Article Text

Abstract
With the advent of immunotherapy as one of the keystones of the treatment of our patients with cancer, a number of atypical patterns of response to these agents has been identified. These include pseudoprogression, where the tumor initially shows objective growth before decreasing in size, and hyperprogression, hypothesized to be a drug-induced acceleration of the tumor burden. Despite it being >10 years since the first immune-oncology drug was approved, neither the biology behind these paradoxical responses has been well understood, nor their incidence, identification criteria, predictive biomarkers, or clinical impact have been fully described. Immune-based Response Evaluation Criteria in Solid Tumors (iRECIST) guidelines have been published as a revision to the RECIST V.1.1 criteria for use in trials of immunotherapeutics, and the iRECIST subcommittee (of the RECIST Working Group) is working on elucidating these aspects, with data sharing a current major challenge to move forward with this unmet need in immuno-oncology.
- immunotherapy
- review
- therapies, investigational
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
It is more than a decade now since the first immune-oncology drug (IO) was approved by the Federal Drug Administration (FDA), and, since then, this class of agents has become the standard of care in many cancers. During the development of ipilimumab, investigators reported unusual response patterns with patients who appeared to have initial disease progression going on later to have deep and durable tumor shrinkage.1 Based on those data, response criteria were developed to capture pseudoprogressive disease to IO drugs,2 which were initially based on WHO criteria and used bidimensional measurements. Later, variations assessments were based on unidimensional measurements, but continued to add new lesions into the sum of measures from baseline.3 4
Concerns were also raised about a second potentially unique response pattern termed hyperprogression disease (HPD), where patients developed rapidly progressive disease shortly after initiating treatment with IO drugs.5
Response Evaluation Criteria in Solid Tumors (RECIST) were developed and implemented >20 years ago,6 7 and have become the standard criteria for response-based end points. The RECIST Working Group (RWG) strategy for developing and validating response criteria is to create a data warehouse of individual patient data. A major initiative was undertaken for targeted anticancer agents, as modified response criteria had also been proposed in the late 1990s for these agents. However, the RWG was able to demonstrate that RECIST V.1.1 performed satisfactorily here as well, and no revisions were needed.7 8
A modified RECIST V.1.1 for immune-based therapeutics (termed iRECIST) was developed in 2016, and published in 20179 by an international multidisciplinary group, created by the RWG when attempts to generate a data warehouse to formally test response criteria for IO drug trials were stymied by the protocol-specific nature of response criteria in use at the time. The iRECIST recommendations concern the collection and management of data after RECIST V.1.1 defined progression. iRECIST defines when treatment past progression (TPP) is reasonable or justified and limits the duration of TPP in the face of continued progression. iRECIST collects data on new lesions separately in a manner consistent with RECIST V.1.1 itself, rather than adding new lesions to the sum of measurements at baseline, to ensure that data are fully analyzable.
Despite the enormous success of immunotherapies, in 2022, there remains no clear consensus on many areas pertaining to response, including those as whether modified response criteria are required, what those criteria should be, how HPD is defined, nor whether response patterns are unique to select classes of immune active agents. Efforts to overcome these challenges have been hindered by many issues predominantly related to inconsistency in designs, data collection and analyses, and the resulting inability to easily pool data and test and then validate criteria. Overall, a major challenge remains in encouraging the sharing of data from completed trials; while academic groups remain committed to these efforts, the collaboration of commercial entities is critical. The RWG continues to create warehouses as well as evaluating radiomics and genomic data to continue to test, optimize, and validate any required changes to standard RECIST. In addition to the iRECIST initiative, the RWG is actively engaged in collecting data and testing and validating definitions of HPD.
The objective of this article is to review the current definitions, current research, and associated challenges to determine the incidence and impact of both pseudoprogression and HPD, and the need for specific immune response criteria to capture them. However, given the iRECIST guideline was published approximately 5 years ago, we also evaluated the uptake of the iRECIST guideline and other immune criteria in clinical trials of immunotherapy agents, to understand the frequency that trial sponsors are using the available tools.
Pseudoprogression and immune-modified response criteria
Since the initial reports of unusual response patterns in patients treated with IO drugs, including pseudoprogression, researchers have sought to understand its true prevalence and to understand whether immune-modified response criteria are required for clinical trials with response-based end points. Of course, understanding how common pseudoprogression is would be pertinent even for clinical trials with survival as an end point, if protocols mandate the agent being tested to be discontinued at the first sign of RECIST V.1.1 progression, presuming continued treatment with the immune checkpoint inhibitor might affect outcomes. The two most used criteria are immune related RECIST (irRECIST) and immune RECIST (iRECIST).
