploring the mechanisms by which cGAS-STING is activated is a promising option for the defense of tumors.
Here, we demonstrated that GSDMD and GSDMD-N were upregulated in HCC and indicated poor prognosis. Knockout of GSDMD impaired hepatocarcinogenesis and upregulating GSDMD-N had the opposite effect. GSDMD-N upregulation inhibited activation of cGAS pathway by promoting the efflux of K+ and transactivated programmed death ligand-1 (PD-L1) expression by Ca2+/ histone deacetylases (HDACs)/signal transducer and activator of transcription 1 (STAT1) axis. Clinically, patients with HCC with positive expression of GSDMD/ PD-L1 had worse prognosis, and positive GSDMD and negative pTBK1 indicated worse outcome. Moreover, High Mobility Group Box 1 (HMGB1)/toll-like receptor 4 (TLR4)/caspase-1 pathway contributed to the upregulation and cleavage of GSDMD. The combined application of GSDMD inhibitor dimethyl fumarate (DMF) and anti-programmed cell death protein-1 (PD-1) abrogated HCC progression.
Materials and methods
Clinical samples
For survival analysis, two independent cohorts of patients with HCC at Tongji hospital of Tongji Medical College (Wuhan, China) were used. The details of the two cohorts were addressed as previously described.13 In addition, 10 normal liver tissues, 50 pairs of fresh HCC tissues and adjacent non-tumor tissue samples and 30 paired metastatic and matched primary HCC tissues were collected after surgical resection and were used for further investigations.
Animals and animal models
All mice were maintained in animal facilities under standard conditions. C57BL/6J wild-type (WT) mice, GSDMD-/- mice, and Alb-Cre mice administered with an adeno-associated virus (AAV) vector that expressed the GSDMD-N (AAV9-FLEX-GSDMD-N) mice were used in this study. Fourteen-day-old GSDMD-/-, AAV9-GSDMD-N and WT mice administered 25 mg/kg diethylnitrosamine (DEN; Sigma Aldrich) via intraperitoneal (i.p.) injection following the administration of 0.5 µL/g carbon tetrachloride (CCl4) dissolved in corn oil via i.p. injection once per week for 20 weeks16 or administration of thioacetamide (TAA) for 40 weeks17 to induce hepatocarcinogenesis. For the inhibition of GSDMD, the mice were treated with DMF (po, daily, 50 mg/kg).18 300 µg anti-PD-1 antibody or isotype antibody (Bio X cell, West Lebanon, New Hampshire, USA) was injected intraperitoneally every 3 days. 20 mg/kg RU320521 was used to inhibit cGAS in vivo. For in vivo signal detection of liver tumor, D-luciferin (PerkinElmer) at 100 mg/kg was injected intraperitoneally into the mice, and bioluminescence was detected using a Lago X optical imaging system (SI Imaging) as previously described.13 At the end of experiment, the mice were sacrificed, and their serum, kidneys, livers and lungs were dissected and prepared for standard further examinations.
Cell treatment
Established HCC cell lines were seeded in six-well plates and allowed to attach overnight. Then these cells were treated with recombination human HMGB1 (Sigma), and/or ERK, JNK, P38 and PI3K pathway inhibitors.
Statistical analysis
All values were recorded as the mean±SD. All experiments were repeated with three or more independent biological replicates. Statistical significance between the means of two groups was determined using Student’s t-tests (normal distribution), Mann-Whitney U tests (abnormal distribution) or Wilcoxon signed-rank test (matched pairs). The statistics of means of multiple groups were performed using one-way analysis of variance (ANOVA) or two-way ANOVA. Immunohistochemical score was analyzed by χ2 test. The cumulative recurrence and survival curves were shown by the Kaplan-Meier method and the statistical significance was determined by log-rank test. Multivariate analysis was performed by Cox regression analysis. Correlations were performed by using a Pearson correlation test. Statistical analysis was justified as appropriate among all figures. P values<0.05 were considered to be statistically significant. All of the data were triplicated. Statistical values were calculated with SPSS software (V.20.0) or GraphPad Prism V.8.0 software.
Other materials and methods applied in this research were available in online supplemental material 1.
Supplemental material
Results
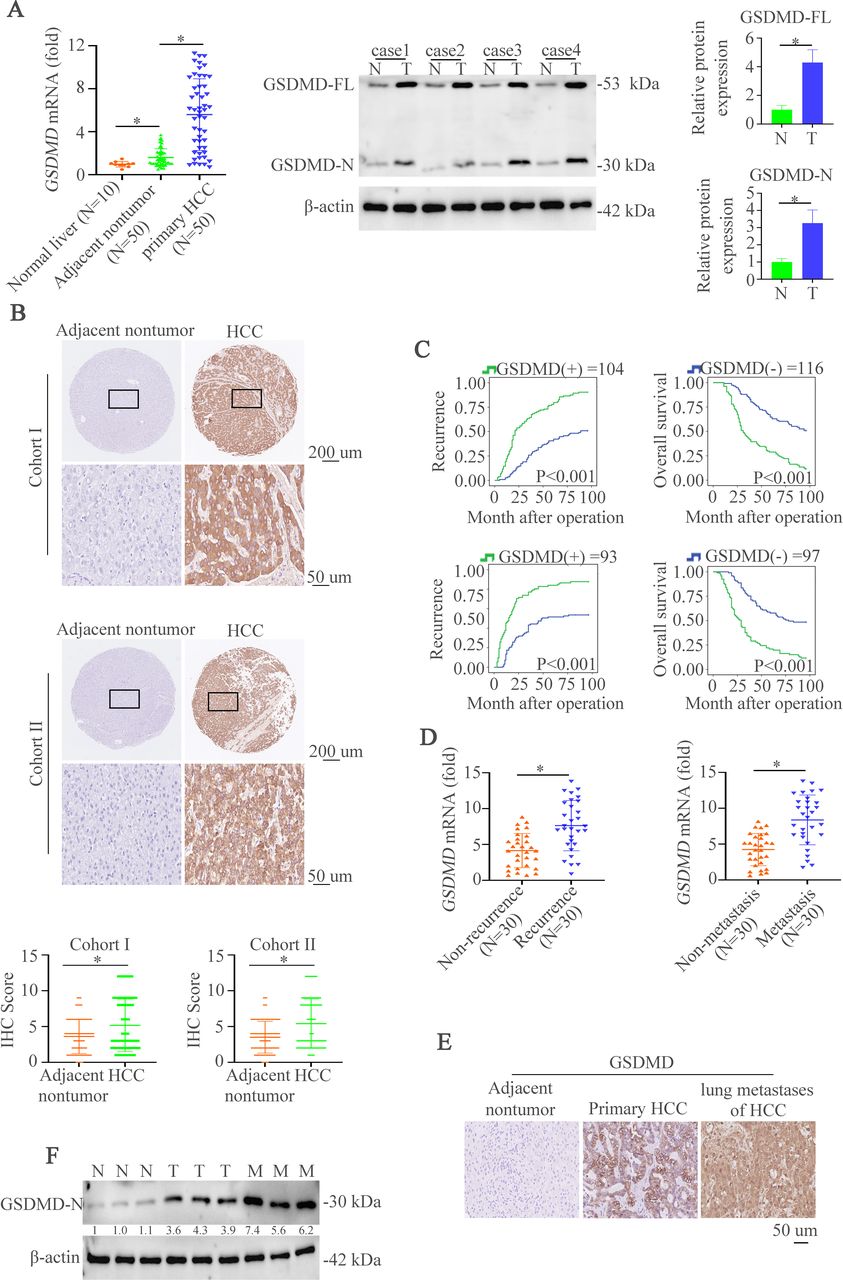
GSDMD and its cleaved GSDMD-N are upregulated in HCC tissues and indicate grim clinical prognosis
To characterize the importance of GSDMD in HCC, we first evaluated the messenger RNA (mRNA) expression of full-length GSDMD (GSDMD-FL) in liver biopsies from 10 normal liver tissues and 50 paired HCC specimens. HCC tissues displayed an upregulation of GSDMD mRNA level compared with non-tumor tissues and normal liver tissues (figure 1A, left). GSDMD-FL and its cleaved production GSDMD-N were elevated in HCC tissues compared with the non-tumor tissues (figure 1A, right). Consistently, immunohistochemistry (IHC) assay was used to show GSDMD expression in two cohorts of HCC tissue microarray. The results demonstrated that staining of GSDMD was stronger in primary HCC tissues than paired non-tumor tissues (figure 1B and online supplemental figure S1A). Furthermore, patients with positive GSDMD expression had shorter overall survival time and higher recurrent rate than those with negative GSDMD expression (figure 1C). Positive GSDMD expression was positively correlated with microvascular invasion, poor differentiation, and high tumor nodule-metastasis (TNM) classification (online supplemental table S1). Multivariate analysis suggested that GSDMD expression was an independent and significant risk factor for recurrence and reduced survival (online supplemental table S2). Metastasis and recurrence have proved to be the main reasons for the poor outcome.3 Therefore, we then detected the expression of GSDMD mRNA in metastatic and non-metastatic HCC tissues or recurrent and non-recurrent HCC tissues. The results indicated the upregulation of GSDMD mRNA in metastatic and recurrent HCC tissues compared with matched groups (figure 1D). Consistently, IHC staining in metastatic and paired HCC tissues revealed the same pattern of changes (figure 1E). Furthermore, western blotting exhibited that GSDMD-N expression in metastatic HCC tissues was higher than paired HCC tissues (figure 1F). We then detected the expression of GSDMD and GSDMD-N in HCC cells. The result showed that the expression of GSDMD and GSDMD-N in HCC cells was higher than normal liver tissues and normal liver cell (online supplemental figure S1B). Considering these observations, GSDMD is a promising prognostic biomarker in patients with HCC.
