Article Text

Abstract
Myeloid immune cells are frequently present in the tumor environment, and although they can positively contribute to tumor control they often negatively impact anticancer immune responses. One way of inhibiting the positive contributions of myeloid cells is by signaling through the cluster of differentiation 47 (CD47)/signal regulatory protein alpha (SIRPα) axis. The SIRPα receptor is expressed on myeloid cells and is an inhibitory immune receptor that, upon binding to CD47 protein, delivers a ‘don’t eat me’ signal. As CD47 is often overexpressed on cancer cells, treatments targeting CD47/SIRPα have been under active investigation and are currently being tested in clinical settings. Interestingly, the CD47/SIRPα axis is also involved in T cell-mediated antitumor responses. In this perspective we provide an overview of recent studies showing how therapeutic blockade of the CD47/SIRPα axis improves the adaptive immune response. Furthermore, we discuss the interconnection between the myeloid CD47/SIRPα axis and adaptive T cell responses as well as the potential therapeutic role of the CD47/SIRPα axis in tumors with acquired resistance to the classic immunotherapy through major histocompatibility complex downregulation. Altogether this review provides a profound insight for the optimal exploitation of CD47/SIRPα immune checkpoint therapy.
- macrophages
- phagocytosis
- adaptive immunity
- immunotherapy
- immunity, innate
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The cancer immunity cycle refers to the sequence of immune-related events that must be initiated to efficiently kill tumor cells in patients with cancer. First, cancer-specific antigens are released by (necrotic) cancer cells, which are subsequently engulfed and processed by dendritic cells (DCs). T cells that have been primed and activated by DCs in the lymph nodes will recognize and bind to these neoantigens presented on major histocompatibility complex (MHC) class I molecules with their complementary T cell receptor (TCR). Activated cytotoxic T cells will locate to the tumor site, recognize the tumor cells as foreign and kill the tumor cells.1 However, in patients with cancer, the immunity cycle is not adequately equipped to eliminate cancer cells.1 2
The upregulation of immune checkpoints is one mechanism of suppressing the anticancer immune response. Programmed death receptor 1 (PD-1)/programmed death ligand 1 (PD-L1) is one of the most well-studied immune checkpoints on T cells, and signaling through PD-1 on T cells negatively affects T cell function, such as T cell activation, cytokine production, division and survival.3 Other immune checkpoints have been identified, such as cytotoxic T-lymphocyte antigen 4 (CTLA-4), T-cell immunoglobulin and mucin-domain containing-3 (TIM3), lymphocyte activating 3 (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT),4 and their role in adaptive immunity has been described extensively and is a topic of active investigation. Importantly, myeloid cells are also negatively regulated within the anticancer immunity cycle, for example through the cluster of differentiation 47/signal regulatory protein alpha (CD47/SIRPα) axis.
CD47 is a glycoprotein that was first identified in 1990 as a protein that associates with integrins, hence its original name integrin-associated protein.5 CD47 is a member of the immunoglobulin superfamily consisting of an extracellular N-terminal single immunoglobulin V-like domain that contains multiple glycosylation sites, five membrane-spanning regions and a C-terminal intracellular domain.6 Among other proteins, such as SIRPy, thrombospondin-1 (TSP-1) and integrins, CD47 interacts with SIRPα.7–10 SIRPα consists of three extracellular immunoglobulin-like domains, followed by a single transmembrane segment and intracellular signaling domain with immunoreceptor tyrosine-based inhibition motifs (ITIM).11 12
While CD47 is expressed on virtually every cell type (see Protein Atlas ENSG00000196776-CD47),5 13 SIRPα is primarily expressed on the myeloid cell compartment, including monocytes, macrophages, granulocytes and subsets of DCs. Moreover, SIRPα can be found on T cells, intestinal epithelial cells, CD34 and CD133 positive hematopoietic progenitor cells and neurons.14–19 Interestingly, during progression from normal colon epithelium to colon carcinoma, SIRPα expression is upregulated on stromal cells.20
For SIRPα engagement to occur, the N-terminal SIRPα binding site of CD47 needs to undergo a post-transcriptional modification induced by glutaminyl-peptide cyclotransferase-like protein (QPCTL).21 22 Specifically, the glutamine residues located at the N-terminal part of the CD47 protein will be modified by QPCTL into a pyroglutamate group (pGlu-CD47), and this modification is required for CD47 to bind to SIRPα.21 Upon CD47 interacting with SIRPα, the intracellular ITIM domain of SIRPα becomes phosphorylated, leading to recruitment and activation of src homology region 2 domain-containing phosphatases, which negatively affect signal transduction pathways,23 such as inhibiting the phagocytosis of ‘self’ cells by cross-linking actin filaments, which prevents the phagocytic synapse formation on myeloid cells.24–26
Phagocytosis is a process in which a target cell is recognized, engulfed, processed and digested by a phagocyte, such as a macrophage. Phagocytosis depends on the balance of prophagocytic interactions, such as calreticulin/low density lipoprotein receptor-related protein 1(LRP1), self-ligand receptor of the signaling lymphocytic activation molecule family member 7 (SLAMF7), antibody/Fc receptor and inhibitory interactions (CD47/SIRPα, PD-1/PD-L1, MHC/leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1)) (reviewed in Feng et al27). Often, only removing the CD47/SIRPα ‘don’t eat me’ signal is not sufficient to induce phagocytose of the (cancer) target cell.28 Therefore, tumor-opsonizing antibodies have been combined with CD47/SIRPα blocking agents to provide a strong ‘eat me’ signal. Using this two-hit approach, the cells that highly express CD47 as well as the tumor-associated antigen will be preferentially targeted.28
Signaling through the CD47/SIRPα axis plays a role in various homeostatic processes, such as maintenance of erythrocytes,24 29 innate immune cells30 31 and T cells.32 33 Moreover, cancer cells28 34–36 and virus-infected and bacteria-infected cells can upregulate CD47 protein expression to prevent immune-mediated elimination.37 38 For example, during the progression of healthy colon epithelium to colon carcinoma, CD47 expression is upregulated on the epithelial cell compartment.20 CD47 blockade promotes myeloid-mediated elimination of cancer cells in preclinical models.34–36 Furthermore, blocking the interaction between CD47 and SIRPα promotes cancer cell elimination in vitro and in vivo28 34 36 39 40 as a monotreatment modality or in combination with immune-activating, tumor-opsonizing antibodies, such as rituximab or trastuzumab.21 40–44
Early clinical trials of anti-CD47 Hu5F9-G4 alone or in combination with rituximab showed promising results in patients with non-Hodgkin’s,45 46 with a total of 50% of patients with an objective response and 36% with a complete response.45 In the phase I ASPEN-01 clinical trial, evorpacept (CD47-binding domain of SIRPα fused to an inactive IgG Fc domain) was found to be safe as a monotherapy and combined with tumor-opsonizing antibodies trastuzumab in patients with solid cancer.47 Currently, there are multiple treatment modalities targeting the CD47/SIRPα axis alone or in combination with tumor opsonizers awaiting or undergoing clinical investigations, such as antibodies targeting CD47, including Hu5F9-G4 (magrolimab; NCT03248479, NCT04599634) and recombinant SIRPα-Fc (TTI-621; NCT02663518).