Published reports to date suggest a wide variation in the reported rates of pseudoprogression, ranging from 1% to 16%, although in most trials they appear to be low. Analyses conducted by the FDA applying iRECIST suggest a low impact on response rates.10 Similar pooled analyses of trials which used irRECIST also confirm some impact on response rates but did not impact to a significant degree the interpretation of progression-free survival (PFS)11 or overall survival (OS).12 Given the potential importance of pseudoprogression on outcome, we sought to investigate the frequency of uptake of iRECIST by clinical trial sponsors in the years after the guideline was published.
Assessment of the uptake of iRECIST
We undertook a study to determine the uptake of iRECIST in clinical trial protocols. Our primary objective was to evaluate the proportion of trials testing immune-based therapies in solid tumors that included iRECIST defined response registered on clinicaltrials.gov between March 2017 and October 2019. While we collected data on other immune criteria that were used, we focused on the iRECIST guideline given the aim of iRECIST was to attempt to standardize immune criteria, and the objective of the iRWG is to understand if the iRECIST criteria are required, or if RECIST V.1.1 is sufficient. We also planned to evaluate whether iRECIST was used as a secondary end point (as per iRECIST recommendations), to assess factors associated with the use of iRECIST criteria in response reporting including the cancer type, phase of trial, and sponsor of trial; and assess the use of other immune-related response criteria.
Methods
Trials of immunotherapy agents in patients with solid tumors that included a response-based end point/s (including objective response rate and PFS), were eligible if they were registered on clinicaltrials.gov between March 2017 and October 2019. Search terms included pembrolizumab, nivolumab, atezolizumab, durvalumab, avelumab, ipilimumab, tremelimumab. Programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1), cytotoxic T-lymphocyte antigen 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin and mucin domain 3 (TIM-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain immunoglobulin suppressor of T cell activation (VISTA), B7-H3, A2aR, CD73, OX40, glucocorticoid-induced tumour necrosis factor receptor-related protein (GITR), inducible T cell costimulator (ICOS), 4-1BB, CD27, CD70, CD40, killer Ig-like receptors (KIR), CD47, and arginase inhibitors. Trials which were suspended, withdrawn, terminated, or had an unknown status were excluded. Studies examining hematological malignancies were excluded since standardized criteria other than RECIST, such as the international working group response criteria in lymphoma, are used to evaluate response. All autologous T cell therapy including chimeric antigen receptor-T cells, tumor-infiltrating lymphocytes, intratumoral T cells, dendritic cells, natural killer cells were excluded. Studies involving the use of cancer vaccines alone were also excluded, unless in combination with pembrolizumab, nivolumab, atezolizumab, durvalumab, avelumab, ipilimumab, or tremelimumab given the lack of data on atypical response patterns, as were studies involving the use of cytokines alone.
The use of iRECIST response as a primary or secondary end point in assessing response was recorded to determine how the guideline is being applied. In addition, information on the specific drugs, cancer type, phase of trial, and the sponsor was collected (table 1) to explore for any associations with the uptake of iRECIST criteria. As the registry (clinicaltrials.gov) may not include information of the response tool included in the protocol, a random subset of academic non-cooperative group study chairs for trials that did not include iRECIST were selected and study chairs or trial sponsors were contacted to confirm if iRECIST was used in the trial (yes/no), and if yes, was iRECIST used as a secondary or exploratory end point. If no, information on alternative immune response criterion was sought. We also contacted several academic cooperative groups, but not all responded so data are not included. The proportions and characteristics of studies employing iRECIST were summarized using descriptive statistics. Prespecified variables influencing the use of iRECIST including the cancer type, phase of trial, and type of sponsor were tested using a multivariate logistic regression.
Summary of trials including iRECIST as tool to evaluate immune response
Results
Of 2622 trials that were initially identified for review, 1080 trials were excluded as they did not meet the prespecified eligibility criteria (273 cancer type; 356 non-cancer or non-immunotherapy trials; 386 did not include response evaluation; 65 other reasons).
Seventy-four per cent (n=1144 of 1542) trials reported using RECIST V.1.1. In comparison, immune response criteria were specified in 12% (n=179), of which 76% used iRECIST and 24% used other immune criteria. The characteristics by sponsor, phase of trial, design of trial are summarized in table 1. When analyzed by year, the uptake of iRECIST has increased (figure 1) from 2% in 2017 to 9%–13% between 2018 and 2019, with a corresponding decrease in the use of other immune response criteria.