Supplemental material
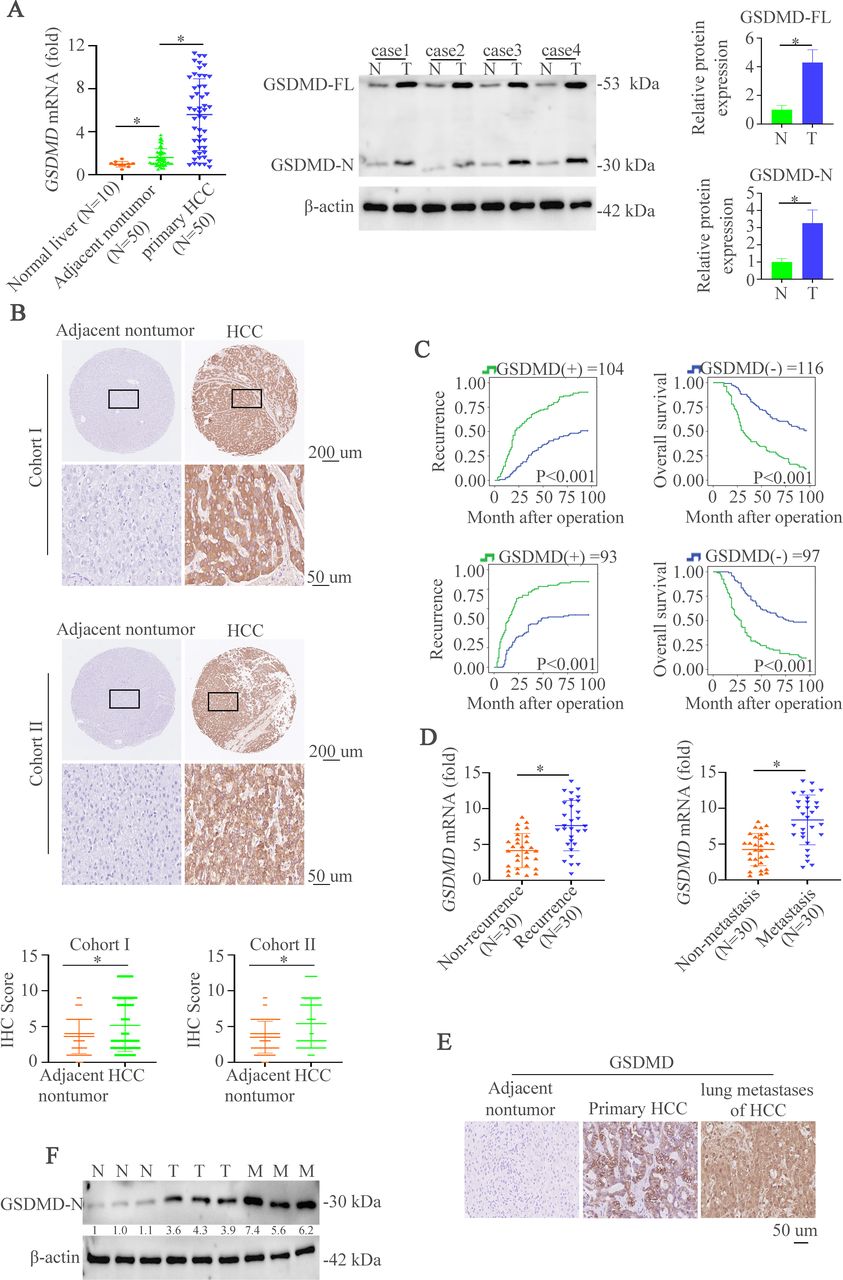
GSDMD and its cleaved GSDMD-N are upregulated in HCC tissues and indicate grim clinical prognosis. (A) RT-qPCR was used to detect GSDMD expression in normal liver tissues (n=10) and 50 paired HCC tissues. Western blotting was applied to detect expression of GSDMD-FL and GSDMD-N expression in paired HCC tissues. (B) Representative IHC images showed GSDMD expression in HCC and adjacent non-tumor tissues in two HCC cohorts. (C) Kaplan-Meier illustrated overall survival time and recurrent rate with positive or negative GSDMD expression in two independent HCC cohorts. (D) RT-qPCR was used to detect GSDMD expression in with or without recurrent patient with HCC samples (n=30) and 30 paired metastatic samples. (E) IHC showed GSDMD expression in adjacent non-tumor, HCC or metastatic tissues. (F) Western blotting exhibited GSDMD-N expression in adjacent non-tumor, HCC or metastatic tissues. GSDMD, gasdermin D; GSDMD-FL, full-length GSDMD; GSDMD-N, gasdermin-N domain; HCC, hepatocellular carcinoma; IHC, immunohistochemistry.
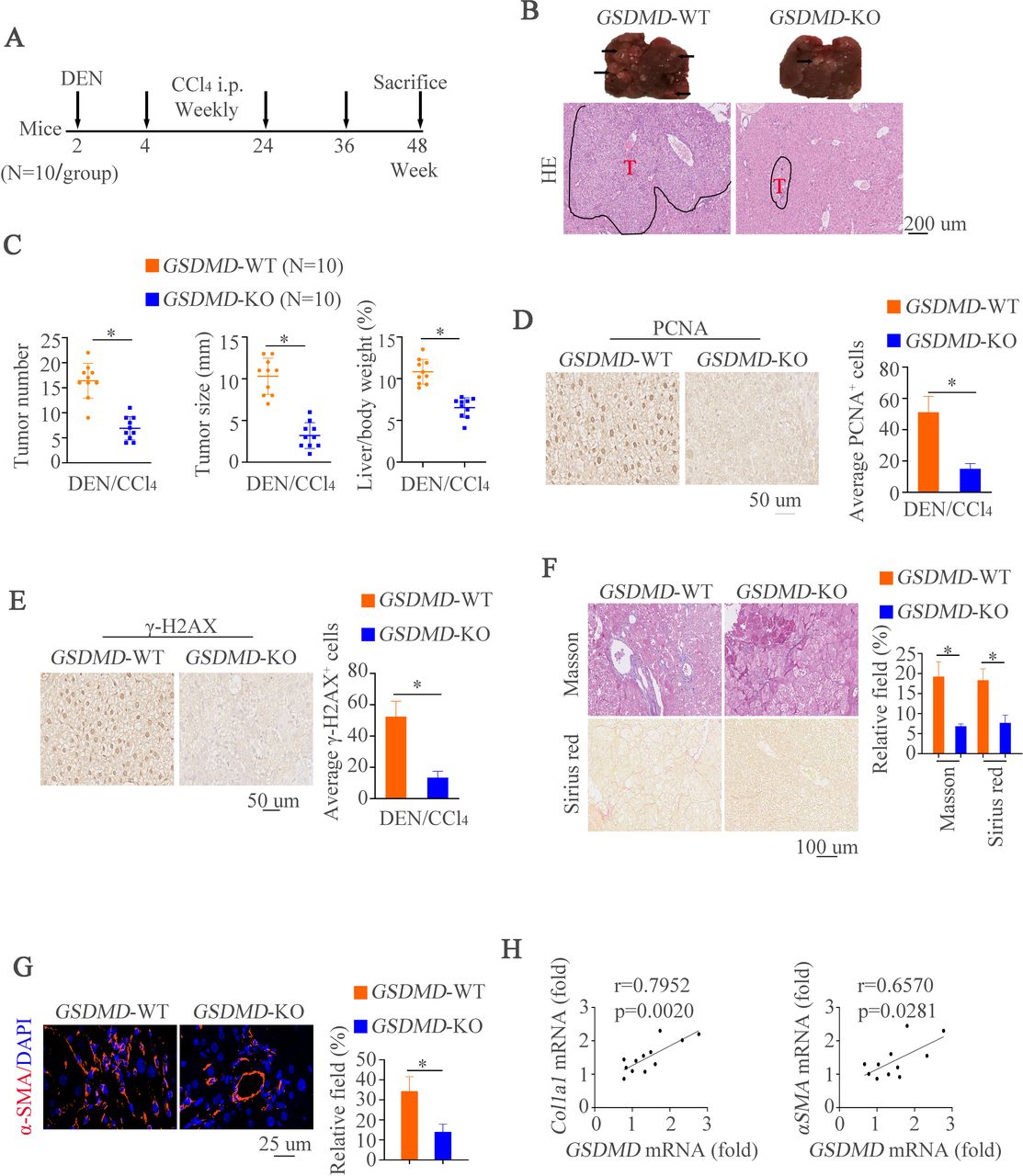
Deletion of GSDMD impairs hepatocarcinogenesis and overexpression of GSDMD-N promotes hepatic tumorigenesis in mice
To further characterize the effects of GSDMD on HCC in vivo, we first established a classical DEN/CCl4-induced mouse model of HCC with GSDMD knockout (GSDMD-/-) by using an established protocol for chemically induced carcinogenesis17 (figure 2A). Compared with WT mice, we had observed decreased histological lesions in GSDMD-/- mice (figure 2B). Remarkably, macroscopic liver analysis revealed that GSDMD-/- mice developed less and smaller tumors than controls at end time point (figure 2C). To evaluate hepatocyte proliferation in pre-neoplastic surrounding liver tissues, we next measured proliferating cell nuclear antigen (PCNA) expression by IHC. The results exhibited that GSDMD deletion could decrease the numbers of PCNA-positive staining in surrounding tissues compared with control groups (figure 2D). It is well documented that DEN could damage DNA,19 20 we then analyzed γH2AX expression by IHC. Decreased number of γH2AX-positive hepatocytes in GSDMD-/- mice was shown compared with WT mice (figure 2E). Since liver fibrosis leads to cirrhosis followed by HCC and CCL4 is a source for inducing liver fibrosis.17 Therefore, histological examinations were used to detect liver fibrosis. Masson and Sirius red staining revealed less collagen fibrils in GSDMD-/- mice (figure 2F). Immunofluorescence (IF) revealed the same pattern of changes by αSMA staining (figure 2G). We next correlated hepatic GSDMD level with pro-fibrotic markers in liver tissue of WT mice after DEN/CCL4 administration. By doing so, observations that GSDMD level correlated with Col1a1 or Acta2 level were shown (figure 2H). These results suggest GSDMD might promote HCC development and progression.