Although several clinical trials with CD47-targeting agents have shown impressive preliminary results, therapy-induced toxicity cannot be neglected, specifically anemia.45 48 CD47 antibodies can induce red blood cell (RBC) agglutination.43 49 50 This process of hemagglutination accelerates clearance of RBCs. The phase I evaluation of CC-90002, an anti-CD47 antibody, was recently published (NCT02641002),48 where the researchers performed a dose-escalation and dose-expansion study in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. CC-9002 enabled macrophage-specific tumor killing in preclinical in vivo models. However, monotherapy activity was not observed in the patients, and considerable toxicity was found in combination with the presence of antidrug antibodies (ADAs) in all patients, which resulted in discontinuation of the study. For the Hu5F9-G4 CD47 antibody, a priming dose was shown to effectively reduce anemia in patients with advanced cancer, with ADAs detected in 9.6% of patients.46 Thus, balancing antitumor efficacy with mitigating potential toxicity is a critical concern in the clinical application of therapeutic CD47 antibody therapy. Alternative strategies to block the CD47/SIRPα checkpoint, such as bispecific antibodies, anti-SIRPα antibodies and QPCTL inhibitors, may display decreased toxicities with high antitumor efficacy.43 50 51
The pioneering studies in the field of CD47/SIRPα were often conducted in immunocompromised animal models that lack B cells, T cells and natural killer (NK),34 36 40 posing the question whether there is a role of the adaptive immune system in the efficacy of CD47/SIRPα blockade. Recently, syngeneic models were implemented to unravel the role of the adaptive immune system and revealed that the innate checkpoint CD47 also induces T cell-mediated adaptive anticancer immune responses.52–55
In this perspective, we will discuss the role of CD47/SIRPα in relation to adaptive T cell responses and the potential therapeutic role of targeting the CD47/SIRPα axis in tumors with acquired resistance to classic immunotherapy via MHC downregulation.
Effect of CD47/SIRPα (blockade) on adaptive anticancer T cell responses
One of the hallmarks of an anticancer immune response is the dependency on T cells to recognize and kill tumor cells.1 2 It is becoming increasingly clear that upon CD47/SIRPα pathway inhibition, T cells can contribute to tumor control. These observations can either be a direct effect of the CD47/SIRPα pathway on T cells, or an indirect effect, for example through the ability of myeloid cells to activate and/or recruit CD8+ T cells.
Preclinical research showed that the CD47/SIRPα axis is involved in T cell homeostasis and/or activation.15 32 56–60 One of the first studies demonstrating that T cell immunity can be activated upon CD47/SIRPα blocking therapy in cancer was published in 2013 by the group of Weissman,52 who showed that macrophages can prime CD8+ T cell responses in mice upon CD47 blockade. The requirement of T cells in the efficacy of CD47/SIRPα blocking therapy was investigated in syngeneic tumor-bearing mice, which were injected subcutaneously with immunogenic CD47-expressing mouse B cell lymphoma cell line A20 or colon cancer cell line MC38.54 Administration of mouse anti-CD47 (MIAP301) or high-affinity SIRPα variant Fc fusion protein significantly reduced tumor growth. Tumor reduction was absent in BALB/c nude mice lacking a thymus upon CD47 blockade, suggesting that T cells are required. The use of T cell subset-depleting antibodies showed in this tumor model that CD8+ T cells, but not CD4+ T cells, were essential for the efficacy of CD47 inhibition in tumor-bearing mice.54 Multiple other studies have validated that CD8+ T cells are required for the efficacy of CD47/SIRPα blockade. Blockade of the CD47/SIRPα axis using an anti-SIRPα antibody (MY-1, IgG2a) inhibits renal cell carcinoma (RENCA) tumor growth in BALB/c mice,61 and an increase of T cells, and specifically CD8+ T cell subsets, in the tumor microenvironment was observed. Growth inhibition was abolished in CD8-depleted BALB/c mice. Furthermore, Granzyme B expression by tumor-infiltrating CD8+ T cells after radiation was higher in SIRPα-deficient mice when compared with SIRPα-proficient mice,62 and cytotoxic T cells isolated from SIRPα-deficient tumors were more able to kill tumor cells in vitro. Depletion of CD8+ T cells in this setting resulted in higher mortality of mice.62 In a similar fashion, the antitumor effect of CD47 blockade in irradiated melanoma or fibrosarcoma tumors in mice was shown to be dependent on CD8+ T cells.63 Furthermore, Manguso et al performed an in vivo screen with a clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9-mutagenized B16 melanoma cell line in mice that were devoid of T cells (Tcra−/−), in wild-type (WT) mice vaccinated with GVAX and in mice receiving GVAX with PD-1 blockade.64 They found in their model that CD47 on the tumors cells is important for T cell-mediated tumor evasion. Thus, CD8+ T cells contribute to tumor control in (cancer) mouse models where the CD47/SIRPα axis has been blocked.