Uptake of immune-based Response Evaluation Criteria in Solid Tumors (iRECIST) from March 2017 to December 2019*. *Data extracted from clinicaltrials.gov. No incorporation included if no immune response criteria was listed in clinicaltrials.gov.
We contacted 10% of non-cooperative group study chairs of immunotherapy trials that did not report the use of iRECIST (or other immune criteria) either in clinicaltrials.gov or trial publications in order to determine if iRECIST had actually been included in the trial protocol, and if so as a primary, secondary, or exploratory end point. Of 81 chairs contacted, 22 responded with 5 (23%) responding that iRECIST was actually included as a response end point. Therefore, it is likely that the implementation of iRECIST is higher than we have estimated. We also observed an increasing uptake up until 2019.
The proportion of clinical trials using iRECIST rather than other immune-related criteria has increased, indicating more standardization across studies over time, which was one of the objectives of the iRECIST guideline. Furthermore, iRECIST was incorporated in both academic and industry sponsored trials, and across phase I, II, and III trial designs.
iRECIST and pseudoprogression conclusions
Our study demonstrated a minority of clinical trials incorporated immune response criteria up until 2019, however, of the trials that did, the use of iRECIST has increased and it may be that uptake has continued to increase since 2019. Given the potential impact of pseudoprogression, but equally the need to avoid patients being exposed to an ineffective drug, understanding the benefit of the iRECIST guideline compared with RECIST V.1.1 is important.
No immune-modified response criteria, including iRECIST, have yet been validated and the iRWG continues to collect data in order to test whether modifications of RECIST V.1.1 are in fact required. However, a recent pooled analysis of trials submitted to the FDA showed that response rates to PD-1/PD-L1 inhibitors, whether measured by RECIST vV.1.1 or iRECIST, were very similar but remained useful in subgroups with atypical response patterns.10 Certainly, the additional scans required in iRECIST may be a barrier due to increases in cost as well as radiation exposure for patients, and other initiatives being investigated such as radiomic and tumor burden evaluations, do also increase cost and complexity. RECIST was developed as simple, easy to implement set of response criteria across large international sites allowing consistent interpretation of results and to date has performed well even for new classes of anticancer therapies such as targeted agents; whether modifications are needed for IO drugs remains unclear.
However, major challenges exist with fully evaluating the need for revisions to the RECIST V.1.1 criteria for use in trials of immunotherapeutics; as the iRECIST guidelines were only published in 2017, most larger pivotal trials are only maturing now, have not been published, and thus are not suitable to be shared in order to formally test whether immune-modified response criteria are indeed required, or are suitable for early clinical trials where response-based end points are critical for the ‘go-no go’ decision, given the increased complexity and costs. Furthermore, data acquisition remains a challenge as described later.
Hyperprogression: unusual response and adverse effect?