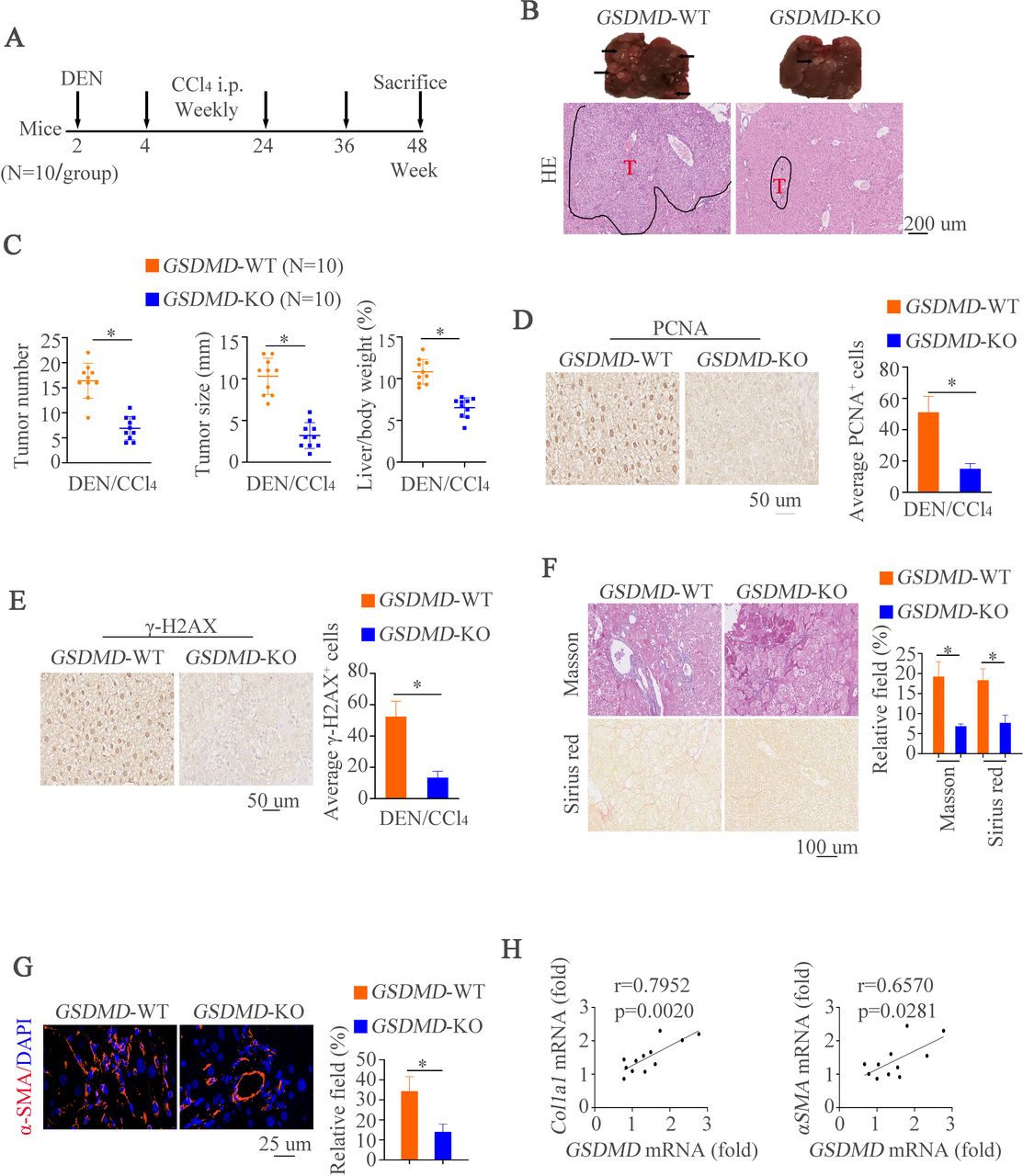
Deletion of GSDMD impairs hepatocarcinogenesis and overexpression of GSDMD-N promotes tumorigenesis in mice. (A) A schematic diagram for procedure of hepatocarcinogenesis induced by DEN and CCL4. (B) Representative liver and H&E images showed liver tumors of both genotypes at indicated time points. (C) Tumor numbers, maximal size and liver weight of tumors from both groups. (D) Representative IHC images illustrated proliferative hepatocytes at indicated time points measured by PCNA positive nucleus staining. (E) Representative IHC images exhibited damaged hepatocytes at indicated time points measured by γH2AX staining. (F) Masson and Sirius red staining were used to show collagenous fiber in different groups. (G) αSMA staining in liver tissues. (H) Correlation of GSDMD mRNA expression with mRNA expression of Clo1a1 and ACTA2 in non-tumor areas of hepatic tissues from WT or GSDMD-/- animals treated with DEN and CCl4. CCl4, carbon tetrachloride; DEN, diethylnitrosamine; GSDMD, gasdermin D; GSDMD-N, gasdermin-N domain; IHC, immunohistochemistry; mRNA, messenger RNA; PCNA, proliferating cell nuclear antigen; WT, wild type.
Our above works have demonstrated the importance of GSDMD-N in HCC. In order to further study the effects of GSDMD-N on HCC, an additional hepatocarcinogenesis model was used. Alb-Cre mice were treated with the vector AAV9-FLEX-GSDMD-N or the control vector AAV9-FLEX-control (online supplemental figure S2A). H&E staining showed more lesions in mice administered with AAV9-FLEX-GSDMD-N (online supplemental figure S2B). Furthermore, increased tumor number and tumor size were observed in animals with AAV9-FLEX-GSDMD-N (online supplemental figure S2C). Similarly, PCNA or γH2AX-positive hepatocytes were largely elevated in mice with AAV9-FLEX-GSDMD-N (online supplemental figure S2D). IF revealed that GSDMD-N upregulation promoted expression of αSMA (online supplemental figure S2E). The positive correlation analysis between GSDMD level and pro-fibrotic markers was exhibited in online supplemental figure S2F. All of these works demonstrate the importance of GSDMD-N in hepatic tumorigenesis.
Supplemental material
GSDMD exerts tumorigenic promotive effects in fibrosis-associated HCC
To strengthen the promotive impacts of GSDMD on HCC in vivo and to further detect its potential effects on fibrosis-associated hepatocarcinogenesis, we next performed another HCC model. TAA was administered to induce fibrosis-associated hepatocarcinogenesis by drinking water,21 starting at week 8 of age, and animals were harvested 40 weeks post-TAA administration (online supplemental figure S3A). GSDMD-deficient animals exhibited smaller tumors and less tumors number post-TAA versus controls (online supplemental figure S3B-C) and akin to the DEN/CCl4 model, this was associated with attenuated PCNA and γH2AX+levels (online supplemental figure S3D). Pathological staining shown by Masson and Sirius red demonstrated the decreased fibrosis level (online supplemental figure S3E). IF showed the decreased expression of αSMA (online supplemental figure S3F). The expression of GSDMD level was associated with colla1 or αSMA level (online supplemental figure S3G). Taken together, these data encompassing different murine models unequivocally suggests that GSDMD and GSDMD-N promotes HCC tumorigenesis.