CD47/SIRPα inhibition can induce T cell responses indirectly via the myeloid cell compartment
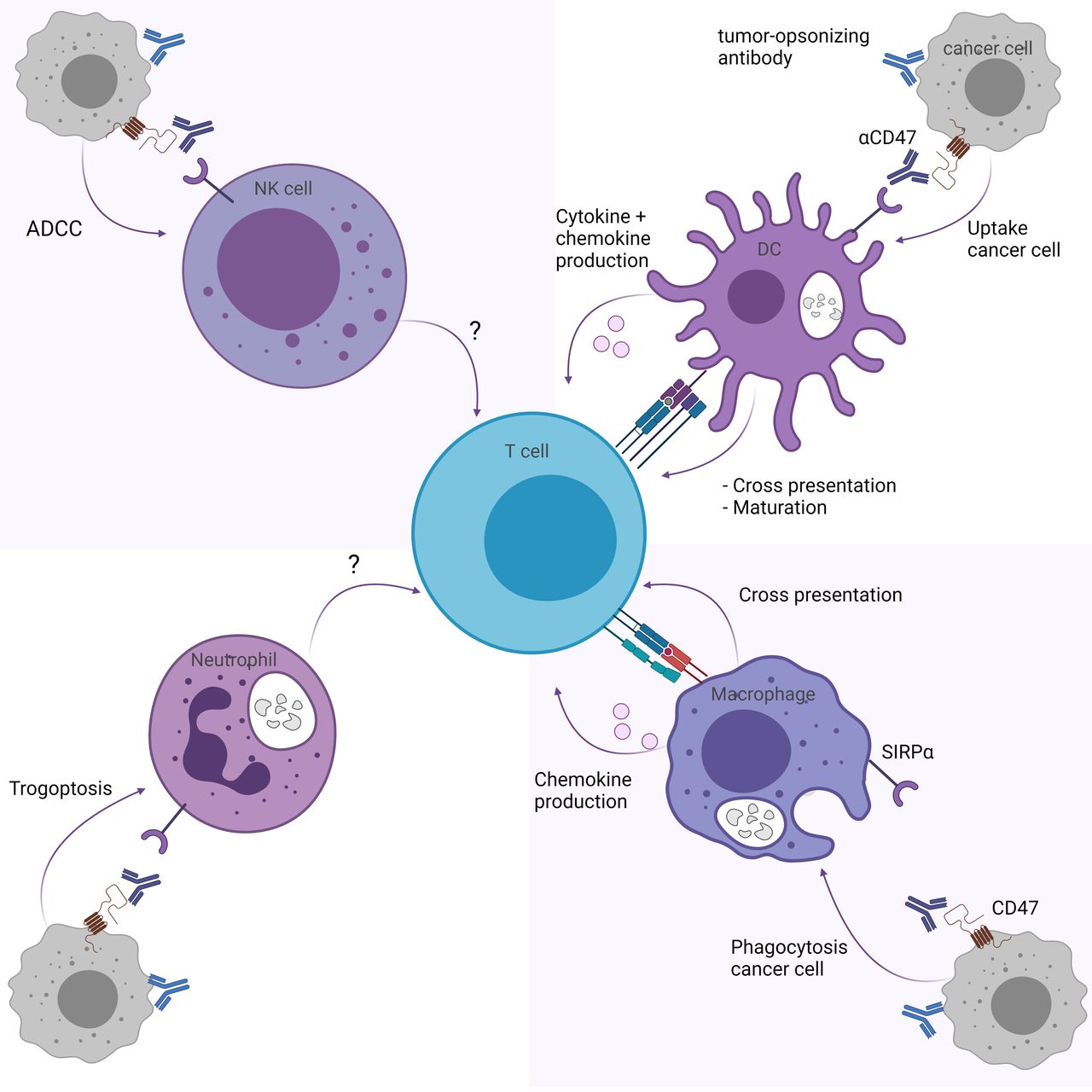
CD47/SIRPα pathway inhibition may influence T cell responses through myeloid immune cells, including DCs, macrophages, neutrophils and NK cells (figure 1). Here we describe the effect of CD47/SIRPα blockade on T cell responses for each myeloid subset.

CD47/SIRPα blockade indirectly enhances T cell responses through myeloid cells. CD47/SIRPα blockade enhances T cell responses through (upper right) the cross-priming and maturation abilities of DCs and production of T cell response promoting cytokines and chemokines, and (lower right) the cross-presentation and chemokine production of macrophages. While trogoptosis by neutrophils (lower left) and ADCC by NK cells (upper left) is promoted through CD47/SIRPα blockade, it is unknown whether this affects T cell responses directly. Figure created with BioRender.com. ADCC, antibody-mediated cellular toxicity; CD47, cluster of differentiation 47; DCs, dendritic cells; NK, natural killer; SIRPα, signal regulatory protein alpha.
Dendritic cells
DCs are professional antigen-presenting cells that can prime and activate T cells.65 Therefore, the role of DCs was assessed in the context of CD47 and SIRPα.53 54 Depletion of DCs using diphtheria toxin-mediated removal of CD11c-DTR bone marrow chimeras abolished the reduction of MC38 tumor volumes in C57BL/6 mice treated with anti-CD47,54 indicating that DCs are important cells in the efficacy of CD47/SIRPα blockade. Of note, CD11c is a marker that is also present on a subpopulation of macrophages and therefore the effects observed may in part be due to the loss of macrophages.38
Blocked SIRPα signaling on DCs results in increased cross-priming of OT-I CD8+ T cells and autologous T cells, as measured by increased interferon-gamma (IFNy) Elispot which improved tumor control after anti-CD47 treatment. Gauttier et al found that the classic DC type 2, but not type 1, significantly enhanced cross-priming of OT-I CD8+ T cells in the presence of P84 and MY1-G1 anti-SIRPα antibodies,55 indicating that not all DC subsets may be equally involved. Mechanistically, DCs can regulate the amount of degradation of engulfed tumor mitochondrial DNA through NADPH oxidase 2 (NOX2)-mediated regulation of the acidity levels in their phagosomes. By preventing SIRPα signaling on DCs, NOX2 is no longer inhibited by SIRPα, and higher pH levels are maintained through NOX2. Mitochondrial DNA can subsequently reside longer inside the phagosomes of DCs, increasing the chance that cytosolic DNA-binding protein cyclic GMP-AMP synthase (cGAS) will bind to the tumor DNA and becomes activated.53 54 66 Activated cGAS can then induce the stimulator of interferon genes (STING) pathway that leads to a type I IFN response, which in turn is required for DCs to cross-prime CD8+ T cells in the presence of anti-CD47 therapy.53 54 Although macrophages are more potent in their phagocytic abilities, they appear to lack the ability to regulate their pH levels in this manner.53
Beyond the fact that SIRPα signaling reduces the cross-priming capacity of DCs to activate cytotoxic CD8+ T cells,53 54 SIRPα ligation on activated human DCs using CD47-Fc results in a reduced cytokine production (interleukin (IL)-12, tumor necrosis factor (TNF)-α, IL-6), as well as reduced expression of maturation markers on these DCs.67 Indeed, SIRPα engagement on Staphylococcus aureus Cowan I-activated human monocyte-derived DCs by CD47-Fc prevents, but not fully blocks, their maturation in an IL-10 independent manner.68 These SIRPα-engaged, activated semimature DCs have a reduced ability to migrate and secrete chemokines, such as T cell-attracting chemokines chemokine (C-X-C motif) ligand (CXCL)9 and CXCL10, but retain their endocytosis ability. Furthermore, these cells were less capable of priming human-naïve CD4+ T cells in an in vitro coculture setting.68 Thus, SIRPα downstream signaling on DCs induced by CD47 binding may result in semimatured DCs less able to migrate to the tumor-draining lymph nodes and less efficient to induce T cell responses.68 Therefore, blocking CD47/SIRPα interactions may benefit T cell responses indirectly via DC maturation, cross-priming and cytokine production.