HPD was first reported in the literature as the unexpected acceleration of tumor growth after the initiation of treatment with IO drugs, with a consequent negative clinical impact for these patients.13 Due in part to the lack of a standard definition and measurement criteria of HPD, the retrospective nature of most published series, the lack of prebaseline or early on IO treatment imaging that excluded many patients from analysis in those studies, and the diverse kinds of IO treatments and tumor types involved (with the most sensitive ones, such as melanoma, being less likely to be associated with HPD), a high variability in the rate of HPD has been described in these studies ranging from 6% to 43%, with a pooled incidence of 13.4%.14
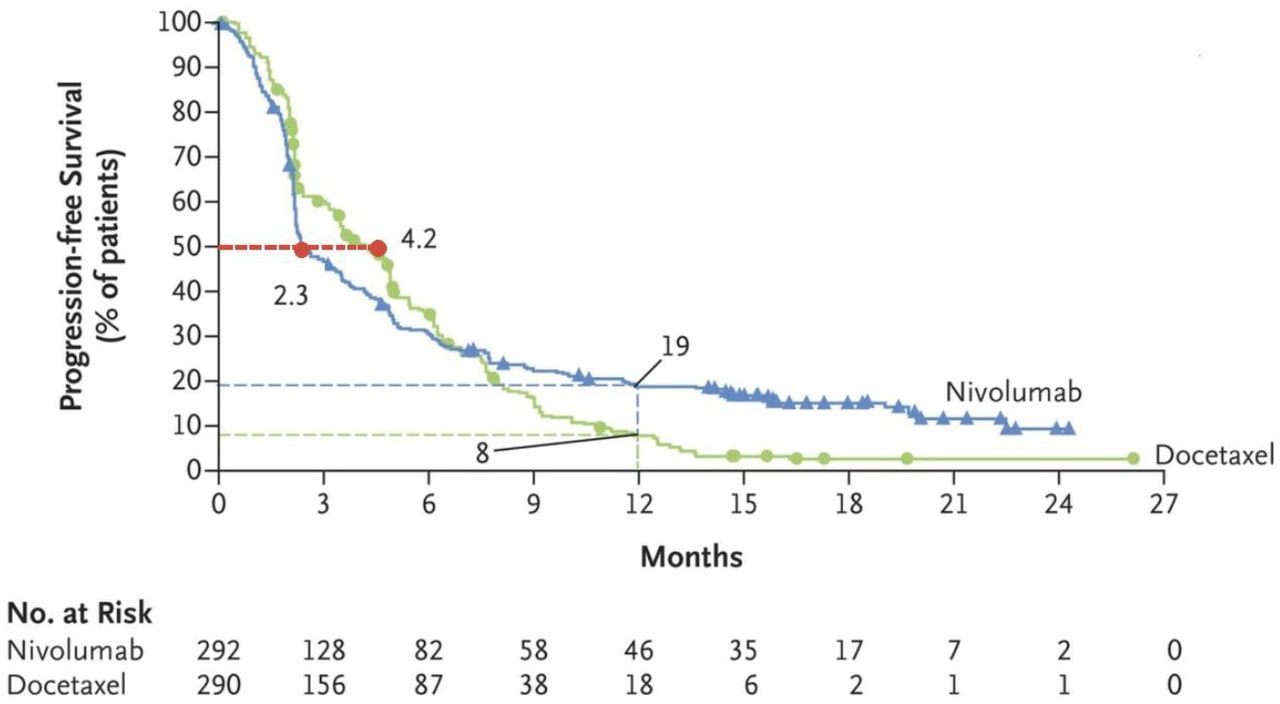
Some investigators initially suggested these observations might be explained merely by overly aggressive tumor biology, independent of IO treatment. However, the cumulative data regarding HPD from different series, systematic reviews, and a meta-analysis over the past few years, as well as the intriguing crossing of Kaplan-Meier actuarial survival curves in randomized studies of the IO investigational arm and the chemotherapy control arm in the early treatment period, such as Checkmate-057 (figure 2),15 together do favor a causal effect of IO drugs on the dramatic acceleration of tumor growth dynamics during IO therapy.16

{kind=link}
{kind=link}
Actuarial survival curves for nivolumab versus docetaxel in non-small cell lung cancer (Checkmate-057). Progression-free survival data for the nivolumab group showed initially worse outcome possibly related to early tumor growth and potentially hyperprogression disease. Adapted from: Borghaei et al.15
Notably, although previously reported anecdotally in non-IO trials, the incidence appears far higher with IO drugs. In a multicenter, retrospective study comparing the incidence of HPD in patients with lung cancer receiving PD-1/PD-L1 inhibitors and in those treated with single-agent chemotherapy, HPD occurred in 14% of patients treated with immunotherapy (n=406), which was almost triple the incidence than patients receiving chemotherapy that met the HPD criteria (n=59, 5%).17 Critically, even though many clinical trials of IO drugs have demonstrated an OS benefit, these data do suggest that a subset of patients may actually be harmed by IO drugs, and the identification of predictive factors is essential so that these patients can be treated with other effective therapies. Furthermore, if HPD occurs typically early in treatment—often in the first 1–2 cycles—it may not be sufficient to merely stop the IO drugs, and identification of patients most likely to experience HPD is important.
Establishing a standardized criterion to identify HPD is a high unmet need in immunotherapy because, if prospectively confirmed in large-scale studies, this phenomenon would imply the occurrence of a paradoxical aggravation of cancer growth induced by immune drugs in a meaningful percentage of our patients. The lack of a proper definition for HPD, and the major conceptual and practical differences between the various measurement criteria, make it extremely challenging to determine its precise prevalence and impact.14
There is consensus that the evaluation of prebaseline CT scans is important in order that a differential tumor growth dynamic pre-immunotherapy and on-immunotherapy can be demonstrated (the hallmark of HPD), although patients who present ab initio with advanced disease may not have prior scans, and it is not considered appropriate to delay the initiation of standard therapies merely to document tumor growth dynamics.