Supplemental material
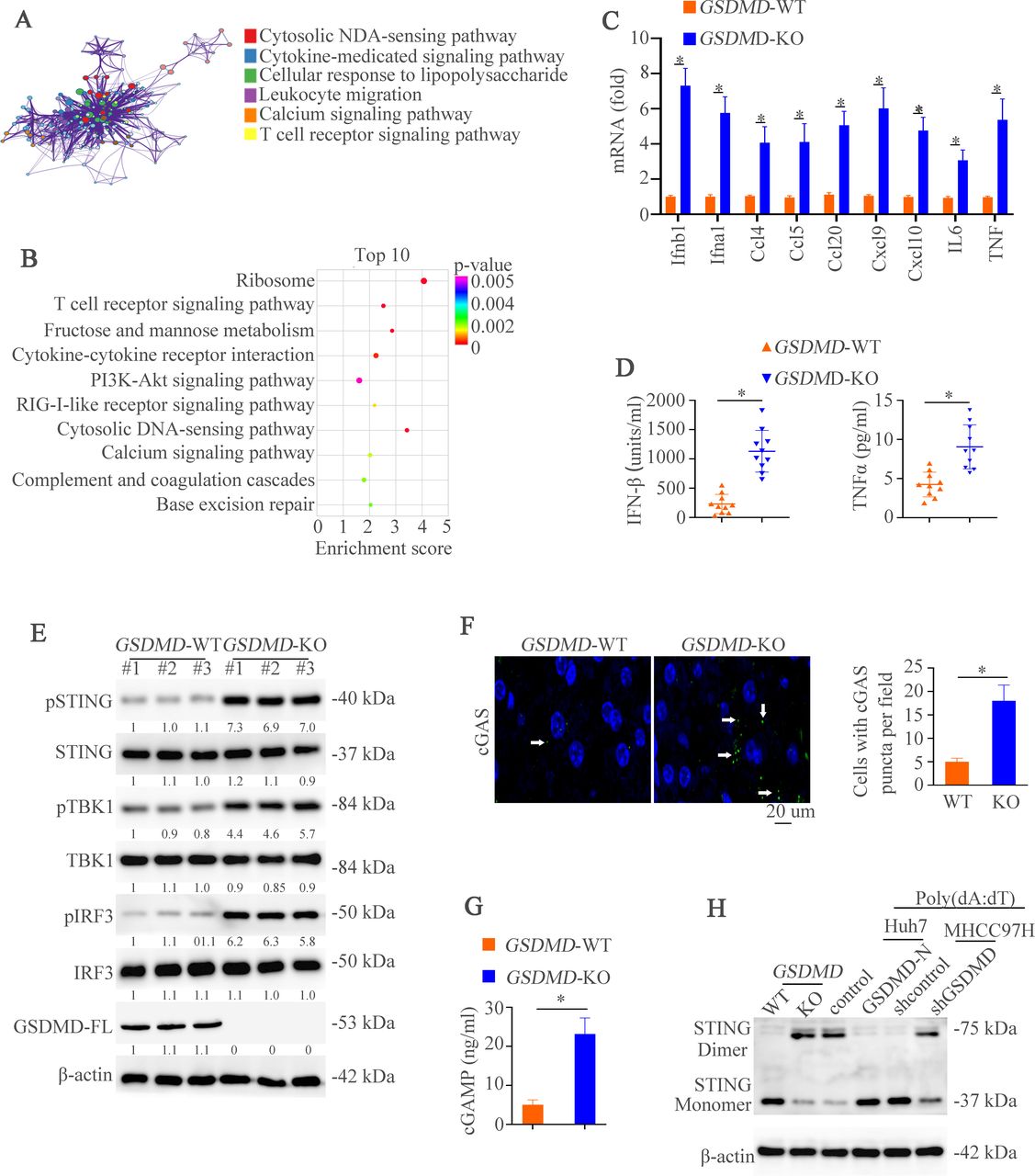
GSDMD deficiency promotes the activation of cGAS pathway in hepatocellular carcinoma. (A) The Metascape analysis stated main changed pathways between WT and GSDMD−/− mice. (B) Top 10 pathways between WT and GSDMD−/− mice were exhibited. (C) RT-qPCR analysis of indicated genes in liver tissues sorted from mice in WT and GSDMD−/− mice. (D) Protein levels of IFN-β and TNF-α was detected by ELISA in different groups. (E) Western blotting was used to analyze phosphorylated (p-)STING, STING, pTBK1, TBK1, pIRF3, IRF3, GSDMD-FL, and β-actin. (F) IF images illustrated punctate staining in different groups. (G) cGAMP amount in WT and GSDMD−/− mice was measured by UPLC/MS analysis. (H) STING monomers and dimers in lysates of WT and GSDMD−/− mice, and Huh7-control, huh7-GSDMD-N, MHCC97H-shcontrol and MHCC97H-shGSDMD stimulated with poly(dA:dT) were assessed by non-reducing polyacrylamide gel electrophoresis and western blotting. cGAS, cyclic GMP-AMP synthase; GSDMD, gasdermin D; GSDMD-FL, full-length GSDMD; IFN, interferon; TNF, tumor necrosis factor; UPLC/MS; ultra performance liquid chromatography/tandem mass spectrometry; WT, wild type.
GSDMD deficiency promotes the activation of cGAS pathway in HCC
To further dissect the mechanisms by which GSDMD induced HCC progression, we performed RNA sequencing (RNA-seq) analysis by using liver tissues of WT and GSDMD−/− mice with DEN/CCl4 treatment (online supplemental figure S4A). The Metascape 22and Kyoto Encyclopedia of Genes and Genomes analysis showed the main changes pathways between WT and GSDMD−/− mice (figure 3A–B). One of the top 10 pathways was cytosolic DNA-sensing pathway which promoted type I IFNs.23 In order to verify whether this pathway was involved in GSDMD-induced HCC, we first detected the downstream genes of cGAS pathway. RT-qPCR analysis verified significantly increased mRNA expression such as IFN-b1 and IFN-a1 associated with cGAS-dependent pathway (figure 3C). The ELISA showed that downregulation of IFN-β and tumor necrosis factor (TNF)-α was found in GSDMD-deficient mice (figure 3D). Next, we used the HCC cells to verify these results. The Huh7 and MHCC97H cells were selected to establish two stable cell lines: Huh7-GSDMD-N upregulating GSDMD-N and MHCC97H-shGSDMD targeting GSDMD (online supplemental figure S1C-E). GSDMD-N upregulation impaired the expression of IFN-β and TNF-α. However, GSDMD downregulation had inverse effects (online supplemental figure S4B).
Supplemental material

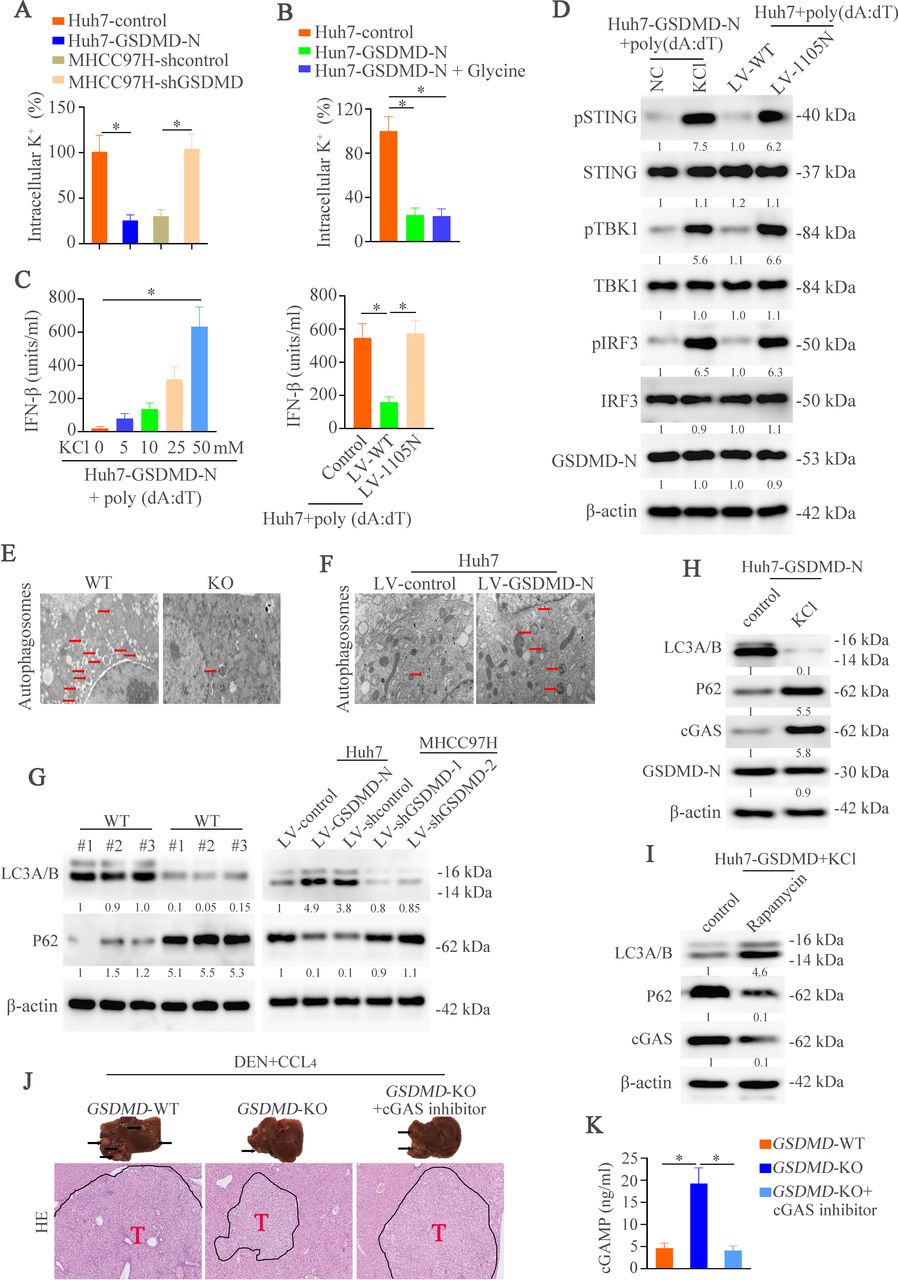
GSDMD promotes output of intracellular K+ which regulates IFN-β expression via autophagy. (A–B) Intracellular K+ was assessed by APG4 staining in indicated groups. (C) IFN-β secretion in indicated cell stimulated with poly(dA:dT) was detected by ELISA. (D) Western blotting analyzed pSTING, STING, pTBK1, TBK1, pIRF3, IRF3, GSDMD-N, and β-actin in Huh7-GSDMD-N with or without KCl and Huh7 transfected with LV-WT or LV- I105N stimulated with poly(dA:dT). (E,F) Transmission electron microscope was shown in different groups. (G) Western blotting exhibiting LC3A/B, P62 and β-actin in different groups. (H) Western blotting exhibiting LC3A/B, P62, cGAS and β-actin in Huh7-GSDMD with or without KCl. (I) Western blotting exhibiting LC3A/B, P62, cGAS and β-actin in Huh7-GSDMD with or without Rapamycin. (J) Representative liver and H&E images illustrated liver tumors of three groups at the indicated time points. (J) cGAMP amounts in indicated group mice as measured by UPLC/MS analysis. CCl4, carbon tetrachloride; cGAS, cyclic GMP-AMP synthase; DEN, diethylnitrosamine; GSDMD, gasdermin D; GSDMD-N, gasdermin-N domain; IFN, interferon; LC3, light chain 3; UPLC/MS, ultra performance liquid chromatography/tandem mass spectrometry; WT, wild type.