Macrophages
Macrophages are phagocytic cells with antigen-presenting features that have the capacity to activate T cells.69 As a high frequency of macrophages may be present in tumor microenvironments,70 the effect of CD47/SIRPα signaling blockade on macrophage-mediated T cell responses was assessed. In vivo depletion of macrophages significantly reduced the efficacy on tumor control of anti-SIRPα (MY-1) in syngeneic renal cell carcinoma-bearing BALB/c mice,61 indicating that macrophages are one of the effector cells in vivo. Tseng et al52 assessed the role of macrophages in the efficacy of CD47 blockade and its effects on T cell-mediated immune responses. Macrophages were subjected to anti-CD47 B6H12 antibodies and ovalbumin (OVA)-expressing cancer cells to induce antibody-dependent cellular phagocytosis. These macrophages were then cocultured with OVA-specific CD8+ T cells, resulting in an increased proliferation rate of T cells in the presence of anti-CD47 due to enhanced phagocytosis by the macrophage. Additionally, macrophages were subjected to OVA-expressing cancer cells with anti-CD47 antibody or IgG control and injected into mice that had received adoptive transfer of OT-I-specific CD8+ T cells. After challenging these mice with an OVA-expressing tumor, mice that received anti-CD47-treated macrophages were protected from tumor growth, while the IgG-treated control tumors grew out.52 Similarly, using multiple isotypes of bispecific antibody targeting CD47 and epidermal growth factor receptor (EGFR), human macrophages were capable of cross-presenting EGFR overexpressing K562 cells to cytomegalovirus (CMV)-specific autologous CD8+ T cells.71 Thus, CD47 blockade provides a vulnerability in cancer cells that enhances antibody-mediated phagocytosis by macrophages, which can in turn better cross-prime tumor antigens to T cells and thereby promote T cell responses. Blocking SIRPα signaling on myeloid cells using anti-SIRPα (MY1-IgG1 or P84-IgG1) in 4T1 breast cancer-bearing mice additionally resulted in the induction of chemotaxis-related genes on myeloid cells. Less secretion of chemokines might explain why T cells are less efficiently recruited to the tumor site after they have been activated.55 Furthermore, it seems that differential expression of SIRPα on human monocytes and macrophages isolated from patients with follicular lymphoma affects their function. CD14+ SIRPα high cells suppressed T cell function, while CD14− SIRPα low and negative cells activated T cell function.16 It thus appears that the CD47/SIRPα axis can influence macrophage-mediated anticancer T cell responses, potentially through reduced ability to phagocytose and cross-prime T cells when CD47 is blocked on a cancer cell or by reduced secretion of T cell-attracting chemokines when SIRPα signaling is blocked on the myeloid cells.
Neutrophils
Neutrophils are innate immune cells that have antitumorigenic properties through antibody-mediated cellular toxicity (ADCC).72 Matlung and colleagues72 described the specific mechanism of action of neutrophil-mediated ADCC, also known as trogoptosis. Neutrophils form CD11b/CD18 integrin-mediated conjugations with the opsonized target cell, and signaling downstream of the Fc receptor in neutrophils results in the lytic death of the target cells through internalization and fragmentation of the target cell membrane.72 Blockade of CD47/SIRPα interaction improves neutrophil-mediated trogoptosis induced by IgG42 43 71 72 as well as IgA73 opsonizing antibodies. Mechanistically, in the absence of CD47/SIRPα signaling, kidlin3 is no longer inhibited in the neutrophil and can therefore positively regulate CD11b/CD18 activation and strengthen the interaction required for trogoptosis induction.74 The question remains whether neutrophils that have engulfed target-cell fragments can activate CD4+ and/or CD8+ T cells. It has been reported that neutrophils that phagocytosed IgG-opsonized erythrocytes acquire antigen-presenting properties, such as the upregulation of MHC class II and costimulatory molecules, resulting in enhanced IFNγ cytokine secretion and proliferation of CD4+ T cells.75 However, in this model, the addition of anti-CD47 did not enhance the antigen-presenting capacity of neutrophils. Further research is warranted to determine whether neutrophil-mediated trogoptosis can prime T cell responses. Interestingly, neutrophils were found to be required to eliminate QPCTL knockout tumor cells and thus form important effector cells that are regulated by SIRPα in vivo. This was shown when human FcαRI transgenic BALB/c mice were injected with a 1:1 ratio of WT and QPCTL knockout murine Ba/F3-transformed cells expressing human Her2, after which they were subjected to opsonizing Her2 antibodies (IgA1) in the presence or absence of neutrophil depletion treatment (anti-Ly6G). While the anti-Her2 antibody alone caused a significantly lower ratio of QPCTL knockout tumor cells to WT tumor cells, when compared with untreated mice, this effect was largely abolished in the absence of neutrophils.21 Similarly, CD47 knockout or pharmacologically depleted (anti-CD47 B6H12 F(ab’)2) CD47 enhances the removal of Her2-expressing SKBR3 breast cancer cells that are opsonized with trastuzumab by neutrophils in vitro.41 Taken together, neutrophils are important effector cells that are highly effective when CD47 is blocked on tumor cells. Further research is required to elaborate on the role of these cells in contributing to T cell responses.