Changes in tumor growth dynamics can be captured based on two similar, although different, parameters (table 2). On one hand, the analysis of ‘tumor growth rate’ (TGR) combines the sums of target lesions and the time between tumor evaluations, allowing for a dynamic and quantitative evaluation of tumor three-dimensional volume changes18; this assessment had been initially used to define HPD related to IO treatment, by exploring the difference in TGRs before and on IO drugs. An alternate method, which also compares tumor growth dynamics before and during IO administration, also uses TGR but simplified as the measurements of the ‘tumor growth kinetics’ (TGK) based only on changes in the bidimensional, RECIST-based, sum of the longest diameters of the target lesions over time.19 A final method that has been proposed uses tumor dynamics on treatment only, regardless of pretreatment tumor growth changes; these delineate aggressive tumor growth but cannot differentiate between treatment-induced HPD versus treatment-independent aggressively growing disease.20 Given the difficulty with accessing prebaseline imaging for patients presenting with advanced disease or being referred to tertiary care centers, the latter method may still be of value to define HPD, especially in the context of randomized clinical trials with a non-IO drug comparator.
Selected hyperprogression disease definitions
Although it has become clear that immune checkpoint inhibitors (ICI) drugs are associated with HPD, after more than a decade of usage in standard clinical practice, there are still no standard radiological criteria for the evaluation of HPD (table 2). This is not merely of academic interest for early clinical trialists, but an urgent issue for patients, oncologists and regulatory authorities, the clarification of which has been stymied by rigid data sharing policies.
Several smaller, usually single center studies have attempted to address this key topic. A retrospective single site study comparing four selected HPD criteria in their own series of consecutive patients on IO treatments showed the use of the Le Tourneau method to assess HPD to be the preferred one, as it captures adequately this atypical progression and TGK is more convenient to use than TGR.21 HPD resulted in the clinical deterioration of patients as reflected in shorter PFS and lower likelihoods of receiving additional, salvage therapies, compared with other patients who had disease progression as their best response but no HPD. Worse clinical outcome for patients with HPD compared with patients with natural progressive disease have also been demonstrated in other two studies: one assessing 5 different definitions of HPD in a multicenter cohort series of 405 consecutive patients with lung cancer,22 and the other one being a systematic review and meta-analysis evaluation of 24 studies including 3109 patients and evaluating all described HPD criteria.14 Although these data are suggestive, there remains no validated definition of HPD. One major obstacle has been the inability of academic investigators to share data due to restrictive contracts with commercial entities, and the reluctance of many investigators to increase the complexity and costs of a clinical trial by adding an additional timepoint (prebaseline) requiring formal RECIST measurements.
HPD is still not properly captured in clinical trials nor during IO treatment. As noted above, reasons include the lack of a validated definition as well as the practical challenges in accessing prebaseline CT scans if previously on another therapeutic study that has contractual limitations on the usage of imaging or if IO drugs are used in the adjuvant or in the frontline setting. Moreover, in order to measure pre-immunotherapy and on-immunotherapy TGR or TGK, practical guidelines on when to define target and non-target lesions (at prebaseline or baseline?) are not available; in a therapeutic study, response is based on a comparison to baseline imaging and prebaseline scans may have different target and non-target lesions. Technical aspects of the different definitions of HPD, in relation to the potential use of non-target lesions or to the size of baseline diameter sizes, might lead to underestimation or overestimation of HPD.14
HPD needs to be formally evaluated, including the incidence, the impact on outcomes and ability to access other potentially effective therapies, and, most importantly, any predictive factors that might allow the identification of patients at highest risk. Regulatory agencies, pharmaceutical companies, oncologists and patient associations would then need to develop safety, reporting and management protocols to protect patients from the potential harmful effects of HPD. Actions might include: incorporating mandatory early HPD assessment in IO clinical trials; specific reporting of HPD as a grade 3–5 treatment-related adverse event; ensuring patients are adequately informed including in the informed consent document about the potential for HPD-related toxicity; incorporating HPD as a new category of immune-related tumor response (which is being assessed currently by the iRECIST subcommittee); and including this HPD information in the product labels of approved IO drugs.