After binding dsDNA, cGAS promotes the production of the second messenger cGAMP and then binds to STING, which initiates IFN-β transcription by triggering the phosphorylation of TBK1 and IRF3.14 To explore which step was affected by GSDMD, downstream of cGAS signaling in WT and GSDMD−/− mice was examined. GSDMD knockout induced phosphorylation of STING, TBK1 and IRF3 compared with controls (figure 3E). These results hinted that cGAS activation itself was intended to be affected by GSDMD. After binding DNA, cGAS forms oligomerization which can be visualized by punctate staining.14 24 25 To define this, IF was used to detect cGAS puncta formation. The puncta were higher in the GSDMD knockout groups than controls (figure 3F). Consistent with oligomerization of cGAS, the quantity of cGAMP in GSDMD-KO group was higher than in WT group (figure 3G). Similarly, GSDMD downregulation in MHCC97H cells also exhibited elevated cGAMP level. However, upregulation of GSDMD-N in Huh7 cells lowered the cGAMP level (online supplemental figure S4C). Furthermore, the non-reducing polyacrylamide gel electrophoresis analysis demonstrated that the number of STING dimers was higher in GSDMD-knockout mice or poly(dA:dT)-treated MHCC97H cells with GSDMD knockdown than controls. However, poly(dA:dT)-treated Huh7 cells with GSDMD-N upregulation exhibited lower STING dimer expression (figure 3H). Taken together, these data illuminates that cGAS is a target of GSDMD which suppresses IFN-β production
GSDMD suppresses cGAS activation and type I IFNs production by promoting autophagy via outputting K+ in HCC
We next defined the mechanisms how GSDMD suppressed cGAS function in HCC. We first tried to analyze whether damaged cells could regulate IFN-β production. Glycine could delay the swelling and rupture of cells and has no effects on membrane pores and metabolic activity.7 26–28 Therefore, glycine was used to inhibit cell death. Huh7 cells treated with glycine which exhibited resistance to GSDMD-induced decrease of cell vitality, did not produce higher amounts of IFN-β than controls (online supplemental figure S4D). The interleukin (IL)-1β and IL-18 are released via the membrane pores in a GSDMD-dependent manner. Therefore, it is possible that IL-1β and/or IL-18 may regulate the IFN-β production through an autocrine or paracrine fashion. To test this hypothesis, we compared poly(dA:dT)-induced IFN-β production in Huh7 cells with anti-IL-1β or IL-18 treatment. The results showed no significant change on IFN-β release between these groups (online supplemental figure S4E). These results rule out the roles of cell lysis, IL-1β or IL-18 in GSDMD-regulated IFN-β production.
GSDMD-induced forming of membrane pores permit ionic fluxes which are an early event before cell lysis.7 Recent study demonstrates that ionic perturbations govern cellular functions.29 Taking all of this into consideration, we hypothesized that ionic fluxes may play a role in GSDMD-induced regulation of cGAS-dependent IFN-β responses. Calcium has been reported to flux across the membrane pores.30 We first detected the function of calcium in IFN-β responses triggered by GSDMD. The result demonstrated that calcium chelator, BAPTA-AM, had no effect on IFN-β response to cytosolic DNA (online supplemental figure S4F). K+ is the most abundant ion in intracellular milieu, which may cross the plasma membrane. First, we assessed whether GSDMD triggered K+ efflux by using the specific potassium stain, APG4.31 32 We observed that the intracellular K+ dropped significantly in Huh7-GSDMD-N cells and downregulation of GSDMD inhibited the efflux of K+ (figure 4A). We then detected the K+ efflux under the HCC cells treated with glycine and the result illustrated that K+ efflux was diminished under the presence of glycine (figure 4B). This result might hint that the decrease of intracellular K+ was not due to cell lysis. A study has found that K+ efflux was inhibited by increasing extracellular concentration of K+ via adding KCl.33 We first found that K+ concentration had no effect on the HCC cell viability (online supplemental figure S4G). We then used the TLR2 ligand Pam3CSK434 to stimulate the Huh7-GSDMD cells with or without KCl . The results found that KCl itself might not induce IFN-β after the stimulation of Pam3CSK4 (online supplemental figure S4H). The incubation of poly(dA:dT)-treated Huh7-GSDMD-N cells with increasing concentrations of KCl increased IFN-β secretion (figure 4C left). These works demonstrate that GSDMD attenuates cGAS-induced IFN-β secretion by promoting K+ efflux. An isoleucine-to-asparagine mutation at position 105 (I105N) in the N terminus of GSDMD compromises its capacity to oligomerize, forming pores and ionic fluxes.35 We stably transfected Huh7 cells with WT or I105N mutant GSDMD and found that this mutation largely increased IFN-β secretion (figure 4C right). The western blotting also verified that supplement of KCl and transfection of GSDMD with I105N mutant could effectively inactivate the cGAS pathway (figure 4D). All of these results suggest that K+ efflux is necessary for GSDMD-induced IFN-β secretion.
Researches have reported the connection of cGAS and autophagy,36 37 and efflux of K+ could promote the autophagy.38 39 Furthermore, in the liver tissues, we found the decreased autophagosomes in the GSDMD–/– mice compared with WT mice by transmission electron microscope (TEM) (figure 4E). Therefore, we hypothesized that GSDMD might regulate the cGAS pathway through autophagy. Then, we used HCC cell lines to detect autophagosomes by TEM. We found that overexpression of GSDMD-N increased the number of autophagosomes. However, GSDMD knockdown impaired autophagy (figure 4F). Microtubule-associated protein light chain 3 (LC3) and P62 served as good markers of autophagy process.40 41 The western blotting exhibited decreased LC3 expression and increased expression of P62 and cGAS under the knockout of GSDMD. The similar results were identified in HCC cell lines (figure 4G). These results indicated that GSDMD might regulate the autophagy.
To further evaluate whether GSDMD increased autophagy through releasing K+, we supplemented the potassium chloride in Hun7-GSDMD cells. The results demonstrated that K+ supplement decreased the expression of LC3 and upregulated P62 level. We then detected whether autophagy was involved in cGAS level. In Huh7-GSDMD cells with potassium chloride, autophagy was decreased (figure 4H). However, the autophagy agonist decreased cGAS expression (figure 4I). All of these results illustrated that GSDMD dependent K+ efflux inhibited activation of cGAS pathway through autophagy.
cGAS inhibitor effectively reverses GSDMD knockout-induced inhibition of hepatic tumorigenesis
To evaluate the importance of cGAS in GSDMD-induced HCC in vivo, we then treated the GSDMD-KO mice with cGAS inhibitor RU320521.42 Compared with GSDMD-KO mice, we found larger histological lesions when treated with cGAS inhibitor (figure 4J). Macroscopic liver analysis showed more and larger tumors in GSDMD-KO mice treated with cGAS inhibitor (online supplemental figure S5A). GSDMD knockout significantly improved the liver function. In contrast, cGAS inhibitor deteriorated liver function (online supplemental figure S5B). Similarly, we found that cGAMP level was decreased after the cGAS inhibitor treatment (figure 4K), which was consistent with the protein expression of cGAS downstream (online supplemental figure S5C). Finally, the expression of type I IFNs was decreased with cGAS inhibitor (online supplemental figure S5D). These results illustrate that GSDMD might promote HCC development through inhibiting cGAS pathway.