NK cells
NK cells influence T cell responses through secretion of cytokines and killing of target cells, resulting in increased cross-presentation of tumor cells by DCs and subsequent activation of T cell immunity.76 NK cells have inhibitory receptors that recognize the peptide-binding region of MHC class I molecules and thereby prevent the activation of NK cells. Loss of MHC class I on cancer cells results in NK cell activation and targeted killing through secretion of cytolytic compounds, such as granzymes and perforins.76 Naïve human and mouse NK cells express little to no SIRPα40 77; however, SIRPα expression is induced on mouse NK cells and primary human NK cells when exposed to IL-2, IL-15 and/or IFNα cytokines.78 Murine-induced embryonic stem cells that were deficient in MHC-I were eliminated by IL-2 or IL-15 activated mouse NK cells. However, the overexpression of CD47 protects these target cells from being targeted by SIRPα signaling on NK cells. The addition of CD47 blockade preventing SIRPα signaling on NKs to this setting stimulates activated NK-mediated removal of mouse-induced embryonic stem cells in vitro. In vivo depletion experiments revealed that CD47 blockade, which prevented SIRPα-mediated inhibitory functions of immune cells, was largely dependent on the presence of macrophages and not necessarily of NK cells.78 Contrary to this, renal cell carcinoma tumor-bearing BALB/c mice treated with anti-SIRPα MY-1 showed an increase in tumor-infiltrating NK cells compared with the untreated, and these cells were in part responsible for the observed reduction in growth of the tumor mass.61 The question still remains whether NK cells significantly contribute to the CD47 blockade efficacy in humans in general and specifically in indirectly inducing an adaptive immune response.78
CD47/SIRPα pathway directly affects T cell responses
While CD47/SIRPα blockade can indirectly enhance anticancer T cell responses via the myeloid cell compartment, it was shown that T cells themselves can express CD4779 and SIRPα.18 This suggests that the CD47/SIRPα pathway might also directly influence T cell responses and indicates that the situation may be more complex than anticipated.
CD47 is transiently expressed by antigen-specific CD4+ T cells
CD47 is expressed on T cells9 56 and the role and expression of CD47 were examined on CD4+ T cells during an immune response. BALB/c mice were immunized with complete Freund's adjuvant (CFA)-OVA 1 day after they received adoptive transfer of carboxyfluoroscein succinimidyl ester (CFSE)-labeled, OVA-specific CD4+ T cells,79 and the surface expression levels of CD47 on these cells were monitored over time. SIRPα-Fc, which binds to the QPCTL-mediated pGlu-CD47, was used to measure CD47 expression levels and showed that CD47 was transiently expressed on antigen-specific CD4+ T cells throughout the immune response (figure 2). The majority of cells have a high SIRPα-Fc binding capacity, which is lost during the first days after immunization and regained when the immune response ends.79 The cells that lose the ability to bind SIRPα-Fc are referred to as CD47low cells. These CD47low cells displayed low expression of CD127 and CD62L, indicative of an effector memory phenotype. A small proportion of OVA-specific CD4+ T cells remained positive for SIRPα-Fc (CD47high). Adoptive transfer experiments of effector CD44high T cells showed that only the CD47high cells, and not the CD44high CD47low or CD44low naïve T cells, were able to mount a memory response. This indicates that CD44high CD47high CD4+ T cells are precursors of memory T cells.79

Expression of pGlu-CD47 and pan-CD47 on CD4+ OVA-specific T cells during an immune response. Pan-CD47 expression does not alter during the immune response, while pGlu-CD47 surface levels decrease during the proliferation phase. Only a small proportion of cells maintain pGlu-CD47, which gives rise to memory T cells. Figure created with BioRender.com. CD, cluster of differentiation; OVA, ovalbumin.
A similar transient binding of SIRPα to CD47 was observed on human CD4+ T cells. Naïve T cells had a high capacity of SIRPα-Fc binding, effector T cells had a decreased SIRPα-Fc binding, and memory T cells had a high SIRPα-Fc binding capacity, similar to IL-2-stimulated T cells. This reduced SIRPα-Fc binding was not detected when TCR-activated CD4+ T cells were stained with two pan-anti-CD47 mAbs (B6H12, 2D3),80 indicating that surface expression of CD47 was not changed. Similarly, others found that SIRPα binding was upregulated in Concanavalin A (ConA)-stimulated human T cells, whereas surface expression of CD47 was not changed.9 Logtenberg et al21 showed that QPCTL inhibition reduces SIRPα-Fc binding to CD47, but not the overall level of CD47 measured by pan-anti-CD47 (B6H12, 2D3). Whether QPCTL is responsible for the observed effects on altered SIRPα binding on T cells is yet unknown. While CD47 expression is preserved on memory CD4+ T cells, it has also been implicated in the contraction phase of the immune response. Using an adoptive transfer model of antigen-specific naïve CD4+ T cells proficient or deficient for CD47 in WT mice, it was observed that the CD47-deficient CD4+ T cell population showed no contraction, unlike the population of CD47-proficient cells.80 This suggests that CD47 plays a role in CD4+ T cell contraction, potentially through induction of cell death.
Beyond SIRPα, CD47 also binds to integrins, TSP-1 and SIRPy, which increases the complexity of analyzing the effects of CD47 on T cells. Association of CD47 with β2 integrin results in a conformational change of adhesion molecules on T cells that facilitate their trans-endothelial migration.81 TSP-1 can bind to CD47 on CD3-activated T cells, resulting in T cell apoptosis in a caspase-independent manner33 82 83 and in suppression of T cell activation.84 85 SIRPy is highly expressed on human T cells (CD4+ as well as CD8+ T cells)9 and binds CD47 with approximately 10× lower affinity than SIRPα.9 86 CD47/SIRPy signaling appears to positively influence T cell responses as it promotes T cell activation86 87 and trans-endothelial migration of T cells.88 Blocking CD47 may therefore also impair the binding of other CD47 ligands that potentially directly (negatively) impact T cell responses.9 Of note, rodents lack SIRPy and are therefore not a good model to study the CD47/SIRPy interaction on T cells.9
Together, the data indicate that the capacity of SIRPα binding to CD47 is transiently regulated on antigen-specific CD4+ T cells, and most likely on CD8+ T cells, during an immune response and is linked to both contraction and formation of memory CD4+ T cells. However, potential involvement of other CD47 ligands, such as integrins, TSP-1 and SIRPy, cannot be excluded. Whether QPCTL underlies the conformational change of CD47 and whether small molecule inhibition of QPCTL positively or negatively affects memory formation and the contraction of T cells remains unknown.