As up to 40% of patients do not have accessible prebaseline scans,14 including patients that are treatment naïve (ie, first line for metastatic disease, and as adjuvant therapy), the identification of reliable biomarkers, other than conventional imaging, is high priority, both to identify HPD and other unusual responses during IO therapy, and to identify predictive factors. None has been seen so far and the biological rationale of HPD is still uncertain.23 Older age, higher number of metastatic sites, or molecular alterations such as murine double minute gene (MDM)2/MDM4 amplification or epidermal growth factor receptor have been postulated as being associated with HPD.24 Radiomics, allowing the evaluation of total tumor burden, and circulating tumor DNA or circulating tumor cells (CTC) burden dynamics may allow better definition and evaluation of HPD.
In the absence of more formal recommendations at present, patients treated with IO therapy should be carefully followed at the beginning of their treatment, when the risk of HPD is highest. Early onset of worsening cancer-related symptoms that suggests HPD should lead to earlier imaging re-evaluation so that patients with HPD can be switched immediately to other treatment. This is particularly relevant in the adjuvant setting.
Conclusions and next steps
Response-based end points, usually based on RECIST, such as response, progression, and time-dependent end points such as PFS or relapse-free survival remain important tools in the development of new anticancer therapies. Although initially developed for clinical trial usage, they are also increasingly used in clinical practice to guide treatment decisions in a less subjective manner.
The advent of highly effective IO therapies has brought new challenges with the identification of novel, or at a minimum, a higher incidence of, atypical patterns of response. Standardization and validation of the definition of these counterintuitive patterns of response, that is, pseudoprogression and HPD, observed in patients with cancer treated with IO drugs—especially the immune checkpoint inhibitors, is a critical, and yet unresolved issue in IO. These response patterns, especially HPD, may not be captured within standard response criteria and may have meaningful consequences in the treatment of our patients as well as from the perspective of clinical drug development.
Although there has been progress in developing and testing criteria to capture pseudoprogression and HPD, these have to date generally been limited to single-center or single drug/company evaluations, and it remains unclear whether any are universally applicable. Where it has been possible to combine data from multiple sources, for example, in the publications by the FDA, limitations include the number of different immune response criteria that were used, which are disparate, as some add new lesion measurements to the sum or measures identified at baseline while others do not, and vary in the number of lesions assessed as well as whether bidimensional or unidimensional measurements are collected.
Because of these challenges, 5 years ago the RWG developed and released the iRECIST recommendations in an attempt to standardize data collection and management. Shortly thereafter, the Hyperprogression Working Group of the Task Force on Methodology for the Development of Innovative Cancer Therapies25 was incorporated into the RWG iRECIST subcommittee. The iRECIST subcommittee is a multidisciplinary group of experts from the academia and the pharmaceutical industry. It is actively engaged on collecting data to enable the testing and validation of any modification to RECIST as well as validating the definition of HPD.
The RWG has successfully created two large data warehouses to test and validate RECIST and RECIST V.1.1. Although data sharing and collection is always challenging and requires the careful development of mutual agreements addressing the usage, including commitment not to re-analyze individual trials, it has been extremely challenging to do this for IO drugs, which has significantly delayed progress in this critical area to the disadvantage of the scientific community engaged in developing new anticancer therapies, and most importantly patients who are at risk of having to discontinue effective therapies (pseudoprogression), or be given or continue IO drugs that may shorten survival (HPD). Repeatedly, academic investigators have been unable to share anonymized data to address these important questions because of overly restrictive contractual obligations to pharmaceutical companies, despite clear patient consent. Similarly, major pharmaceutical companies who were active partner of RWG initially and who have adopted RECIST-based end points, are now unable or unwilling to consider such collaboration for IO drugs. RWG is also exploring novel approaches to elucidate whether an anticancer drug is working, such as radiomics, enhanced MRI, PET scanning, deep learning algorithms, and liquid biopsies, and developing collaborations; two international expert workshops are being conducted in early 2022 addressing these areas of interest.
The RWG is actively pursuing other mechanism of data sharing including, for example, sharing the information of a random subset of the total patients participating in a given clinical trial (to prevent re-analyses of the study) or a federated approach to data sharing—where the data do not travel from where it is located but the software analytical tool does. Ultimately, all interested parties—patients, academia, pharma, and regulators must have a shared understanding of the key importance of these questions in order to ensure successful collection of data so that better tools to evaluate and manage atypical response patterns can be developed and validated.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @j_lramon, @SallyCMLau
JLR-P, SS and SL contributed equally.
Contributors First three authors contributed equally to this work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.