Supplemental material

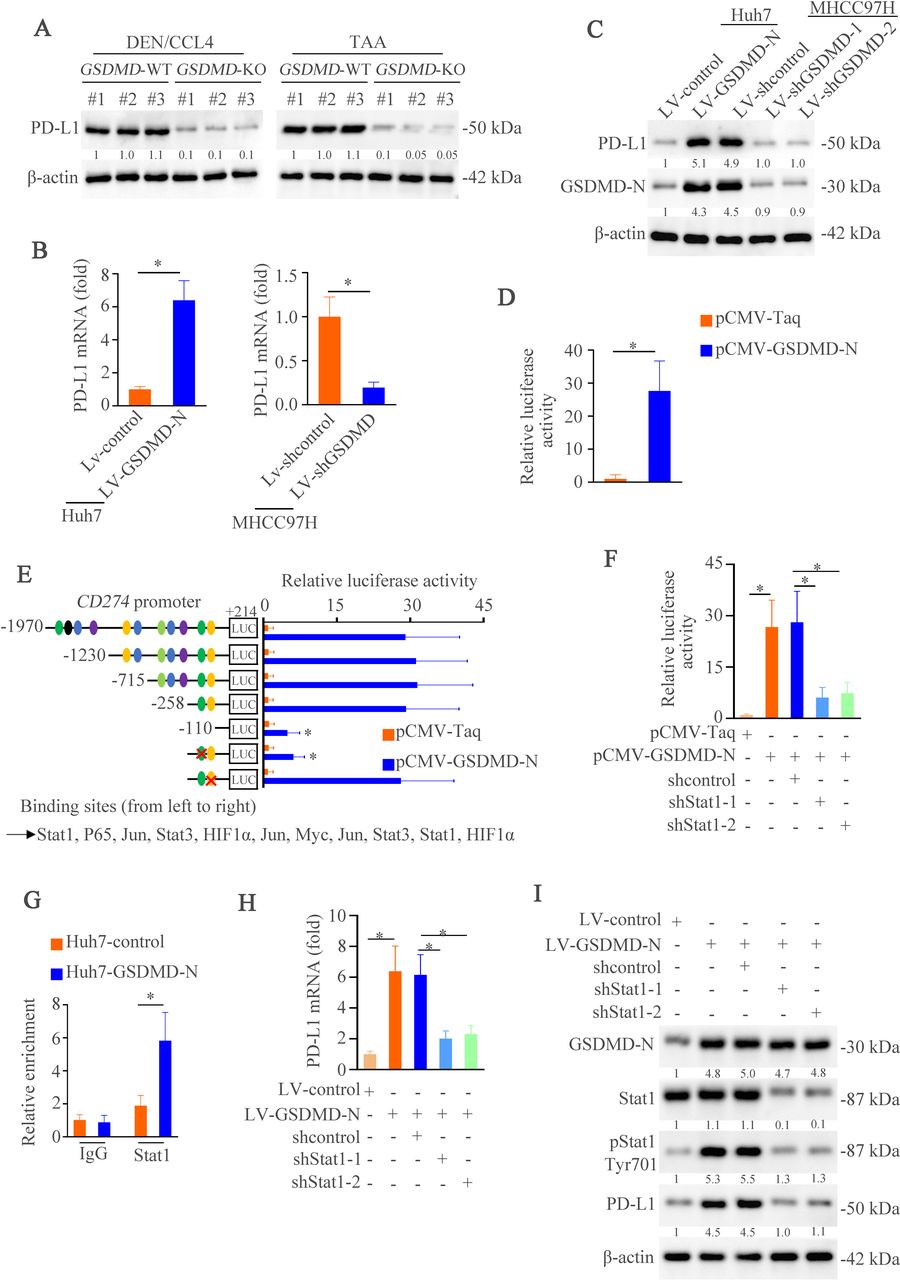
GSDMD upregulates PD-L1 expression through inducing phosphorylation of STAT1. (A) Western blotting analyzed PD-L1 expression in WT and GSDMD–/– group treated with diethylnitrosamine/carbon tetrachloride or thioacetamide. (B) The mRNA expression of PD-L1 in Huh7-control, Huh7-GSDMD-N, MHCC97H-shcontrol and MHCC97H-shGSDMD. (C) The expression of PD-L1 and GSDMD-N in indicated groups was measured by western blotting. (D) CD274 promoter activity was detected after the overexpression of GSDMD-N in Huh7 cells. (E) Truncated and mutated CD274 promoter sequences and pCMV-GSDMD-N were cotransfected into Huh7 cells for testing luciferase activity. (F) CD274 promoter activity was detected after overexpression of GSDMD-N in Huh7 cells transfected with or without shSTAT1. (G) chromatin immunoprecipitation assays showed binding of STAT1 on CD274 promoter in hepatocellular carcinoma cells. (H) RT-qPCR analysis of PD-L1 mRNA after overexpression of GSDMD-N in Huhn7 cells transfected with or without shSTAT1. (I) Western blotting analyzed STAT1, pSTAT1, PD-L1, GSDMD-N and β-actin after overexpression of GSDMD-N in Huh7 cells transfected with or without shSTAT1. CCL4, carbon tetrachloride; DEN, diethylnitrosamine; GSDMD, gasdermin D; GASMD-N, gasdermin-N domain; mRNA, messenger RNA; PD-L1, programmed death ligand-1; TAA, thioacetamide.
GSDMD upregulates PD-L1 expression through Ca2+/ HDACs / STAT1 pathway
Our above works have demonstrated that T cell receptor signaling pathway was regulated by GSDMD (figure 3A–B). PD-1 is one of the most vital receptors comprised in T cell receptor signaling pathway and PD-L1 is a special ligand which counteracts T cell-activating signals and induces durable tumor progression.43 44 Our previous study also demonstrates the importance of PD-L1 in HCC metastasis.13 Therefore, we hypothesized that GSDMD might regulate PD-L1 expression. In both HCC models, we found that GSDMD knockout downregulated PD-L1 expression (figure 5A). Similarly, PD-L1 expression was increased in Huh7-GSDMD-N cells and decreased in MHCC97H-shGSDMD cells compared with control cells in both mRNA and protein levels (figure 5B–C). These data hinted that PD-L1 could be regulated by GSDMD in a transcription-dependent way. In order to verify this premise, we detected CD274 (encoding PD-L1) promoter activity induced by GSDMD-N through dual-luciferase reporter system. GSDMD-N transfection was markedly increased CD274 promoter activity (figure 5D). Bioinformatics analysis identified several putative transcription factors binding sites within CD274 promoter. Through serial deletion mapping and point mutagenesis, we found that STAT1 binding site was essential for GSDMD-N-induced CD274 promoter activation (figure 5E). In order to further verify this result, we detected the CD274 promoter activity and found that knockdown of STAT1 impaired promoter activity induced by GSDMD-N overexpression (figure 5F). A chromatin immunoprecipitation (ChIP) assay further confirmed the direct binding of STAT1 to CD274 promoter (figure 5G). After downregulating STAT1, we observed decreased expression of PD-L1 in both mRNA and protein (figure 5H–I). These results suggest that GSDMD-N promotes the expression of PD-L1 through STAT1.
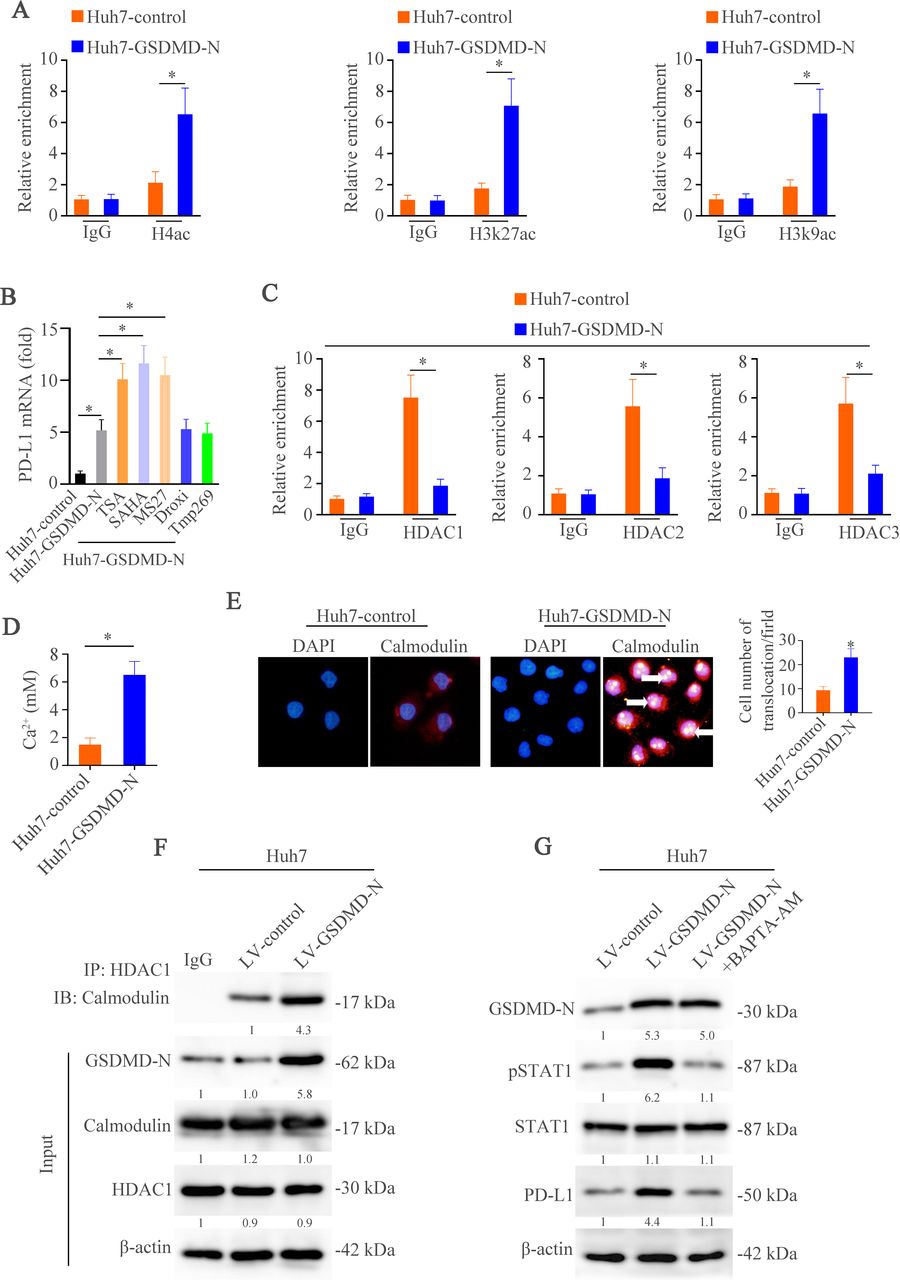
We then determined to define how GSDMD-N promoted STAT1 to access to CD274 promoter. Transcription factors binding to gene promoters are dependent on chromosomal structural changes regulated by histones.45 Therefore, we used the ChIP analysis to assess if epigenetic regulation was occurred in our study. The results showed direct binding of H4ac, H3k27ac and H3k9ac to CD274 promoter under upregulation of GSDMD-N rather than trimethylated (me3) histones: H3K9me3 and H3K27me3 (figure 6A, online supplemental figure 6A). Furthermore, among a panel of inhibitors of HDAC, only Trichostatin A (TSA) and Vorinostat (SAHA) (pan–class I and class II HDAC inhibitors) as well as Entinostat (MS-275) (selective class I HDAC inhibitor) rather than other inhibitors promoted PD-L1 mRNA level (figure 6B). The ChIP assays further confirmed direct binding of HDACs especially HDAC1 to CD274 promoter (figure 6C). These findings demonstrate that GSDMD might epigenetically regulate histones acetylation, which promotes the binding of STAT1to PD-L1 promoter and induces PD-L1 transcription.