CD8+ T cells express CD47 and a subset of CD8+ T cells express SIRPα
CD47 is highly expressed on CD8+ T cells located in tumorous and adjacent tissue obtained from patients with esophageal carcinoma.89 Furthermore, ConA-stimulated human peripheral blood mononuclear cells (PBMCs) (that activate both CD4+ and CD8+ T cells) showed that SIRPα binding was increased, while surface expression of CD47 was not changed.9 Whether CD47 expression is similarly regulated on CD8+ T cells during the immune response and has a function on CD8+ T cells, as was observed for antigen-specific CD4+ T cells, is still an unexplored field.79 80
It was long believed that SIRPα expression was restricted to the myeloid compartment. However, recently a subpopulation of antigen-specific CD8+ T cells were identified that express SIRPα.18 38 These SIRPα+ CD8+ T cells were found in mice chronically infected with Friend retrovirus or lymphocytic choriomeningitis virus (LCMV) Clone 13 and were also present in hepatitis C-infected humans. SIRPα expression highly correlated with PD-1 expression, and SIRPα+ CD8+ T cells displayed both activating and inhibitory receptors. Functionally, antigen-specific SIRPα+ CD8+ T cells display an enhanced ability to proliferate, secrete IFNy and show increased cytolytic potential compared with their SIRPα− counterparts during acute and chronic viral infection. The status and function of CD47 on these CD8+ T cells are currently unknown. Blockade of CD47 by an antibody or using SIRPα-deficient T cells in this LCMV model did not affect T cell functionality,38 indicating that in this model CD47 blockade is not dependent on SIRPα signaling on CD8+ T cells. Interestingly, PD-L1 blockade significantly increases the T cell granulation marker CD107 on virus-specific SIRPα+ cytotoxic T cells.18 It would be interesting to investigate whether SIRPα+ T cells are also present and functional in patients with cancer, a disease in which chronic antigenic stimulation and exhaustion of T cells in the tumor microenvironment also occur. One could hypothesize that tumor-specific SIRPα+ T cells bind the tumor cells via CD47/SIRPα axis and thereby further enable the T cell to kill the tumor cell through its enhanced cytotoxic potential. On the other hand, like PD-1 expression, SIRPα may be upregulated on activated T cells to limit T cell function. It would therefore be worthwhile to explore if CD47 or SIRPα blockade has a positive or negative effect on these SIRPα+ cytotoxic T cells. In the situation where SIRPα expression on T cells facilitates T cell killing, CD47/SIRPα blockade may not be beneficial, whereas blocking the potential negative regulator SIRPα may be advantageous in terms of directly promoting adaptive anticancer immune responses.
Together, SIRPα marks a subset of cytotoxic T cells with enhanced functionality in chronic virus-infected humans and mice. The presence and potential function of these CD8+ T cells in patients with cancer remain unknown.
CD47/SIRPα pathway inhibition combined with PD-1/PD-L1 axis blockade improves tumor phagocytosis and attracting T cells
CD47/SIRPα blockade as a monotherapy has shown promising results in specific preclinical and clinical studies. However, CD47/SIRPα blockade is more effective when potentiated with additional therapies, such as tumor-specific, ADCC-inducing antibody therapy. There is also a rationale to combine innate immune checkpoint CD47/SIRPα blockade with immune checkpoint PD-1/PD-L1 blockade. First, macrophages isolated from colorectal cancer-bearing mice and humans express PD-1. PD-1 signaling on tumor-associated macrophages was inversely correlated to their ability to phagocytose,90 and PD-L1 deficiency on tumors promotes the ability of PD-1+ macrophages to phagocytose target cells. Second, both PD-L1 and CD47 are induced by oncogene MYC,91 which is a driver oncogene in multiple cancer types, and by hypoxia-inducible factors (HIF)-1/hypoxia.92 Therefore both these inhibitory markers may be present in MYC-driven or hypoxic tumors. Chemotherapy-induced expression of both CD47 and PD-L1 via HIF-1α/2α suppresses both innate as well as adaptive immune cells, thereby facilitating therapy-induced immune resistance.92 Third, resistance to vaccination immunotherapy was associated with an impaired infiltration of proinflammatory myeloid cells in mice and humans,93 indicating that myeloid cells are under certain circumstances required for optimal (therapy-induced) CD8-mediated immune responses. Fourth, SIRPα expression follows the expression pattern of PD-1 on virus-specific CD8+ T cells isolated from LCMV-infected mice.18 Lastly, PD-L1 blockade enhances the degranulation of virus-specific SIRPα+ cytotoxic T cells,18 which may also be applicable in anticancer immunity. Thus, blocking both CD47 and PD-1/PD-L1 signaling simultaneously may have synergistic effects in promoting an antitumor response, and therefore studies have focused on the combination of both checkpoint blocking agents in cancer-bearing mice.
Anti-PD-L1 and anti-SIRPα (MY1-G1) synergistically inhibited tumor growth of MC38 in syngeneic mice compared with mice treated with each therapy separately, and improved survival rates were observed in the combination-treated mice compared with untreated Hepa1.6 hepatocellular carcinoma-bearing mice.55 Similarly, MC38 tumor growth in C57BL/6 mice was inhibited by CD47-targeting CV1-Fc and anti-PD-L1 atezolizumab as separate treatments. However, the greatest reductions were observed when a bispecific antibody that targets both CD47 and PD-L1 was used.94 This effect was partly abrogated when either macrophages or CD8+ T cells were depleted in this setting.94 Not only cells that are sensitive to CD47/SIRPα blockade, but also cells that are insensitive to this treatment appear to benefit from combination therapy. CT26-bearing mice were subjected to anti-SIRPα (My-1), anti-PD-1 or a combination thereof. While CT26 is insensitive to anti-SIRPα alone (MY-1), the combination with anti-PD-1 led to significant growth inhibition of CT26 cancer cells compared with mice treated with anti-PD-1.61 The mechanism that underlies growth inhibition was not studied in more detail. In a similar fashion, a significant reduction of B16F10 mouse melanoma tumor size, a tumor insensitive to both therapies separately, improved survival significantly compared with PD-L1 therapy alone.95 Thus, it appears tumor-bearing mice can benefit from CD47/SIRPα blockade combined with PD-1/PD-L1 blockade.