Supplemental material

GSDMD upregulates PD-L1 expression through Ca2+/HDACs/STAT1 pathway. (A) ChIP assays were used to illustrate binding of H4ac, H3K27ac and H3K9ac on CD274 promoter in HCC cells. (B) RT-qPCR analysis of PD-L1 mRNA in the Huh7-control, Huh7-GSDMD-N and Huh7-GSDMD-N with inhibitors of HDACs. (C) The binding of HDAC1, HDAC2 and HDAC3 on CD274 promoter in HCC cells was detected by ChIP. (D) The Ca2+ level was measured in Huh7-control and Huh7-GSDMD-N cells. (E) Immunofluorescence was used to detect calmodulin translocation in Huh7-control and Huh7-GSDMD-N cells. (F) Co-IP was used to show interact of HDAC1 and calmodulin in Huh7-control and Huh7-GSDMD-N cells. (G) Western blotting analyzed STAT1, pSTAT1, PD-L1, GSDMD-N and β-actin in indicated HCC cells. ChIP, chromatin immunoprecipitation; GSDMD, gasdermin D; GSDMD-N, gasdermin-N domain; HCC, hepatocellular carcinoma; HDAC, histone deacetylase; mRNA, messenger RNA; PD-L1, programmed death ligand-1.
Next, we determined to find out how GSDMD regulated HDACs. In the figure 3A–B, we found that calcium signaling pathway was regulated by GSDMD and researches have demonstrated that Ca2+ could regulate HDACs pathway through calmodulin.46 Therefore, we first detected the Ca2+ influx. The data showed the increase of Ca2+ influx in the Huh7-GSDMD-N cells compared with the controls (figure 6D). Calmodulin binds Ca2+ with high affinity and directs itself subcellular distribution.47 Therefore, IF was performed to detect the location of calmodulin. The results exhibited that GSDMD-N upregulation promoted massive nuclear translocation of calmodulin (figure 6E). Furthermore, as determined by coimmunoprecipitation, activated calmodulin bound to HDAC1 at CD274 promoter (figure 6F). Calcium chelator, BAPTA-AM, decreased the expression of PD-L1 induced by GSDMD-N/STAT1 pathway (figure 6G, online supplemental figure S6B). These results suggest that GSDMD-N upregulation-induced calcium influx promotes nuclear translocation and activation of calmodulin, which in turn removes HDACs from CD274 promoter and opens chromatin for STAT1 binding and CD274 transcription.
According to above works, we found that two target genes were involved in GSDMD-induced hepatic tumorigenesis. The decreased release of IFN-β induced tumor progression and upregulation of PD-L1 promoted immune evasion of tumors. Therefore, we thought both of them play an important role in hepatic tumorigenesis. In order to verify this hypothesis, we performed subcutaneous xenograft mice model. The mice were treated with phosphate buffered saline, IFN-β or IgG, anti-PD-L1 by intratumor injection. The results showed that IFN-β overexpression and anti-PD-L1 treatment decreased tumor growth (online supplemental figure S6C,D). IHC staining for Ki67 in xenografts exhibited lowered proliferation of tumor cells induced by IFN-β injection and anti-PD-L1 treatment (online supplemental figure S6E). Although the results showed that the anti-PD-L1 treatment lowered the tumor growth more than IFN-β under the basis of measurements, there were no statistically significant values. Therefore, we conclude that both target genes had important roles in GSDMD-induced hepatic tumorigenesis.
GSDMD expression is positively associated with PD-L1 expression and negatively associated with pTBK1 expression in human HCC tissues
We above work demonstrated that GSDMD could regulate the expression of pTBK1 and PD-L1 and then we profiled the expression of pTBK1 and PD-L1 by IHC staining in two independent HCC cohorts. Representative IHC images of GSDMD, pTBK1 and PD-L1 expression were shown in online supplemental figure 7A. IHC score found that GSDMD expression was positively associated with PD-L1 upregulation. However, GSDMD expression was negatively associated with pTBK1 in both HCC cohorts (online supplemental figure 7B). Our previous work has found that positive PD-L1 expression indicated poor prognosis.13 In contrast, HCC tissues with positive pTBK1 displayed a lower aggressive tumor phenotype (online supplemental table S3) and were associated with a better prognosis (online supplemental figure 7C). Furthermore, patients with co-expression of GSDMD/ PD-L1 had the worse prognosis (online supplemental figure 7D). However, patients with positive GSDMD and negative pTBK1 expression had the worse prognosis (online supplemental figure 7E).
Supplemental material
HMGB1 contributes to GSDMD-FL upregulation through TLR4/ERK/P65 pathway and cleaves GSDMD-FL through activating caspase-1
The molecular mechanism of GSDMD-FL upregulation in HCC is still unclear. Several studies have found that caspase-1 could cleave GSDMD-FL into GSDMD-N5 and HMGB1 could promote caspase-1 activation.48 Therefore, we assumed that HMGB1 might induce GSDMD upregulation and cleave c-terminal in HCC. To verify this hypothesis, we first cultured Huh7 cells with recombinant human HMGB1. RT-qPCR and western blotting demonstrated that HMGB1 stimulated expression of GSDMD-FL and GSDMD-N in a dose-dependent manner (figure 7A and online supplemental figure 8A). These results demonstrate that HMGB1 could promote transcription and cleave GSDMD-FL.
Supplemental material

HMGB1 contributes to GSDMD-FL upregulation through TLR4/ERK/P65 pathway and cleaves GSDMD-FL through activating caspase-1. (A) Huh7 cells were treated with recombinant human HMGB1 for 24 hours. GSDMD expression was detected by western blotting. (B) Truncated and mutated GSDMD promoter sequences in Huh7 cells transfected with plasmids of shcontrol or shP65 under HMGB1 treatment. (C) Western blotting analyzed P65, pP65, GSDMD-FL and β-actin in indicated HCC cells. (D) Western blotting analyzed TLR4, P65, pP65, GSDMD-FL and β-actin in indicated HCC cells. (E) ERK, JNK, p38, or PI3K pathway inhibitors were applied in Huh7 cells under HMGB1 treatment. Expression of GSDMD-FL, total and phosphorylated ERK, JNK, p38, and AKT was detected by western blotting. (F) Caspase-1 was detected in Huh7 cells treated with HMGB1 by western blotting. (G) GSDMD-N was detected in Huh7 cells treated with HMGB1 and/or Z-YVAD-FMK. GSDMD, gasdermin D; GSDMD-FL, full-length GSDMD; HCC, hepatocellular carcinoma; HMGB1, High Mobility Group Box 1; TLR4, toll-like receptor 4.
To decipher how HMGB1 stimulated GSDMD-FL expression, we first found that HMGB1 treatment enhanced GSDMD promoter activity (online supplemental figure 8B). We constructed a serial of reporter gene plasmids with truncated and mutated GSDMD promoter fragments. A deletion from −521 to −107 bp significantly decreased HMGB1-induced GSDMD promoter activity, suggesting that this region was crucial for HMGB1-induced GSDMD promoter activation. One presumptive P65 and STAT1, two specificity protein 1 binding sites in this sequence were identified. Mutation of P65 binding site largely impaired HMGB1-mediated GSDMD promoter activation whereas mutation of other binding sites in this sequence had little effect (figure 7B). In order to further verify the importance of P65 in HMGB1-induced GSDMD transcription, promoter activity was measured. P65 knockdown impaired promoter activity and mRNA expression of GSDMD induced by HMGB1(online supplemental figure S8C,D). The ChIP assay verified that binding of P65 and GSDMD promoter was decreased under P65 knockdown (online supplemental figure 8E). The western blotting showed that P65 knockdown decreased HMGB1-induced GSDMD-FL expression (figure 7C). HMGB1 has been proved to activate the signaling pathway through TLR2, TLR4 and Rage.49 Therefore, we then determined to detect which receptor was involved in GSDMD-FL expression. We found that only knockdown of TLR4 but not TLR2 and Rage impaired GSDMD-FL expression treated by HMGB1 (figure 7D and online supplemental figure S6F). Several pathways including ERK, JNK, P38 and PI3K have been activated by HMGB1. Which pathway contributed to HMGB1-induced GSDMD-FL expression was unclear. In order to address this question, we treated Huh7 cells with pathway inhibitors under the treatment of HMGB1, the results showed that block of ERK pathway largely abolished the GSDMD-FL expression. All of these data demonstrated that HMGB1 contributes to GSDMD-FL expression through TLR4/ERK/P65 pathway (figure 7E). Our above works also found that HMGB1 could cleave GSDMD-FL to form GSDMD-N (figure 7A). Therefore, we then tried to find how HMGB1 promoted the cleavage of GSDMD-FL. Researches have illustrated that HMGB1 might activate caspase-1 which could induce formation of GSDMD-N. Accordingly, we hypothesized that HMGB1-caspase-1 pathway contributed to the cleavage of GSDMD-FL. The western blotting showed that HMGB1 activated caspase-1 in a dose-dependent manner (figure 7F). Caspase-1 inhibitor Z-YVAD-FMK impaired GSDMN-N expression (figure 7G). These above results suggest that HMGB1 promotes cleavage of GSDMD-FL through activating caspase-1.