Further assessments were conducted to investigate whether T cells may be relevant effector cells induced by this form of combination treatment. Depletion of CD8+ T cells showed a partial dependency of those cells for the observed growth reductions achieved by the CD47/PD-L1 bispecific antibody.94 Additionally, histochemical analysis of the tumor microenvironment of Hepa1.6-bearing mice showed a significant increase of tumor-infiltrating T cells in mice that were treated with anti-PD-L1 and anti-SIRPα compared with anti-PD-L1-treated mice.55 Thus, CD47/SIRPα blockade combined with PD-1/PD-L1 blockade synergistically inhibits tumor growth in vivo, which may be in part explained by the enhanced T cell attraction and/or recruitment inside the tumor.
Because of the promising preclinical in vitro and in vivo data, there are currently multiple clinical trials combining CD47/SIRPα blockade together with other forms of checkpoint blockade, such as nivolumab or pembrolizumab (table 1). Recently, a phase I clinical trial was published that showed that the combination of evorpacept (CD47-binding domain of SIRPα fused to an inactive IgG Fc domain) with pembrolizumab (anti-PD-1) was safe to use in solid tumors.47 It will be very interesting to unravel whether the combination of both CD47/SIRPα and PD-1/PD-L1 blockade has synergic effects in patients with advanced cancer.
Registered clinical trials that investigate the combination of CD47/SIRPα blockade together with T cell immune checkpoint PD-1/PD-L1 blockade
CD47/SIRPα blockade in targeting intrinsic immune resistance to CD8-mediated immunotherapy
Primary resistance to CD8+ T cell-mediated adaptive responses can occur due to the intrinsic properties of cancer cells. Cancer cells may also develop acquired resistance under the pressure of (immune)therapy, which will together be referred to as intrinsic immune resistance. Intrinsic immune resistance is often facilitated by the downregulation of MHC class I on the surface of cancer cells.1 96 97 Indeed, downregulation of MHC class I and II and β2 microglobulin was found in PD-1/PD-L1-resistant cancer cells compared with the parental cells98 99 and in patients with cancer refractory to PD-1/PD-L1 blockade through mutations in IFN receptor signaling and antigen processing pathways.97
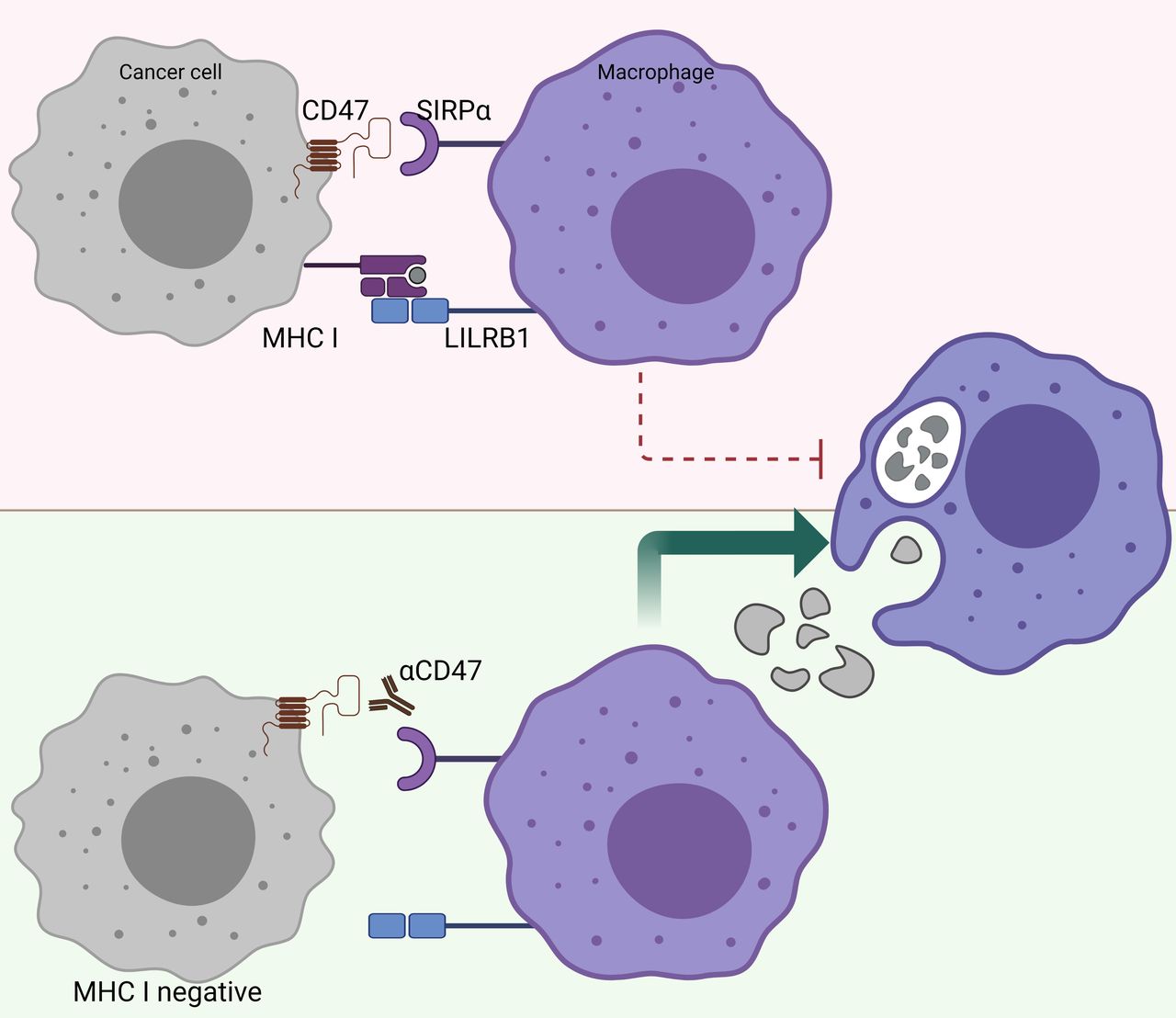
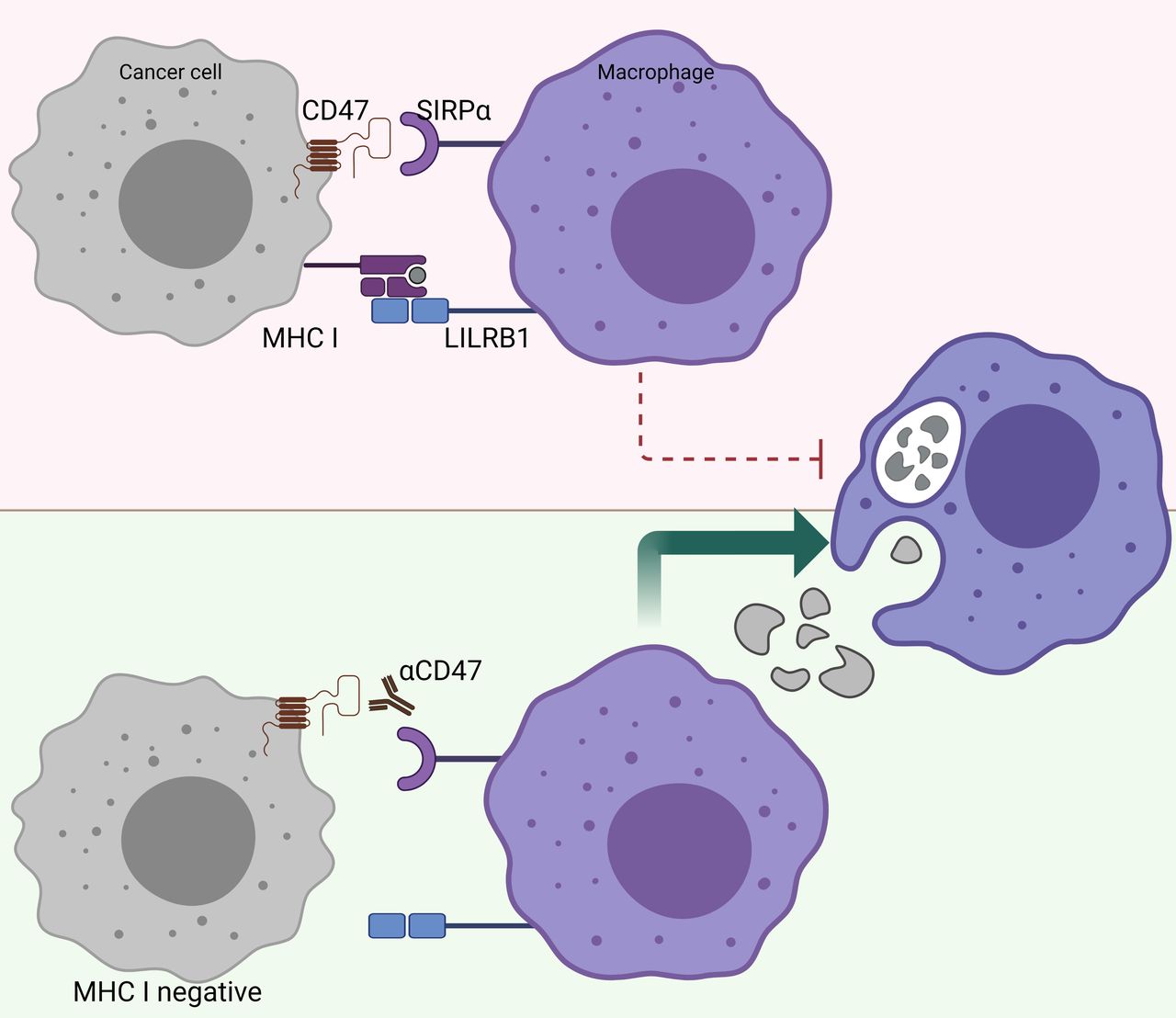
There is a reverse correlation between the sensitivity to phagocytosis induced by anti-CD47 and the expression levels of MHC class I.100 In other words, tumors that highly express MHC class I are more resistant to phagocytosis induced by anti-CD47 (figure 3). Further research revealed that MHC class I is recognized by LILRB1 on human macrophages, which results in a phagocytic inhibitory signal.100 Indeed, MHC-deficient, human-induced pluripotent stem cells are selectively removed by macrophages in vitro and in vivo, and overexpression of CD47 in these target cells prevents