To investigate whether HMGB1/GSDMD axia could promote HCC tumorigenesis in mouse models. We performed subcutaneous xenograft mice model to study the role of HMGB1/GSDMD pathway on tumorigenesis. The results showed that HMGB1 increased tumor growth, whereas GSDMD inhibition suppressed tumor growth (online supplemental figure S8F,G). IHC staining for Ki67 in xenografts showed increased proliferation of tumor cells induced by HMGB1, which was reversed by GSDMD knockdown (online supplemental figure S8H). In the vitro, we found that HMGB1 could promote GSDMD expression and impaired the expression of cGAS. In order to confirm these results in clinical specimens, we used IHC staining for detecting HMGB1, cleaved caspase-1, GSDMD, pSTAT1, IFN-β and CD8 expression. Representative IHC images were shown in online supplemental figure S8I. The results found that the expression of HMGB1, cleaved caspase-1 and pSTAT1 was positively associated with GSDMD upregulation, and IFN-β and CD8 expression were negatively associated with GSDMD upregulation. These results illustrated the importance of HMGB1/GSDMD in hepatic tumorigenesis.
Combined treatment of GSDMD inhibitor DMF and anti-PD-1 abolishes hepatic tumorigenesis and HCC metastasis
According to the above works, we hypothesized that block of GSDMD could promote IFN-β production and impair PD-L1 expression, which induced HCC cells death and improved the function and amount of CD8+ T cells. DMF is proved to react with GSDMD and prevent GSDMD interaction with caspases, limiting its processing, oligomerization, and capacity to induce cell death.18 The downregulation of PD-L1 might improve anti-PD-1 therapy.43 Therefore, we assumed that combination of GSDMD inhibitor DMF and anti-PD-1 could block HCC tumorigenesis and metastasis through improving IFN-β production and immunosuppressive microenvironment. The sketch of design was shown in online supplemental figure S9A. Bioluminescent images found effectiveness of combination treatment in blocking hepatic tumor progression (figure 8A, online supplemental figure S9B). No significant changes in average mice body weight or toxicity in liver and kidney were observed (online supplemental figure S9C). H&E staining also verified that combined therapeutic pattern strikingly decreased HCC number and size (figure 8B,C). Decreased lung metastasis burden also validated the efficiency of combined therapy (figure 8D). The combination treatment also prolonged survival time of mice (online supplemental figure S9D). To characterize the mechanism under this combination, we examined infiltration of CD8+ T cells in HCC tumors. The combination treatment dramatically increased CD8+ T cells infiltration compared with control group or single agent alone shown by flow cytometer (figure 8E). The results illustrated that combination treatment promoted infiltration and activation of CD8+ T cells, and decreased the number of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) (figure 8F, online supplemental figure S9E-G). These results suggest that double blockade of GSDMD and PD-1 improves antitumor immune response and inhibits HCC tumorigenesis and metastasis.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combined GSDMD inhibitor DMF and PD-1 treatment abolishes hepatic tumorigenesis and HCC metastasis. (A) Representative bioluminescent images in mice liver. (B) Representative H&E images showed liver tumors of different groups at indicated time points. (C) Tumor numbers, maximal size and liver weight of tumors from both groups. (D) H&E staining of metastatic lung nodules was shown in different groups at indicated time points. (E) flow cytometer for CD8+ T cells in HCC tissues of HCC model after indicated treatment was displayed. (F) Immunofluorescence showed infiltration and activation of CD8+ T cells in different groups. (G) A schematic illustration elaborated the role of GSDMD on HCC metastasis. HMGB1 upregulated GSDMD expression through the ERK/P65 pathway. GSDMD overexpression inactivated cGAS pathway and induced programmed death ligand-1 expression through K+ influx and Ca2+ efflux respectively. The combined treatment of anti-PD-1 and GSDMD inhibitor DMF suppressed GSDMD-induced HCC metastasis. cGAS, cyclic GMP-AMP synthase; DMF, dimethyl fumarate; GSDMD, gasdermin D; GSDMD-N, gasdermin-N domain; HCC, hepatocellular carcinoma; HMGB1, High Mobility Group Box 1; PD-1, programmed cell death protein-1.
Discussion
HCC is most common in liver cancer and accounts for one of the most cancer-associated deaths. Though understanding of pathophysiology and oncogenes of this disease has improved, this knowledge is yet to be translated into clinical practice. Dominant mutational drivers in HCC, such as TERT, TP53 and CTNNB1, remain undruggable.3 Therefore, finding druggable targets that contributes to HCC progression is urgent.
Pyroptosis is a caspase-1 dependent form of cell death, which is morphologically and mechanistically distinct from other forms of cell death50 and participates in host defense and human diseases including HCC.51 GSDMD is discovered to have pore-forming activity and is well studied as effector for pyroptosis.5 However, the roles of GSDMD itself in HCC remain unclear. Considering the importance of inflammation in HCC, we hypothesized that GSDMD might induce HCC progression. In this study, we found that GSDMD-FL and GSDMD-N were elevated in HCC especially in metastatic HCC tissues. Positive GSDMD-FL indicated poor outcome and might be an independent biomarker.
cGAS pathway detects intracellular double strand DNA and triggers an innate immune reaction to promote product of type I IFN response which functions in infection, autoimmunity, sterile inflammatory responses and cellular senescence.52 In this study, we found that GSDMD formed membrane pore and induced efflux of K+ which blocked cGAS activation and induced IFN-β production. Autophagy in this study was found to connect K+ and cGAS activation. Nowadays, works reveal that cGAS presents in nucleus and plasma membrane, and such subcellular compartmentalization, which is linked to regulate DNA repair, to activate nuclear factor kappa B subunit 1 (NF-κB) and mitogen activated kinase-like protein (MAPK) pathway and to induce autophagy and lysosome-dependent cell death.52 Further works should focus on these new roles in human diseases.
PD-L1 is ligand of PD-1 and is a major co-inhibitory checkpoint signaling that controls T cells activity. Blocking PD-L1/PD-1 pathway has consistently shown remarkable antitumor effects in patients with advanced cancers. However, this response rates remain low in HCC.53 Therefore, exploring regulatory mechanisms that PD-L1 could bring substantial benefits to patients who receive immune treatment is important. In this study, we found importance of GSDMD in regulating PD-L1 expression. Mechanically, we showed that GSDMD promoted Ca2+ influx and induced binding of pSTAT1 in PD-L1 promoter. Meanwhile, Ca2+ influx induced interacting of calmodulin and HDACs, which promoted pSTAT1 to access into PD-L1 promoter. These works demonstrated the role of GSDMD in regulating PD-L1 expression. Clinically, we found that GSDMD expression was positively associated with PD-L1 expression and negatively associated with pTBK1 expression in human HCC tissues.
In order to find upstream that induced GSDMD-FL expression and cleavage, we focused on HMGB1 which induces HCC progression.54 55 HMGB1 is reported to activate caspase-148 that can induce cleavage of GSDMD-FL. Therefore, we hypothesized HMGB1 might be an upstream of GSDMD upregulation in HCC. Actually, we found that HMGB1 contributed to GSDMD-FL expression through ERK/P65 pathway and cleaved GSDMD-FL into GSDMD-N through caspase-1. Finally, we determined to find therapeutic approaches targeting GSDMD-induced HCC progression. The combined GSDMD-N inhibitor and anti-PD-1 effectively blocked GSDMD-induced HCC progression and metastasis through changing microenvironment and promoting HCC cell death. In conclusion, we demonstrated that HMGB1-induced expression and cleavage of GSDMD promoted HCC tumorigenesis through blocking cGAS pathway and upregulating PD-L1 expression. Treatment of GSDMD inhibitor and anti-PD-1 is a promising approach in abolishing hepatocarcinogenesis and HCC metastasis.
In conclusion, in this study, we identified oncogenic role of GSDMD in HCC. The regulation of GSDMD indicated poor prognosis and GSDMD promoted hepatic tumorigenesis. Mechanically, GSDMD upregulation activated cGAS pathway through K+ efflux and induced PD-L1 expression through Ca2+/HDACs/STAT1 pathway. Clinically, GSDMD expression was positively associated with PD-L1 expression and negatively associated with pTBK1 expression in human HCC tissues. Furthermore, HMGB1/TLR4/ERK/P65 pathway contributed to GSDMD-FL upregulation and cleaved GSDMD-FL through activating caspase-1. Finally, we found that combined treatment of DMF and anti-PD-1 abolished hepatic tumorigenesis and HCC metastasis.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involving human studies using liver tissue samples was approved by Shandong Provincial Hospital Affiliated to Shandong First Medical University 2021-571. Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TL performed the experiments. XX provided assistance in immunohistochemistry, HE staining, plasmid construction, and animal experiments. WY, ML and HX provided assistance in collecting tissue samples, clinical statistical analysis, and analyzing data. QH designed the study and wrote the paper.
Funding This work was supported by Clinical Science and Technology Innovation Program of Jinan (202134039), National Natural Science Foundation of China (81472311) and Incubation Fund of Shandong Provincial Hospital (2021FY014). We acknowledge the patients who agreed to participate in this clinical trial, as forward trajectory of science hinges on their support. And we also thank the Metascape for help.55
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.