this macrophage-mediated elimination.78 This finding may have great implications for the treatment of patients who become resistant to immunotherapy, such as PD-1/PD-L1 blockade, due to loss of MHC class I expression.97 Further investigations are warranted to unravel whether CD47/SIRPα may be a suitable treatment for patients who acquire resistance to other forms of immune checkpoint blockade. Taken together, CD47/SIRPα blocking modalities may be a suitable therapy for patients who have acquired classic immune resistance due to loss of MHC class I expression.
{kind=link}
{kind=link}
{kind=link}
MHC-I-deficient tumors are sensitized to CD47/SIRPα blockade. The inhibitory receptor LILRB1, which binds surface MHC I, shows synergy with other inhibitory receptor pathways, including the CD47/SIRPα axis. Figure created with BioRender.com. CD47, cluster of differentiation 47; LILRB1, Leukocyte immunoglobulin-like receptor subfamily B member 1; MHC-I, major histocompatibility complex I; SIRPα, signal regulatory protein alpha.
Conclusion
Immunotherapies that target the CD47/SIRPα axis have shown very promising preclinical outcomes, and results from early clinical testing are also highly encouraging. Specifically, combination strategies that include blockade of the CD47/SIRPα axis are attractive routes to enhance the therapeutic efficacy needed for disease elimination in patients, as evidenced by the number of clinical trials.
Preclinical data show that T cells can be required for the efficacy of CD47/SIRPα blockade, which may be due to direct effects on T cells or indirectly through other immune cell subsets. Currently, the effects of CD47/SIRPα checkpoint blockade on T cells are unknown in a clinical setting. For optimal exploitation of this myeloid checkpoint, it is important to fully understand the role of T cells in relation to CD47 and SIRPα checkpoint inhibition and further investigation is warranted.
Acquired resistance to classic CD8+ T cell-mediated immunotherapies is an expanding clinical problem directly affecting their success and thus the survival of patients. As these patients have no alternative therapies left, targeting acquired resistance is a clear unmet clinical need. It is of high interest that therapy-resistant tumors, through downregulation of MHC class I, are specifically vulnerable to CD47/SIRPα interfering modalities. An optimal usage of CD47 or SIRPα checkpoint therapy for acquired resistant tumors is therefore likely to increase the breadth of clinical application.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors AvD researched data for the article. AvD, SHVdB and FS jointly discussed the data and cowrote the article.
Funding AvD and FS received funding from the Dutch Cancer Society (KWF grant 12629).
Competing interests FS is inventor on a patent application that covers manipulation of the CD47/SIRPα axis via QPCTL.
Provenance and peer review Not commissioned; externally peer reviewed.